
Abstract
A new series of pyridine-2-one and pyrazole derivatives were designed and synthesized based on cyanoacrylamide derivatives containing 2,4-dichlro aniline and 6-methyl 2-amino pyridine as an aryl group. Condensation of cyanoacrylamide derivatives 3a–d with different active methylene (malononitrile, ethyl cyanoacetate cyanoacetamide, and ethyl acetoacetate) in the presence of piperidine as basic catalyst afforded the corresponding pyridinone derivatives 4a–c, 5, 9, and 13. Furthermore, the reaction of cyanoacrylamide derivatives 3a–d with bi-nucleophile as hydrazine hydrate and thiosemicarbazide afforded the corresponding pyrazole derivatives 14a,b and 16. The newly designed derivatives were confirmed and established based on the elemental analysis and spectra data (IR, 1H NMR, 13C NMR, and mass). The in vitro antibacterial activity was evaluated against four bacterial strains with weak to good antibacterial activity. Moreover, the results indicated that the most active derivatives 3a, 4a, 4b, 9, and 16 might lead to antibacterial agents, especially against B. subtilis and P. vulgaris. The DFT calculations were performed to estimate its geometric structure and electronic properties. In addition, the most active pyridinone and pyrazole derivatives were further evaluated for in silico physicochemical, drug-likeness, and toxicity prediction. These derivatives obeyed all Lipinski’s and Veber’s rules without any violation and displayed non-immunotoxin, non-mutagenic, and non-cytotoxic. Molecular docking simulation was performed inside the active site of Topoisomerase IV (PDB:3FV5). It displayed binding energy ranging from -14.97 kcal/mol to -18.86 kcal/mol with hydrogen bonding and arene–cation interaction. Therefore, these derivatives were suggested to be good antibacterial agents via topoisomerase IV inhibitor.
Graphical abstract

Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Bacterial infections, which are caused by gram-positive and gram-negative pathogens, are responsible for the majority of hospital-acquired infections and lead to extensive mortality and burden on global healthcare systems [1, 2]. The emergency and widespread of drug-resistant organisms such as methicillin-resistant S. aureus (MRSA), methicillin-resistant S. epidermidis (MRSE), vancomycin-resistant S. aureus (VRSA), extended-spectrum β-lactamase (ESBL)-producing E. coli, and drug-resistant TB (DR-TB) has already increased up to an alarming level in the recent decades and is associated with considerable mortality [3]. The cyanoacetamide derivative contains two electrophilic centers that appeared at the carbon of carbonyl and carbon of the cyanide group. In addition, it includes two nucleophile centers localized on NH of acetamide and methylene group [4]. The advantage of cyanoacetamide enables chemists to study its reaction with common reagents to form a variety of heterocyclic compounds with pharmacological applications [5].
Furthermore, pyridine (C5H5N), an aromatic heterocycle in which a ring shares π electrons, forms one continuous circle of electrons besides the alternate double bonds shared by every atom on the circle [6]. Pyridine rings are present in many natural products, including vitamins such as vitamin B6 and niacin’s, coenzymes like nicotinamide adenine dinucleotide (NAD), and alkaloids as trigonelline [7]. One role of pyridine in medicinal chemistry is to improve water solubility because of its weak basicity, therefore, lead to improved pH [8]. Over 7000 known medications contain pyridine as the main core [9]. Additionally, pyridine is one of the most widely used groups that is reported to have many biological activities, including antitubercular [10], anticancer [11], antiviral [12], and antimicrobial [13]. Besides, the 4-(phenylamino)thieno[2,3-b]pyridine derivatives showed inhibitory activity against Herpes simplex virus type 1 (HSV-1) [14]. Various drugs contain a pyridine nucleus as anti-malarial [15], antidiabetic [16], antioxidants [17], anti-inflammatory [18] cardiotonic (as Milrinone and Olprnone) [19], and anti-amoebic agents [20]. Recently, two pyridinone derivatives, as di-dymellamide A and Trichodin A isolated from marine-derived fungi, exhibited antibiotic activity against clinically relevant microorganism, S. epidermidis, with lower MIC values [21] (Fig. 1). Besides, the pyridine ring fused with benzene to form a quinoline nucleus that is reported as a broad-spectrum antibiotic that is active against both Gram-positive and Gram-negative bacteria as Moxifloxacin, Nadifloxacin, Levofloxacin, Ozenoxacin, and Nalidixic acid. These drugs were mostly demonstrated to be bactericidal and exerted their mode of action to inhibit the bacterial DNA replication or interfere with the synthesis of RNA and, consequently, with protein synthesis [22,23,24] (Fig. 1).

Examples of drugs containing pyridine or pyridinone cores in the literature
On the other hand, the pyrazole system is an important heterocyclic template, and its derivatives are rare in nature due to the difficulty of living organisms to construct N–N bond [25]. The pyrazole nucleus belonged to heterocycles aromatic compounds containing two adjacent nitrogen atoms in 1, 2 positions and therefore called 1,2 diazoles and have pharmacological effects on humans [26]. Also, pyrazole is used as a ligand for transition metal-catalyzed reactions [27] and is necessary as intermediates for synthesizing other pharmaceutical heterocycles [28,29,30]. The production of pyrazole and its derivatives showed various applications in the pharmaceutical and agriculture industries [31, 32]. It was reported that heterocycles containing pyrazole scaffold exhibit antibacterial [33] and anti-inflammatory[34], analgesic [35], antidepressant [36], antiviral [37], and antitumor activities [38]. Also, one of the important heterocycles that attracted good interest is the 5-aminopyrazole derivative used in a wide range of pharmaceutical applications [39].
Based on the previous literature and in continuous efforts to design and synthesize heterocyclic compounds that exhibited biological activities [40,41,42,43,44], the purpose of this work is to design and synthesize new pyridine or pyrazole derivatives from cyanoacrylamide derivatives. Additionally, the antibacterial activity screened against four bacterial strains. Moreover, the most active pyridinone and pyrazole derivatives were selected to determine the in silico physicochemical, drug-likeness, and toxicity prediction. The DFT calculation was performed for exploring geometrical structural and reactivity parameters of the most actively studied compounds. Finally, the molecular docking simulation achieved to determine the binding mode and examine the compounds’ interaction and binding activity inside the active site of Topoisomerase IV as the antimicrobial target.
