
Abstract
The synthesis and structural characterisation (Fourier transform infrared, FTIR spectrometry, scanning electron microscopy, SEM and energy-dispersive X-ray, EDX) of amino-modified silicates (unloaded L1, and aspirin-loaded, L2) are reported. The optimal conditions for the extraction of aspirin from water by the modified silicate material were determined as a function of the mass of the extracting agent and the pH of the aqueous solution. The optimum mass was found to be 0.08–0.10 g with 99.9% removal of aspirin. Maximum extraction of aspirin by the material was observed at pH 4. The kinetics, the removal capacity of the material, as well as its recycling, were investigated. The results indicate that (i) the process is fast and (ii) the removal capacity for the drug is greater than that of previously reported materials and (iii)the modified silicate can be easily recycled. These data along with the low cost involved in the production of the material led to the conclusion that the modified silicate has the required potential for industrial use. Molecular simulation calculations suggest that one unit of aspirin interacts with one unit of the modified silicate L1 through hydrogen bond formation between the amine functional group of the silicate and the oxygen donor atoms of aspirin. Final conclusions are given.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Contamination of drinking water by pharmaceuticals is a topic which has gained considerable interest in recent years. The importance of this research area has been emphasised in the report published by the World Health Organization (WHO) in 2012 which addressed several issues such as the presence of pharmaceuticals in water, their risk to human health, a variety of technological approaches for their removal, as well as prevention methods concluding remarks and recommendations were given. (WHO 2012). An important aspect to consider is that regarding the development of innovative technologies for their removal. Aspirin (acetyl salicylic acid) (Fig. 1) is one of the analgesics most frequently used for reducing pain, inflammation, heart attacks prevention, angina and strokes (Albert 2010; Mundasad 2016). However, its excessive use has led to a high degree of water contamination from point and non-point sources (Nicolaou et al. 2007). Recently, a number of studies have reported the use of different approaches for the removal of aspirin from water. Such approaches included photocatalysis using titanium dioxide (Li et al. 2018), graphene nanoplatelets (Al-Khateeb et al. 2014), electrocoagulation (Ozyonar and Aksoy 2016), and membrane separation technology (Khamis et al. 2011).

Chemical structure of aspirin
Several research groups have used mesoporous silica for removing pharmaceuticals including aspirin from water (Bui and Choi 2009; Punyapalakul and Sitthisorn 2010; Kim et al. 2014; Akhtar et al. 2016).
In an attempt to explore the field of supramolecular chemistry, Danil de Namor et al. (2017) reported the selective interaction of a calix[4]arene derivative, 5,11,17,23-tetra-tert-butyl, 25,27-bis[amino ethoxy]26,28-dihydroxycalix[4]arene with clofibric acid, diclofenac and aspirin. It was demonstrated that the calix[4]-based receptor is a suitable extracting agent for the two former drugs but its extracting ability for aspirin was found to be very weak. Indeed the thermodynamic characterisation of these systems gave a quantitative assessment not only of the selective recognition of the calix[4]-based receptor for clofibric acid and diclofenacb but also about its hosting ability for these drugs (two drug units per unit of receptor) while a 1:1 (drug: receptor) interaction was found for aspirin. Therefore, it was thought that for technological purposes, silica is a naturally occurring material of suitable strength and structure which can be easily modified by the introduction amino functionalities, in its structure as to produce a potential material for the removal of aspirin from water. This paper reports (i) the synthesis and characterisation of modified silicate materials L1 and L2, (ii) FTIR, SEM and EDX of the unloaded and aspirin-loaded modified silicates (iii) the optimal conditions for the removal process (iv) the advantages of the modified silicate relative to previously reported material for removing aspirin from water are discussed.
Materials and methods
Materials
Silica (0.2–0.5 mm), 3-aminopropyltrimethoxysilane (97%), 2-diethylamino ethyl chloride hydrochloride (99.5%) and aspirin were all purchased from Aldrich Chemical Co. Aspirin (99%) was purchased from Sigma Aldrich.
Methanol (HPLC), dried toluene, hydrochloric acid, Ethanol (HPLC), chloroform, ammonium hydroxide, triethylamine and formaldehyde (37%) were purchased from Fisher.
Preparation of modified silicate compounds
This experiment involved the preparation of two modified silicates which would be used to capture and extract acetylsalicylic acid in aqueous solution.
Synthesis of modified silicate, L1 (Ho et al. 2003)
Silica (0.2–0.5 mm size) was activated overnight in an oven at 400° C and then cooled down to room temperature. The activated silica was suspended in freshly refluxed toluene (150 cm3) after which 3-aminopropyltrimethoxysilane (APTMS) (10 cm3, 54.30 mmol) was added. The mixture was refluxed with vigorous stirring under a nitrogen atmosphere for 48 h. The mixture was then cooled to room temperature, filtered and washed with methanol and toluene. The product was dried in a piston dryer at 110 °C for 20 h. Microanalysis was carried out at the University of Surrey for L1: Found %: C, 7.83; H, 1.95; N, 2.09. The synthetic procedure is shown in Scheme 1. (Ho et al. 2003).