Result and discussion
Chemistry
The synthesis of intermediate and the target compounds was outlined from Scheme 1 to Scheme 4 using cyanoacetamide derivatives as a starting material. The cyanoacetamide derivatives are highly reactive molecules and used as precursors or reaction intermediates because it contains two centers of reaction carbonyl and cyanocenters which make it able to react with widespread as bidentate reagents to form a variety of heterocycles [45, 46]. Our research is interested in the synthesis of pyridinone and pyrazole derivatives. According to the previously reported methods, the cyanoacetamide derivatives 1a,b were prepared in a high yield [47, 48]. The cyanoacetamide derivatives 1a, b and different aromatic aldehydes as p-methoxy benzaldehyde and p- tolualdehyde were heated under reflux condition in ethanol containing drops of piperidine gave the corresponding acrylamide derivatives 3a–d (Scheme 1).

Synthesis of acrylamide derivatives 3a–d
The structure of acrylamide derivatives 3a–d approved using spectral analysis. The IR spectra of 3a–d revealed absorption bands for amidic carbonyl (1667–1696 cm−1), cyano (2203–2217 cm−1), and imino (3283–3397 cm−1) groups at the correct frequencies. Additionally, the 1H NMR spectrum of acrylamide derivative 3a in (DMSO-d6) displayed signals at δ 3.87 for methoxy group, and the aromatic protons at δ 7.13, 7.18, 7.49, 7.69, 7.27, and 8.06 ppm as five doublet signals with coupling constant ranged between (J = 8.4–9.0 Hz), and one singlet signal. Moreover, the acrylamide derivative 3a displayed an exchangeable signal at δ 9.86 ppm related to the NH group and the methine proton (–CH = C–) appeared at δ 8.27 ppm. Its 13C NMR spectrum showed specific signals at δ 55.69, 151.56, and 163.03 ppm attributed to the methoxy, carbon attached to methoxy, and carbonyl group (C = O).
Cyclocondensation of acrylamide derivatives 3a, b, d with malononitrile was afforded the corresponding 3,5-dicarbonitrile-2-oxo-pyridine 4a–c. The mechanistic equation of this reaction can be described by attacking the active methylene (CH2) to the carbanion β-carbon of acrylamide, then the NH nucleophile of cyanoacrylamide added to the cyano-group of malononitrile, and then the cyclization occurred (Scheme 2). Spectral data and analytical analysis are in agreement with the postulated structure. The IR spectra showed stretching significance absorption bands for amino, cyano, and carbonyl groups at exact frequencies for 4a–c. 1H NMR spectrum (DMSO-d6) revealed two singlet signals at δ 2.10, 2.32 ppm for two methyl protons, multiple signals between at δ 7.16 and 8.19 ppm due to seven aromatic protons, and exchangeable singlet signal at δ 6.66 ppm related to the amino group (NH2) for pyridin-2-one derivative 4c. Similarly, pyridine-2-one derivative 4a appeared specific signal at δ 3.86 ppm, which referred to the methoxy group (OCH3) due to exposure to 1H NMR (DMSO-d6). The fragmentation pattern of pyridine-2-one derivative 4a is illustrated in chart S1.

Synthesis of 3,5-dicyano-pyridine-2-one derivatives 4a–c and 2-amino-6-cyanopyridin-6-one derivative 5
Furthermore, refluxing of acrylamide derivatives 3b and 3d with ethyl cyanoacetate in ethanol catalyzed with piperidine (3drops) produced ethyl 2-amino-5-cyano-6-oxo-4-(4-methylphenyl)-pyridine-3-carboxylate derivative 5 and ethyl 2-cyano-3-(4-methylphenyl)acrylate derivative 8 [49] rather than pyridine-2-one derivative 6, respectively. The mechanistic route for formation of compound 8 represented as in Scheme 2.
The structure of 5-cyano-4-(4-methylphenyl)-pyridin-6-one derivative 5 was confirmed the result of spectral analyses. The 1H NMR spectrum (DMSO-d6) showed characteristic signals at δ 2.31, 1.51, 3.96 ppm corresponding to methyl and ethoxy group, besides signals between δ 7.22 and 7.64 ppm as three doublet and one singlet signals related seven aromatic protons and broad exchangeable signal at δ 7.26 ppm owning to the amino group (NH2). Moreover, the 13CNMR spectrum (DMSO-d6) assigned signals at δ 1.67.61, 158.35, 155.16 ppm related to two carbonyl groups (2C = O) and carbon attached to the amino group (C-NH2). Besides, three significant signals at δ 69.85, 20.95, and 29.05 ppm were assigned to the ethoxy and methyl groups, respectively.
The reaction of cyanoacrylamide derivative 3a and cyanoacetamide derivative in one molar ratio under reflux condition in ethanolic piperidine afforded the corresponding 2-amino-5-cyano-6-oxo-pyridine-3-carboxamide derivative 9. The IR spectrum displayed characteristic absorption bands at ʋ 3448, 3303, 3174, 2206, and 1697 cm−1 corresponding to NH, amino group (NH2), nitrile (CN), and carbonyl (C = O) groups, respectively. The 1H NMR spectrum of 6-oxo-pyridine-3-carboxamide derivative 9 revealed three significant exchangeable signals at δ 7.77, 7.83, and 7.96 ppm corresponding to amino and two NH groups. Besides, signals at δ 3.84 ppm for the methoxy group and the seven aromatic protons that ranging between δ 7.13 and 8.11 ppm. Additionally, the 13C NMR spectrum displayed characteristic signals at δ 163.13, 162.56, 150.16, and 55.60 ppm for two carbonyl carbons (2C = O), carbon attached to the amino (C-NH2), and methoxy (OCH3) groups. The mass spectra showed a molecular ion peak at m/z = 428.95 with an intensity of 25.52% corresponding to the molecular formula C20H14Cl2N4O3 (Scheme 3).

Synthesis 6-oxo-pyridine-3-carboxamide derivative 9 and 6-hydroxy2-oxo-4-(p-tolyl)-2H-[1,2'-bipyridine]-5-carbonitrile derivatives 13 based on cyanoacrylamide derivatives
Subsequent, reaction of cyanoacrylamide derivative 3d with ethyl acetoacetate in absolute ethanol and in the presence of catalytic amount of piperidine (3 drops) afforded the corresponding 6-hydroxy-2-oxo-4-(p-tolyl)-2H-[1,2'-bipyridine]-5-carbonitrile derivative 13 rather than 5-acetyl-6-hydroxy-2-oxo-4-(p-tolyl)-2H-[1,2'-bipyridine]-3-carbonitrile derivative 12 and 3-cyano-2-oxo-4-(p-tolyl)-2H-[1,2'-bipyridine]-5-carboxylate derivative 10 based on the spectral data and elemental analysis. As described in Scheme 3, this reaction can be proceeded via two pathways: first one, elimination of water molecule to afford 2H-[1,2'-bipyridine]-5-carboxylate derivative 10, while the second pathway, proceed via elimination of ethanol molecule to afford the intermediate 11. The spectra data based on 1H NMR spectra confirmed formation of compound 13. The 1H NMR spectra displayed significant two methyl groups at δ 2.37 and 2.43 ppm, singlet signal at δ 8.35 ppm related to pyridine-H, and one exchangeable singlet signal at δ 10.63 ppm assigned to the hydroxyl group. Additionally, the 13C NMR spectrum exhibited signals at δ 161.22, 156.73, 151.14, 105.49, 23.51, and 21.25 ppm assignable to carbon attached to hydroxy group (C–OH), carbonyl group, C = N, and pyridine-C3, and two methyl groups, respectively. Besides, the mass spectra revealed a molecular ion peak at m/z = 317.72 with an intensity of 20.61% and related to empirical formula C19H15N3O2.
Here, the authors aimed to synthesize pyrazole nucleus from cyanoacrylamide derivatives hoping to add pyrazole nucleus that exhibit a versatile range of biological activities such as antimicrobial [50, 51], antidepressant [52], anti-inflammatory [34], antiviral [53], and antitumor activities [38]. Upon treatment, the acrylamide derivatives 3a–d with hydrazine hydrate and thiosemicarbazide in ethanolic solution afforded the corresponding pyrazole derivatives 14a, b and 16, respectively (Scheme 4). The structure of pyrazole derivatives is confirmed with the usual spectroscopic techniques and elemental analysis. The IR spectra of pyrazole derivatives 14a, b displayed a lack of significant amino group band and strong stretching frequency for carbonyl group at ʋ 1720 cm−1. Additionally, the 1H NMR spectra (DMSO-d6) for the same compounds 14a, b assigned specific signals for (OCH3) and (CH3) protons at δ 3.82 and 2.36 ppm, as well as the pyrazole protons at δ 8.62 for pyrazole derivative 14a, and δ 8.55 ppm for pyrazole derivative 14b. The fragmentation pattern of 5-(4-methoxyphenyl)-3H-pyrazol-3-one 14a can be illustrated in chart S2.
Finally, the pyrazole derivative 16 showed new bands in the IR spectrum related to two amino groups at ʋ 3406, 3290, and 3151 cm−1. Besides, the 1H NMR spectrum showed singlet signal at δ 3.82 ppm of methoxy group, while three exchangeable signals appeared at δ 7.60, 7.89, and 11.33 ppm attributed to two amino (2NH2) and one NH group, respectively. Besides, the seven aromatic protons ranging between δ 6.94–8.11 ppm. Also, 13C NMR spectrum (DMSO–d6) showed signals at δ 177.66, 160.72, 152.47, and 55.26 ppm attributed for thiocarbonyl (C = S), carbon attached to the methoxy (C-OMe), carbonyl group (C = O), and methoxy carbon (OCH3), respectively.

Illustrates synthesis of new pyrazole derivatives 14a, b, and 16 from the reaction of cyanoacrylamide derivatives 3a–d with bi-nucleophile reagents as hydrazine hydrate and thiosemicarbazide
Antibacterial activity with structure–activity relationship (SAR)
All the newly synthesized derivatives were evaluated for their in vitro antibacterial activity against two gram-positive strains as S. aureus (RCMB010010) and B. subtilis (RCMB 015), and two gram-negative strains as E. coli (RCMB010052) and P. vulgaris (RCMB004) by agar well diffusion method according to the previously reported methods [54,55,56]. The antibacterial activity is determined by the zone of inhibition expressed by (mm). Levofloxacin was used as positive control (Table 1). The preliminary assessment results exert varying activity against the tested bacteria pathogens. As can be seen in Table 1, the cyanoacrylamide derivatives 3a–d showed no activity against S. aureus (RCMB010010) and E. coli (RCMB010052), while exhibited weak to good activity against B. subtilis (RCMB 015) and P. vulgaris (RCMB004).
Generally, the 2-cyano-N-(2,4-dichlorophenyl)acrylamide derivatives 3a, b displayed moderate activity against B. subtilis with a zone of inhibition (IZ = 13.83 ± 0.15 and 11.20 ± 0.20) mm. Besides, the 3-(4-methoxyphenyl)acrylamide 3a revealed the strongest activity against P. vulgaris with a zone of inhibition 17.13 ± 0.15 mm, while -3-(4-methylphenyl)acrylamide derivative 3b showed moderate activity with inhibition zone 13.13 ± 0.012 mm. The higher activity of cyanoacrylamide derivative 3b might be related to the presence of methoxy group that exhibited more effective than the methyl group in position three of 3-(aryl)-2-cyanoacrylamide derivatives 3a,b.