Synthesis of modified silicate, L1
Attachment of diethylamine to the silicate, L2 (Blasius et al. 1980)
The 2-(diethylamino) ethyl chloride hydrochloride (2.2833 g, 15 mmol) was placed in a 250 cm3 round bottom flask and a mixture of ethanol and chloroform (100 cm3: 100 cm3) was added, followed by triethylamine (2.1 cm3, 15 mmol) and formaldehyde (2.4 cm3, 30 mmol). The mixture was stirred for one hour followed by addition of modified silica (5 g). This mixture was refluxed for 24 hours at 60–70 °C. Then, it was cooled down and filtered to collect the solid. The resulting compound was then washed with a mixture of ethanol and chloroform and then dried overnight in a piston dryer. Scheme 2 shows the synthetic procedure to produce the modified silicate, L2.

Attachment of diethylamine group (product: N,N-Diethyl-N'-propyl-methanediamine dimethoxysilane), L2
Characterisation of L1 and L2 (loaded and unloaded)
Molecular modelling
A molecular modelling of the interaction of L1 with aspirin was carried out using Argus lab 4.0.1 software. The minimum potential energy was calculated by geometry convergence function using the software and performed according to Universal Force Field (UFF) calculation method.
FTIR analysis
The chemical surface properties of the modified silica compounds were determined by Fourier transform infrared (FTIR) spectrometry, using Agilent Cary 600 Series FTIR spectrometer. FTIR spectra were recorded by averaging 32 scans at a spectral resolution of 4 cm−1 in the wavelength region from 4000 to 600 cm−1.
Scanning electron microscopy and energy-dispersive X-ray (SEM–EDX)
A scanning electron microscope (SEM) JEOL JSM-7100F, equipped with secondary and backscattered imaging coupled with Ultradry energy-dispersive X-ray (EDX) for elemental analysis, was used for morphological investigations. L1 and L2 untreated and treated with aspirin were mounted on aluminium stub, sputtered with thin layer of gold to prevent charging of the surface. Working conditions were 15 keV for accelerating voltage and 10 mm for the detector working distance.
Extraction of aspirin using L1
Effect of the amount of L1 on the extraction of aspirin from aqueous solution
To study the effect of the mass of the material on the uptake process, different masses of L1 (0.01–0.2 g) were added to a fixed concentration of aspirin in water (5.59 × 10–3 mol.dm−3) The mixtures were mixed well by whirlimixer and left overnight in a water bath at 298 K. Each solution was filtered and filtrates were analysed by UV–Vis measurements where the used λmax for the aspirin was 276 nm. The percentage of extraction, % E, is calculated according to Eq. 1.
In this equation, ci and ce are the initial and equilibrium concentrations of aspirin before and after extraction, respectively, in mol.dm−3.
Effect of pH on the uptake of aspirin at 298 K
The effect of pH on the extraction of aspirin by L1 was tested by using a fixed amount of the material (0.08 g) and a volume (10 cm3) of an aqueous solution of aspirin (5.59 × 10–3 mol.dm−3) in the 2–12 pH range. The desired pH of the solution was attainable by adding hydrochloric acid (HCl) or ammonium hydroxide (NH4OH) solution. Buffer solutions of pH 4 and 9.2 were used for calibration. The percentage of extraction, % E, is calculated using Eq. 1.
Kinetics of removal of aspirin from aqueous solution at 298 K
In order to assess the kinetics of the removal processes, separate fixed amounts of L1 (0.08 g) were mixed with aqueous solutions of aspirin (5.59 × 10–3 mol dm−3) at different contact times ranging from 15 to 1080 min at 298 K, while other parameters such as pH (~ 4), temperature (298 K) were kept constant.
Determination of the uptake capacity of the materials