On the contrary, 2-cyano-N-(6-methylpyridin-2-yl)acrylamide 3c, d exhibited weak activity with the zone of inhibition ranged from 8.03 ± 0.15 to 12.13 ± 0.12 mm and changing the methyl by methoxy at aryl group in position three not enhancement the bacterial activity. This decrease in activity in acrylamide derivatives 3c, d might be attributed to the replacement of N-(2,4-dichlorophenyl) with N-(6-methylpyridin-2-yl) moiety. Additionally, the 2-oxo-pyridine-3,5-dicarbonitrile 4a, b showed moderate to potent activity against S. aureus and P. vulgaris with inhibition zones ranged between (12.13 ± 0.15–17.13 ± 0.15) mm and that might be attributed to the presence of two chlorine atoms in N-(2,4-dichlorophenyl) at position one in pyridine nucleus. Moreover, the 3,5-dicarbonitrile-1-(2,4-dichlorophenyl)-4-(4-methylphenyl)pyridine-2-one derivative 4b displayed the highest activity compound against B. subtilis IZ = 17.13 ± 0.15 mm with an activity index of 80.87% compared with Levofloxacin (IZ = 21.18 ± 0.45) mm, while it showed no activity against S. aureus. Additionally, the 2-cyano-N-(2,4-dichlorophenyl)acrylamide derivative 3a and 3,5-dicarbonitrile-1-(2,4-dichlorophenyl)pyridine-2-one derivative 4b demonstrated equipotent activity with the zone of inhibition 17 mm against P. vulgaris. Further, modification of pyridine-2-one derivatives 4a, b to form bi-pyridine derivative 4c in one hybrid structure causes a decrease in the antibacterial activity against all tested strains. Also, the 2-amino-5-cyano-6-oxo-pyridine-3-carboxylate derivative 5 exhibited no activity against the tested strains.
Surprisingly, the 2-amino-5-cyano-6-oxo-pyridine-3-carboxamide derivative 9 showed the most active derivatives against P. vulgaris with a zone of inhibition 18.07 ± 0.12 mm with an activity index nearly to 68% compared with Levofloxacin (IZ = 26.52 ± 0.24). This activity may be due to the presence of 3-carboxamide moiety at position three that bearing 2,4-dichlorophenyl group. Further, 6-oxo-pyridine-3-carboxamide derivative 9 revealed moderate activity against B. subtilis (IZ = 13.07 ± 0.12 mm) and weak activity against S. aureus (IZ = 11.07 ± 0.12 mm) in comparison to Levofloxacin (21.18 ± 0.46 and 25.62 ± 0.31) mm, respectively. The 6-hydroxy-2H-[1,2'-bipyridine]-5-carbonitrile derivative 13 demonstrated moderate activity against P. vulgaris (IZ = 14.90 ± 0.17) mm and S. aureus (IZ = 11.93 ± 0.21) mm, while displayed no activity against B. subtilis and E. coli. Notably, the 5-(Aryl)-3H-pyrazol-3-one derivatives 14a, b exhibited weak activity against the tested strains and that might be due to absent of N-(2,4-dichlorophenyl) or pyridine moieties in their structure. Additionally, the 5-amino-1-carbamothioyl-1H-pyrazole-4-carboxamide derivative 16 showed moderate to good activity against B. subtilis and P. vulgaris with an inhibition zone 11.97 ± 0.15, 16.87 ± 0.12 mm. respectively, and compared with Levofloxacin (IZ = 21.18 ± 0.46 and 26.52 ± 0.24 mm). Besides, 1H-pyrazole-4-carboxamide derivative 16 revealed no activity against E. coli (RCMB010052) strains. However, the pyrazole derivative 16 showed moderate activity with zone of inhibition (14.93 ± 0.31 mm) against S. aureus, but still the highest antibacterial activity member in our study and that might be reflected to the combination between three pharmacophore groups as N-(2,4-dichlorophenyl) in the amide group, 4-methyl phenyl at C3 of pyrazole, and pyrazole moiety.
Finally, the preliminary assay results indicated that the most active derivatives 3a, 4a, 4b, 9, and 16 might lead to antibacterial agents, especially against B. subtilis and P. vulgaris and the SAR study can be summarized as;
-
(1)
Presence of N-(2,4-dichlorophenyl) observed higher antibacterial activity than N-(6-methyl pyridine-2-yl) moiety as described in derivatives 3a–d.
-
(2)
For cyanoacrylamide derivative, the presence of methoxy at para position enhances the antibacterial activity than the methyl group.
-
(3)
Formation of 6-aminopyridin-2-one derivatives 4a–c improved the activity against B. subtilis and P. vulgaris, besides the presence of (4-methoxyphenyl) group in position four in pyridine-2-one derivative as 4a causes inhibition to bacterial growth, especially against S. aureus strain.
-
(4)
Adding the N-(2,4-dichlorophenyl) in the acetamide group as pyrazole derivative 16 improved the antibacterial activity against S. aureus, B. subtilis, and P. vulgaris
Computational and theoretical studies
Geometry optimization
Frontier molecular orbitals (FMOs) can be defined as highest occupied molecular orbitals (HOMO) and lowest unoccupied molecular orbitals (LUMO). The HOMO energy is an orbital that has ability of molecules to donate electrons, while LUMO energy represented the ability of molecule to revive electrons from occupied orbitals. The HOMO and LUMO orbitals are responsible for many molecular properties of molecules, including electronic properties, chemical reactivity, stability, a biological activity that is affected by intermolecular charge transfer [57, 58]. Based on the FMOs theory, it is believed that the antimicrobial properties of molecules are related to their LUMO energy, where molecules with low LUMO energy can accept more electron than those with high LUMO energy, which explains their higher activity [59]. A molecule with a high electron capacity may bind to protein more effectively because it interacts more effectively with the structure of the target protein [60]. The FMOs and chemical reactivity descriptors of the most active derivatives and Levofloxacin (positive control) are summarized in Table 2. Additionally, the pictorial drawing of HOMO–LUMO energies was shown in Fig. 2. Besides, The positive lopes are shown in turquoise color, whereas the negative phase is expressed in pale purple color.

Molecular orbital surfaces and energy bandgap of the most active derivatives and positive control
Furthermore, the HOMO electron density of cyanoacrylamide derivative 3a is localized over all the structure, while the LUMO is represented over the cyanoacrylamide except the 2,4-dichloro phenyl moiety. Additionally, the pyridine derivatives (4a and 4b) of the HOMO and LUMO molecular orbital are distributed over the pyridine-2-one nucleus, except the 2,4-dichloro phenyl moiety. On the other hand, for the 2-amino pyridine-6-one derivative 9 the HOMO orbitals located in the bottom and left of the molecules around the pyridine nucleus and slightly distributed over the 4-tolyl group attached to position 4 in pyridine nucleus, while the LUMO orbital dispersed overall molecule. For 5-amino pyrazole derivative 16, the HOMO and LUMO orbitals were found to be localized over the pyrazole nucleus with a low density over the 4-methoxy phenyl group attached at position three in pyrazole. As a comparison, the HOMO orbital of Levofloxacin localized over the quinolone (benzo[b]pyridine) and piperazine derivatives. In contrast, the LUMO orbital was distributed over all molecule except the piperazine derivative.
As represented in Table (2), all the synthesized compounds displayed low total energy ranging from − 1835.36 to − 2437.74 Hartree compared to Levofloxacin (E Total = − 1262.96 Hartree). Besides, these derivatives showed dipole moment ranging between (3.39–8.02 Debye) compared with Levofloxacin (µ = 10.21 Debye). Furthermore, the energy difference between HOMO and LUMO is the energy bandgap (Δ E) and reflects chemical stability. The cyanoacrylamide derivatives 3a showed the lowest energy bandgap ΔE = 3.86 eV, while the pyrazole derivative 16 revealed the highest energy bandgap of 4.37 eV. Besides, the pyridine derivatives 4a, 4b, and 9 showed nearly equal energy bandgap ~ (4.17–4.20 eV) and were close to Levofloxacin (ΔE = 4.19 eV). Moreover, the ionization potential can be defined as the energy required to remove an electron from a molecule’s ground state.
On the other hand, electron affinity is represented by the energy released when a molecule in the ground state captures an electron. Additionally, in a chemical reaction, the hardness and the softness of a molecule are important descriptors. The synthesized derivatives exhibited high hardness and low softness values and were very close to Levofloxacin except for cyanoacrylamide derivative 3a, which displayed slightly harder and softer than the reset derivatives. Additionally, the electronegativity, chemical potential, and electrophilic index were calculated.
Furthermore, the electrophilicity (ω) is measured by the tendency of the molecule to accept an electron where the organic molecules can be classified as weak electrophiles (ω < 0.8), moderate electrophiles (ω = 0.8–1.5), and strong electrophiles (ω ˃ 1.5). The tested derivatives showed strong electrophilic properties with electrophilic index (ω) ranging between 3.09 and 4.84. the chemical potential (µp) indicates the probability of a chemical reaction occurring; a high value of (less negative) indicates that it is easier to donate electrons (electron donor), while a low value of (more negative) indicates that it is easier to accept electrons (electron acceptor). The synthesized derivatives demonstrated high chemical potential values. Thus, these derivatives are easy to donate an electron to the neighboring groups or biological target and could form different types of interaction inside the active site of the protein target.
In silico molecular docking study
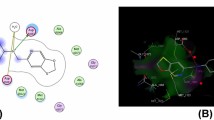
Molecular docking simulation of the most active derivatives 3a, 4a, 4b, 9, and 16 was evaluated to determine the structure orientation, conformation and identify the binding mode of the most active derivatives inside the active site Topoisomerase IV (PDB:3FV5). The docking process was performed using Molecular operating Environmental (MOE) 10.2008, Chemical computing Group Inc, Montreal, Quench, Canada [61,62,63] according to the previously reported method [64]. The docking results are represented in Table S1 and Figs. 3, 4 and 5. The original ligand 1-(4-acetyl-6-pyridin-3-yl-1H-benzimidazol-2-yl)-3-ethylurea represented as 1EU and displayed binding energy S = 23.60 kcal/mol with RMSD = 0.828 oA, through two hydrogen bonds sidechain donor between Asp69 and two NH of urea derivative with bond length 2.49 and 3.18 oA. Besides, one hydrogen bond sidechain acceptor between the residue Arg132 and nitrogen of pyridine with bond length 3.09 oA and strength 13%, as well as one arene–cation interaction between Arg72 and pyridine nucleus is observed (the superimposition 3D of co-crystalized ligand and docking pose showed in Fig. 3). Additionally, the positive control used in antibacterial activity demonstrated binding energy S = − 20.72 kcal/mol. The Levofloxacin fitted inside the active site with two hydrogen bonds side chain donor between the Arg72 and carbonyl of carboxylic group of Levofloxacin with bond length 2.29 and 3.10 oA and strength 40 and 11%. Besides, the residue Asp69 bound with nitrogen of N-methyl piperazine with bond length 3.17 oA and strength 12% (See SI file).

Shows the superimposition 3D of co-crystalized ligand (orange) and docking pose (green) with RMSD = 0.838 oA, inside the active site of topoisomerase IV (PDB:3FV5)

a 2D structure of compound 9 inside the active site of topoisomerase IV (PDB:3FV5), b illustrated the 3D structure and mapping surface of the compound 9 inside the active site topoisomerase IV (PDB:3FV5)

a 2D structure of compound 16 inside the active site of topoisomerase IV (PDB:3FV5), b illustrated the 3D structure and mapping surface of the compound 16 inside the active site topoisomerase IV (PDB:3FV5)
Furthermore, cyanoacrylamide derivative 3a showed binding energy S =− 14.97 kcal/mol, with binding mode, represented two hydrogen bond sidechain acceptors between Arg132 and Arg72 with the carbonyl of amide and nitrogen of cyano with bond length 2.83 and 3.08 oA, respectively. Besides, one arene–cation interaction between Arg132 and phenyl of 2,4-dichlorophenyl derivative is observed. Simultaneously, the pyridine-2-one derivative 4a revealed only one hydrogen bond sidechain acceptor between the Arg132 and nitrogen of cyanide group with bond length 2.85 oA and strength 47%, as well as the tolyl group and dicyanide exhibited hydrophobic interaction. On the contrary, 3,5-dicarbonitrile-1-(2,4-dichlorophenyl)pyridine-2-one derivative 4b showed one hydrogen bond sidechain donor between Glu46 and amino group localized at position six on pyridine-2-one derivative and arene cation interaction between phenyl of 2,4-dichlorophenyl (See SI file). Interestingly, 2-amino-5-cyano-6-oxo-pyridine-3-carboxamide derivative 9 revealed fitted inside the active site and showed the lowest binding energy S = − 18.86 kcal/mol through two sidechain hydrogen bonds: one hydrogen bond acceptor between Thr163 and nitrogen of cyanide and the other between Asn42 and amino group with bond length 3.02 and 2.75 oA, respectively. Besides, arene–cation interaction between the Arg72 and phenyl ring of the 4-methoxyphenyl group is observed (Fig. 4). Moreover, the 1-carbamothioyl-1H-pyrazole-4-carboxamide derivative 16 demonstrated one hydrogen bond sidechain donor between Arg132 and oxygen of methoxy group with bond length 3.07 and strength 21%, as well as two arene–cation interaction between Arg72 and pyrazole nucleus and Arg72 with phenyl of 4-methylphnyl group (Fig. 5).