Batch experiments were carried out at 298 K by varying the concentrations of aspirin (1.00 × 10–3 – 1.00 × 10–2 mol dm−3) with a fixed mass of the material (0.08 g) in order to determine the capacity of the material to extract aspirin from aqueous solution.
The capacity q (mmol g−1) amount of aspirin taken up per unit of mass of material is calculated from Eq. 2.
In Eq. 2, ci and ce are the initial and equilibrium concentrations of aspirin before and after extraction, respectively, in mol.dm−3, \(m\) is the mass of the material used in gram (g) and \(V\) is the volume of the solution in dm3.
Extraction experiments of aspirin in aqueous solutions using batch approach were carried out in duplicate and average values are reported.
Recycling of the loaded silicate
The material was recycled by a pH switching mechanism using HCl 0.1 M. The modified silicate loaded with aspirin (5 g) was placed in a column and treated with an aqueous solution of HCl 0.1 M until the amount of aspirin in the effluent was undetectable. Then, the material was washed with deionised water and dried in an oven at 110 °C and its capacity was determined. This operation was repeated ten times.
Results and discussion
Molecular modelling
The interaction of aspirin was simulated using L1 as a representative example. The aim was to gain suggestions regarding the forces involved in the interaction of the modified silicate and aspirin. Figure 2a, b shows a subtle stability detected through the H-bond formation between the –NH functional group of L1 and the oxygen donor atom of aspirin which indicate a 1:1 binding mode. From the energetic perspective, aspirin-modified silicate interaction is enthalpically stable (− 16.58 kJ.mol−1). These suggestions were corroborated by FTIR analysis as discussed below.

Molecular simulation for the interaction of L1 with aspirin and its computed interaction energy (a). Hydrogen bonds designated by green lines
FTIR analysis
FTIR spectroscopy analysis was carried out for L1 and L2 (unloaded and aspirin-loaded materials) as shown in Fig. 3a, b, respectively.

FTIR spectra of unloaded and aspirin-loaded L1 and L2 materials
For the unloaded material, strong and sharp bands observed in Fig. 3a correspond to Si–O–Si (1040 cm−1), Si–O (800 cm−1) and N–H (1640 cm−1) (Parida et al. 2006; Hernandez-Morales et al. 2012). The peak at 980 cm−1 is attributed to Si–OH bond stretching and the stretching and bending vibrations of the amino propyl functionality (Zhang et al. 2008). The presence of CH2 is reflected by the peak observed at 1380 cm−1.
The shift observed in the NH peaks of the loaded material shown in Fig. 3a indicates the involvement of this functional group on the uptake of aspirin which is in accord with the suggestion given in the molecular modelling. The slight shifts observed in the Si–O–Si and Si–O peaks are attributed to the configurational rearrangement of L1 upon interaction with aspirin.
Figure 3b shows similar peaks positions for L2 functional groups. However, in the presence of aspirin, no significant shifts were observed
Analysis of L1 and L2 unloaded and aspirin loaded by SEM–EDX
The morphology and the surface of L1 and L2 materials untreated and treated with aspirin were analysed using the SEM–EDX technique where micrographs and spectra are presented in Fig. 4. L1 and L2 morphologies (Fig. 4a, c) appear to consist of macro-particles with smooth large surfaces. The morphology of both materials does not seem to be altered by the presence of aspirin. However, an increase in the level of carbon and oxygen peaks in the EDX spectra (Fig. 4b, d) is observed suggesting the presence of aspirin in the materials. Moreover, smaller, white patches were observed on L1 and L2 micrographs indicating the uptake of aspirin by the investigated materials.