Finally, it concluded that the most active derivatives could inhibit bacterial growth through topoisomerase IV inhibitors with binding energy ranging from − 14.97 to − 18.86 kcal/mol compared to co-crystallized ligand S = − 23.60 kcal/mol and Levofloxacin S = 20.72 kcal/mol. Additionally, all the most active derivatives exhibited at least one interaction as the co-crystallized ligand observed, and that showed the similarities in the structure of synthesized derivatives and co-crystallized ligand, and therefore, these derivatives were suggested to be good antibacterial agents via topoisomerase IV inhibitor.
Molecular electrostatic potential (MEP) maps
MEP maps of a molecule are used to provide information about the positions of charge distribution (positive and negative charge) in the molecule and determine the active site for nucleophile and electrophile attack in the molecules for binding to protein in protein substrate interactions. The positive charge appears as blue color, and the negative charge appears as red color. Both of them can bind with the active site through different types of interaction (hydrogen bond donor, H-acceptor, arene–arene, and arene–cation interaction) and therefore confirmed the docking study. According to the previously reported method, the MEP was performed for the most active derivatives 3a, 4a, 4b, 9, 16, and Levofloxacin using MOE 10.2008 [50]. As represented in Fig. 6, the positive charge was localized on NH of anilide derivative, amino of pyridine, the nitrogen of pyridine, amino at C5 of pyrazole, the nitrogen of pyrazole, amino of thioamide derivative, nitrogen of quinoline, and nitrogen of piperazine. On the other hand, the negative charge distributed around the nitrogen of cyano, oxygen of carbonyl at pyridinone, methoxy group, amide group, around chlorine atoms, sulfur of thioamide, and carboxylic group. Both the positive and negative charges are responsible for interaction between the ligand and active site in the docking study.

shows the molecular electrostatic maps of the most active derivatives 3a, 4a, 4b, 9, 16, and Levofloxacin, where the blue color indicates the positive charge and red color related to the negative charge
In silico ADMET study
The most active five derivatives 3a, 4a, 4b, 9, 16, and Levofloxacin were conducted to determine some physicochemical and drug-likeness properties, as well as the surface area represented by angstrom to estimate the drug-likeness of compounds using Molinspiration web tool according to the previously reported method [65, 66]. As shown in Table S2, the most active derivatives 3a, 4a, 4b, 9, 16, and Levofloxacin obeyed Lipinski’s and Veber rule without any violation. Additionally, the most active derivatives have the number of hydrogen bond acceptor groups from four to seven and the number of hydrogen bond donors from one to five. Besides, topological polar surface area TPSA ranged between (62.12 and 121.01 oA2), molecular weight less than 500 Dalton, and partition coefficient MLogP from 3.64 to 4.59.
Subsequently, to determine the potential safety or toxicity of the most active derivatives 3a, 4a, 4b, 9, and 16 were assessed using two web tools as ProTox-II and pkCSM prediction as described previously [67, 68]. From Table S2, and based on ProTox-II prediction, the most active derivatives 3a, 4a, 4b, and 16 belonged to class IV in toxicity classification with lethal dose LD50 ranged between 386 and 1000 mg/kg, while 6-oxo-pyridine-3-carboxamide derivative 9 and Levofloxacin demonstrated LD50 = 5000 and 1478 mg/kg, respectively, and belongs to class V. Additionally, the most active derivatives and Levofloxacin displayed non-immunotoxin, non-mutagenic, and non-cytotoxic with probability ranged from 0.53 to 0.99, except cyanoacrylamide derivative 3a and Levofloxacin that exhibited mutagenic with probability of 0.59 and 0.66, respectively.
Further, all the tested derivatives and positive control revealed inactive against phosphoprotein (Tumor suppressor) p53. Furthermore, the tested derivatives 3a, 4a, 4b, 9, and 16 showed non-hepatotoxic, non-skin sensitization, and no AMES toxicity, except pyridin-2-one 4a that displayed a very low probability of AMES toxicity. Besides, the Levofloxacin and pyrazole derivative 16 predicted to have hepatotoxic properties. Moreover, the most active derivatives 3a, 4a, 4b, 9, 16, and Levofloxacin showed no inhibition to hERG I, while 4a, 4b, 9, 16 exhibited hERG II inhibitors. The tested compounds had max. tolerated doses (human) ranged from -0.026 to 0.446 (log mg/kg/day) lower than Levofloxacin 0.965 (log mg/kg/day). Besides, oral rat chronic toxicity (LOAEL) was predicted to have values ranging from 0.939 to 1.303 lower than Levofloxacin LOAEL = 1.79 (log mg/kg_bw/day). Similarly, the tested derivatives displayed oral rat chronic toxicity (LOAEL) between 2.576 and 2.775 mol/kg compared to Levofloxacin LD50 = 2.59 mol/kg.
Conclusion
The present study involves the synthesis and characterization of some new pyridinone and pyrazole derivatives based on cyanoacrylamide derivatives 3a–d that were prepared previously by reaction of cyanoacetamide with different aldehydes. The synthesized compounds were assessed for their in vitro antibacterial activity by the agar well diffusion method and compared with Levofloxacin as a positive control. The preliminary assessment results exert varying activity against the tested bacteria pathogens from weak to good. Five derivatives 3a, 4a, 4b, 9, and 16 exhibited potent antibacterial activity with a zone of inhibition ranging from 11.07 ± 0.12 mm to 18.07 ± 0.12 mm and displayed the highest activity against B. subtilis and P. vulgaris. The most active derivatives 3a, 4a, 4b, 9, and 16 obeyed all rule of five and Veber’s rule without any violation, and displayed non-immunotoxin, non-mutagenic, and non-cytotoxic. Additionally, the most active derivatives 3a, 4a, 4b, and 16 belonged to class IV in toxicity classification with lethal dose LD50 ranged between 386 and 1000 mg/kg, while 6-oxo-pyridine derivative 9 demonstrated LD50 = 5000 mg/kg belongs to class V. Surprisingly, the tested derivatives 3a, 4a, 4b, 9, and 16 showed non-hepatotoxic, non-skin sensitization, and no AMES toxicity, except pyridin-2-one 4a that displayed a very low probability of AMES toxicity. Additionally, the pyrazole derivative 16 and Levofloxacin predictied to had hepatotoxic properties. Furthermore, the electronic parameters of the most active derivatives and Levofloxacin were analyzed by computing HOMO and LUMO orbitals. Additionally, quantum chemical parameters were evaluated and discussed using frontier molecular orbital analysis to explore the chemical reactivity of the most actives molecules. Finally, molecular docking simulation revealed that these derivatives suggested inhibiting bacterial growth through topoisomerase IV inhibitors with binding energy ranging from − 14.97 to − 18.86 kcal/mol compared to co-crystallized ligand S = -23.60 kcal/mol and Levofloxacin S = − 20.72 kcal/mol. Additionally, the MEP maps were studied to confirm the docking study by determining the position of groups that can bind with the possible sites involved in interactions with protein receptors.
Experimental
Chemistry
Melting points are uncorrected and recorded on digital Gallen Kamp MFB-595 instrument. The IR spectra were recorded on a Shimadzu 440 infrared spectrophotometer (υ/cm−1) using the KBr technique (Shimadzu, Japan). 1H NMR spectra were recorded on a Varian Gemini spectrometer (δ/ppm) 300 MHz and Bruker spectrometer (400 MHz) spectrometer using trimethyl silane (TMS) as internal standard. The 13C NMR spectra were run out at 75 MHz and 13C NMR at (101 MHz, δ/ppm). Additionally, mass spectra were recorded on a Jeol-JMS-600 mass spectrometer. Further, the micro analytical data were obtained from the Micro analytical Research Centre, Faculty of Science, Cairo University. The reactions were monitored by thin layer chromatography (TLC) using TLC sheets with UV fluorescent silica gel Merck 60f254 plates using UV lamp and different solvents as mobile phases. The antimicrobial activity was evaluated at the Regional Center for Mycology and Biotechnology, Al-Azhar University. The 2-cyano-N-(2,4-dichlorophenyl)acetamide (1a) and 2-cyano-N-(6-methylpyridin-2-yl)acetamide (1b) were prepared using ethyl cyanoacetate according to the previously reported methods [47, 48].
Synthesis of 2-cyano-N-(aryl)-3-(aryl)acrylamide (3a–d).
To a solution cyanoacetamide derivative (1a, b) (0.01 mol) in absolute ethanol (20 mL) having little amount of piperidine (3drops), 4-methoxy benzaldehyde or 4-methylbenzaldehyde (0.01 mol) was added and heated under reflux for 3 h. The solid product formed was collected by filtration and recrystallized from ethanol.
2-Cyano- N -(2,4-dichlorophenyl)-3-(4-methoxyphenyl)acrylamide (3a)
Yellow powder (EtOH); Yield 72%; M.p. = 175–177 ºC; IR (KBr, ν/cm−1): 3283 (NH), 3078 (CH-arom.), 2939 (CH-aliph.), 2206 (C≡N) and 1697 (C = O amide) 1H NMR (300 MHz DMSO-d6): δ/ppm 3.87(s, 3H, OCH3), 7.13 (d, 1H, J = 8.7 Hz, Ar–H), 7.18 (d, 1H, J = 9.0 Hz, Ar–H), 7.49 (d, 1H, J = 8.4 Hz, Ar–H), 7.69 (d, 1H, J = 8.7 Hz, Ar–H), 7.27 (s, 1H, A-H), 8.06 (d, 2H, J = 8.7 Hz, Ar–H), 8.27 (s, 1H, methine-H), 9.86 (s, 1H, NH exchangeable by D2O); 13C NMR (76 MHz DMSO-d6): δ/ppm 163.03 (C = O), 151.56 (C-Ome), 141.54 (C = C), 132.88, 131.79, 130.89, 130.18, 130.03, 129.73, 129.10, 128.48, 127.78, 124.21, 116.75, 114.95, 114.50, 55.69 (OMe); Anal. Calcd. for C17H12Cl2N2O2 (346.03): C, 58.81; H, 3.48, N, 8.07; Found: C, 58.98; H, 3.28; N, 8.22.
2-Cyano- N -(2,4-dichlorophenyl)-3-(4-methylphenyl)acrylamide (3b)
Yellow powder (EtOH); Yield 72%; M.p. = 246–248 ºC; IR (KBr, ν/cm−1): 3339 (NH), 3050 (CH-arom.), 2980, 2836 (CH-aliph.), 2217 (C≡N), and 1667 (C = O amide); 1H NMR (300 MHz DMSO-d6): δ/ppm 2.49 (s, 3H, CH3), 7.07 (d, 2H, J = 7.8 Hz, Ar–H), 7.39 (d, 2H, J = 8.7 Hz, Ar–H), 7.62 (s, 1H, Ar–H), 8.11 (d, 2H, J = 8.7 Hz, Ar–H), 8.34 (s, 1H, methine-H), 9.13 (s, 1H, NH, exchangeable by D2O); 13C NMR (101 MHz, DMSO d6) δ/ppm 163.97 (C = O), 151.91, 147.99, 143.98, 139.49, 135.93, 132.03, 130.03, 126.80, 125.44, 124.58, 121.19, 117.31 (Ar-Cs), 115.46 (CN), 102.66, 19.02 (CH3); Anal. Calcd. for C17H12Cl2N2O (330.03): C, 61.65; H, 3.65; N, 8.46; Found: C, 61.92; H, 3.89; N, 8.12.