SEM images and EDX spectra showing the microstructures and the elemental composition of L1 (a); L1 treated with aspirin (b); L2 (c) and L2 treated with aspirin (d). Au peak referred to the gold coating of the samples
The outcome of the IR and SEM–EDX is indicative that the interaction of L1 with aspirin is stronger than that for L2. This is an unexpected result given that the tertiary amine is more basic the primary amine. This must be attributed to the fact that the primary amine in L1 is more available to interact with the drug than the tertiary amine in L2. Therefore, L1 was the material selected for investigating its use as extracting agent for the removal of aspirin from aqueous medium.
Removal of aspirin from water using modified silicates
Many factors need to be considered in the removal of pollutants from water. Among them, the mass of material, the pH of the aqueous solution, the kinetics of the process and the removal capacity of the material for the targeted pollutant were investigated with the aim of establishing the optimum conditions for removing aspirin from water by L1. In addition for commercial purposes recycling of the material, a key parameter was considered.
Determination of optimum amount of L1 for the uptake of aspirin from water at 298 K
Figure 5 shows that as the amount of L1 increases, the extraction percentage (% E) increases until saturation occurs and this is due to the increase in the sites of interaction (amino active sites) in the material. It is concluded that the optimum mass to be used for almost complete removal of aspirin from water by L1 is between 0.08 to 0.10 g.

Effect of masses on the uptake of aspirin from aqueous solution by the L1 at 298 K, Ci = 5.59 × 10–3 mol dm−3, V = 10 cm3 at 298 K
Effect of pH of the aqueous solution on the removal of aspirin by L1 at 298 K
Figure 6 shows the effect of varying the pH of the aqueous solution of aspirin against the % E. At pH lower than the pKa of aspirin, the undissociated drug predominates in aqueous solution. At pH = 3.5, the concentrations of the undissociated and dissociated drug are the same. At pH higher than 3.5, the dissociated species predominates in solution. The results shown in Fig. 6 clearly indicate that hardly any extraction of the pharmaceutical takes place at low pH. This is due to the protonation of the amino group in L1 which limits the availability of the binding sites of the material. As the pH increases and ionic species predominate in aqueous medium, the percentage of aspirin removed by the modified silicate increases until the material is fully saturated and no further changes are observed. Another outcome of this investigation is the fact that at lower pH, there is hardly any extraction of aspirin. This finding led us to explore the possibility of recycling the material via a pH switching mechanism as described in the final part of this paper.

Effect of pH of the aqueous solution on the removal of aspirin from water (5.59 × 10–5 mol dm−3, 10 cm3) by L1 at 298 K
Kinetics of removal of aspirin by L1
The purpose of this experiment is to assess the kinetics of the extraction process which is an important aspect to be considered for industrial use. The experimental data for L1 were fitted to a simple exponential model assuming that the rate of the uptake of the materials is proportional to the distance from equilibrium (i.e. pseudo-first-order process). Figure 7 shows that the obtained rate of equilibrium (k) for aspirin removal was found to be 0.05 with half-life of 14 min for L1. It is therefore concluded that the kinetics of the process is fast.

Determination of the optimum time for the uptake of aspirin by L1 from aqueous solution at 298 K
Determination of the capacity of L1 for the removal of aspirin from water at 298 K Comparison with other materials previously reported
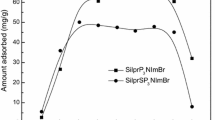
The uptake in mmol.g−1 of L1 against the molar concentration of aspirin is shown in Fig. 8. The results show that the capacity of L1 to remove aspirin from aqueous solution is 1.27 mmol g−1 (228.79 mg.g−1). The results are striking and indicate that L1 is an efficient material for the removal of aspirin from water to an extent that its capacity to remove this drug from water is much higher than those capacities of materials previously reported as shown in Table 1 including those reported by Mphahlele and co-workers (Mphahlele et al. 2015). The materials consisted of polymerised nanotubes containing nitrogen and cyclodextrin (N-NT- CD) and iron (Fe/N-NT-CD). The results showed that the capacities of the materials to extract aspirin were 71.9 and 101 mg.g−1, respectively (Mphahlele et al. 2015). An additional advantage of L1 relative to the latter is the reduction of steps required to synthesise the material and the consequent reduction in cost and time. Graphene nanoplatelets were also used for the extraction of aspirin by Al-Khateeb and co-workers (Al-Khateeb 2014) and a capacity of only 12.98 mg.g−1 was found (Datt et al. 2012). The results show that the capacity of L1 is greater by factors of 2.54, 3.01, 4.22, 3.75 and 3.87 mg.g−1 relative to MS, MS-CC-1, MS-CC-2, MS-PS-1 and MS-PS-2, respectively. These data demonstrate again that the silica-based material reported in this paper is characterised by a much higher removal capacity and lower cost (18 US dollars per 10 g of L1) than those silica-based compounds reported in the literature (Datt et al. 2012). In addition to the number of steps, time consumption and chemicals needed for its preparation are significantly reduced.

Uptake capacity (mmol.g−1) of L1 to remove aspirin from aqueous solution at 298 K
Recycling of L1
Figure 9 is a plot of the removal capacity of the material after recycling the material ten times using 0.1 mol.dm−3 HCl a reduction of 18.6% (53.71 mg/g) in its capacity is observed which means that still after this number of recycles, this material has a much higher capacity (175.09 mg/g) than existing ones. It is quite clear from these preliminary studies that there is a scope for further recycling processes before reaching the capacity of the unrecycled materials reported in the literature.

Capacity of the material (mg.g−1) for aspirin after several recycling
Conclusion
From the above discussion, the following conclusions are drawn:
-
(i)
This paper shows the importance of enriching naturally occurring materials for the development of low-cost methodologies for the removal of pharmaceuticals from water, particularly those which are frequently used and therefore most likely to contaminate water.
-
(ii)
The suggestions given from molecular simulation calculations regarding the possible sites of interactions between the species involved are corroborated by experimental data.
-
(iii)
The need to determine experimentally the capacity of the material to remove the targeted drug in order to proceed with comparative studies with existing ones. This information is not often reported. It was found that the capacity of L1 to remove aspirin from aqueous solution is 1.27 mmol g-1 (228.79 mg g-1). This material has a higher capacity than existing ones and can be easily recycled by treatment with HCl 0.1 M.
-
(iv)
The relevance of assessing the pH effect of the aqueous solution on the extraction process is not only to optimise the process but also for exploring the possibility of recycling the material via a pH switching mechanism. The optimum pH for highest removal of aspirin from water by the modified silica was found to be around 4 at the standard temperature of 298 K.
Data and material availability
The data generated during this research study are available from the corresponding author upon request.
References
Akhtar J, Amin NAS, Shahzad K (2016) A review on removal of pharmaceuticals from water by adsorption. Desalin Water Treat 57:1–19
Albert JS (2010) Lead generation approaches in drug discovery. Wiley, New Jersey
Al-Khateeb LA, Almotiry S, Abdel Salam M (2014) Adsorption of pharmaceutical pollutants onto graphene nanoplatelets. Chem Eng J 248:191–199
Blasius E, Janzen KP, Keller M, Lander H, Nguyen-Tien T, Scholtien G (1980) Austauscher mit cyclischen Polyethern als Ankergruppen-II. Talanta 27:107–126
Bui TX, Choi H (2009) Adsorptive removal of selected pharmaceuticals by mesoporous silica SBA-15. J Hazard Mater 168:602–608
Danil de Namor AF, Al Nuaim M, Villanueva Salas JA, Bryant S, Howlin B (2017) A calix[4]arene derivative and its selective interaction with drugs (clofibric acid, diclofenac and aspirin). Eur J Pharm Sci 110:1–8
Datt A, El-Maazawi I, Larsen SC (2012) Aspirin Loading and Release from MCM-41 Functionalized with Aminopropyl Groups via Co-condensation or Postsynthesis Modification Methods. J Phys Chem C 116:18358–18366
Hernandez-Morales V, Nava R, Acosta-Silva YJ, Macias-Sanchez SA, Pérez-Bueno JJ, Pawelec B (2012) Adsorption of lead (II) on SBA-15 mesoporous molecular sieve functionalized with -NH2 groups. Microporous Mesoporous Mater 160:133–142
Ho KY, McKay G, Yeung KL (2003) Selective adsorbents from ordered mesoporous silica. Langmuir 19:3019–3024
Khamis M, Karaman R, Ayyash F, Qtait A, Deeb O, Manssra A (2011) Efficiency of advanced membrane wastewater treatment plant towards removal of aspirin, salicylic acid, paracetamol and p-aminophenol. J Environ Sci Eng 5:121–137
Kim Y, Bae J, Park J, Suh J, Lee S, Park H, Choi H (2014) Removal of 12 selected pharmaceuticals by granular mesoporous silica SBA-15 in aqueous phase. Chem Eng J 256:475–485
Li L, Ma Q, Wang S, Song S, Li B, Guo R, Cheng X, Cheng Q (2018) Photocatalytic performance and degradation mechanism of aspirin by TiO2 through response surface methodology. Catalysts 8:118–166
Mphahlele K, Onyango MS, Mhlanga SD (2015) Adsorption of aspirin and paracetamol from aqueous solution using Fe/N-CNT/β-cyclodextrin nanocomopsites synthesized via a benign microwave assisted method. J Environ Chem Eng 3:2619–2630
Mundasad S (2016) Asthma pill 'promising' for people with severe symptoms (BBC News). Available at https://www.bbc.com/news/health-36987243. Verified 6 August 2014.
Nicolaou A, Meric S, Fatta D (2007) Occurrence patterns of pharmaceuticals in water and wastewater environments. Anal Bioanal Chem 387:1225–1234
Ozyonar F, Aksoy S (2016) Removal of Salicylic Acid from Aqueous Solutions Using
Various Electrodes and Different Connection Modes by Electrocoagulation. Int J Electrochem Sci 11: 3680–3696.
Parida SK, Dash S, Patel S, Mishra BK (2006) Adsorption of organic molecules on silica surface. Adv Colloid Interfac Sci 121:77–110
O’Neil M J (2013) The Merck Index. An Encyclopedia of Chemicals, Drugs and Biologicals 15th edn. Royal Society of Chemistry, New Jersey.
World Health Organisation (2012) Pharmaceuticals in Drinking Water. Available at https://www.who.int/water_sanitation_health/publications/2012/pharmaceuticals/en/.
Punyapalakul P, Sitthisorn T (2010) Removal of ciprofloxacin and carbamazepine by adsorption on functionalised mesoporous silicates. Int J Environ Chem Ecol Geo Geophys Eng 4:412–416
Zhang X, Wu W, Wang J, Tian X (2008) Direct synthesis and characterization of highly ordered functional mesoporous silica thin films with high amino-groups content. Appl Sur Sci 254:2893–2899
Acknowledgements
The authors thank Prof. John Watts and his staff for invaluable assistance in the use of the SEM equipment available in the Micro Structural Unit and Surface Analysis Laboratory (University of Surrey).
Funding
This research did not receive any specific funding.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflict of interest.
Additional information
Editorial responsibility: Samareh Mirkia.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Danil de Namor, A.F., Al Nuaim, M., Fairclough, G. et al. Amine-modified silica for removing aspirin from water. Int. J. Environ. Sci. Technol. 19, 4143–4152 (2022). https://doi.org/10.1007/s13762-021-03417-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13762-021-03417-9