2-Cyano-3-(4-methoxyphenyl)- N -(6-methylpyridin-2-yl)acrylamide (3c)
Yellow powder (EtOH); Yield: 87%; M.p. = 190–192 ºC; IR (KBr, ν/cm−1): 3397 (NH), 3056 (CH-arom.), 2976, 2922 (CH-aliph.), 2206 (C≡N), and 1688 (C = O amide); 1H NMR (300 MHz DMSO-d6): δ/ppm 2.49 (s, 3H, CH3), 3.87 (s, 3H, OCH3), 7.06 (d, 1H, J = 7.5 Hz, Ar–H), 7.18 (d, 2H, J = 9.0 Hz, Ar–H), 7.76 (t, 1H, J = 7.8 Hz, Ar–H), 7.89 (d, 1H, J = 8.4 Hz, Ar–H), 8.03 (d, 2H, J = 8.7 Hz, Ar–H), 8.31 (s, 1H,methine-H),10.55 (s, 1H, NH, exchangeable by D2O); 13C NMR (76 MHz DMSO-d6): δ/ppm 162.83 (C = O), 161.47 (C-OMe), 156.79 (C = C-N), 150.79 (C = N), 138.61, 132.71, 124.33, 119.40, 116.75, 114.84, 111.18, 103.12, 55.64 (OCH3), 23.52 (CH3); Anal. Calcd. for C17H15N3O2 (293.33): C, 69.61; H, 5.15; N, 14.33 Found: C, 69.32; H, 5.43, N, 14.07.
2-Cyano-3-(4-methylphenyl)- N -(6-methylpyridin-2-yl)acrylamide(3d)
Yellow powder (EtOH); Yield 85%; M.p. = 140–142 ºC; IR (KBr, ν/cm−1): 3396 (NH), 3015 (CH-arom.), 2959, 2920 (CH-aliph.), 2203 (C≡N) and 1698 (C = O amide); 1H NMR (300 MHz DMSO-d6): δ/ppm 2.24, 2.49 (2 s, 6H, 2CH3), 7.06 (d, 2H, J = 7.2 Hz, Ar–H), 7.42 (d, 2H, J = 8.4 Hz, Ar–H), 7.76 (t, 1H, Ar–H), 7.91 (d, 2H, J = 8.4 Hz, Ar–H), 8.32(s, 1H, methine-H),10.72 (s, 1H, NH exchangeable by D2O); 13C NMR (76 MHz DMSO-d6): δ/ppm 161.27 (C = O), 158.05 (C = C-N), 156.76 (C = N), 151.18 (C = CH), 143.72, 138.96, 130.97, 129.32, 119.49, 116.37, 111.24, 106.11, 105.52, 23.71, 21.27 (2CH3); Anal. Calcd. for C17H15N3O (277.33): C, 73.63; H, 5.45; N, 15.15 Found: C, 73.31; H, 5.52, N, 14.97.
Synthesis of 6-amino-1-(aryl)-2-oxo-4-(aryl)-1,2-dihydropyridine-3,5-dicarbonitrile (4a–c)
To a solution of acrylamide derivatives 3a–d (0.01 mol) in absolute ethanol (20 mL) catalyzed with piperidine (3drops), malononitrile (0.01 mol) was added. The reaction mixture was heated under reflux for 6 h. The solid products obtained were collected by filtration and recrystallized from ethanol.
6-Amino-1-(2,4-dichlorophenyl)-2-oxo-4-(4-methoxyphenyl)-1,2-dihydropyridine-3,5-dicarbonitrile (4a)
Brown crystals (EtOH); Yield: 77%; M.p. = 229–231 ºC; IR (KBr, ν/cm−1): 3452, 3213, 3174 (NH2), 3097 (CH-arom.), 2970, 2935 (CH-aliph.), 2225, 2202 (C≡N) and 1678 (C = O amide); 1H NMR (400 MHz, DMSO-d6) δ/ppm 3.81 (s, 3H, OCH3), 6.77 (s, 2H, NH2 exchangeable by D2O), 6.90 (d, J = 7.6 Hz, 1H, Ar–H), 7.09 (d, J = 7.2 Hz, 1H, Ar–H), 7.26 (d, J = 7.6 Hz, 1H, Ar–H), 7.48 (s, 1H, Ar–H), 7.89 (d, J = 7.2 Hz, 2H, Ar–H); 13C NMR (101 MHz, DMSO-d6) δ/ppm 160.48 (C = O), 159.28 (C-NH2), 157.80 (C-OMe), 156.63 (C = C-Ar), 145.47, 135.90, 130.21, 129.45, 128.88, 127.33, 126.16, 116.23, 115.57, 113.80, 113.67 (2CN), 87.41, 75.04 (2C-CN), 55.00 (OCH3); Anal. Calcd. for C20H12Cl2N4O2 (410.03): C, 58.41; H, 2.94; N, 13.62 Found: C, 58.73; H, 3.21, N,13.94.
6-Amino-1-(2,4-dichlorophenyl)-2-oxo-4-(4-methylphenyl)-1,2-dihydropyridine-3,5-dicarbonitrile (4b)
Pale brown crystals (EtOH); Yield 77%; M.p. = 242–244 ºC; IR (KBr, ν/cm−1): 3485, 3421 (NH2), 3087 (CH-arom.), 2982 (CH-aliph.), 2218, 2209 (C≡N), and 1650 (C = O amide); 1H NMR (400 MHz, DMSO-d6) δ/ppm 2.18 (s, 3H, CH3), 6.75 (s, 2H, NH2, exchangeable by D2O), 7.48 (d, J = 8.0 Hz, 2H, Ar–H), 7.53 (d, J = 8.4 Hz, 2H, Ar–H), 7.63 (d, J = 8.4 Hz, 2H, Ar–H), 7.91 (s, 1H, Ar–H); 13C NMR (101 MHz, DMSO-d6) δ/ppm 161.88 (C = O), 159.04 (C-NH2), 157.69 (C = C-Ar), 145.36, 135.78, 134.71, 133.09, 130.72, 130.46, 129.46, 128.73, 128.43, 127.92, 127.83, 127.24, 115.82, 115.18 (2CN), 87.16, 75.16 (2C-CN), 21.81 (CH3); MS m/z (%): 395.67 (M+, 20.12), 343.84 (100%); Anal. Calcd. for C20H12Cl2N4O (395.24): C, 60.78; H, 3.06; N, 14.18 Found: C, 60.32; H, 3.41, N, 14.45.
6-Amino-6'-methyl-2-oxo-4-(4-methylphenyl)-2 H -[1,2'-bipyridine]-3,5-dicarbonitrile (4c)
Brown crystals (EtOH); Yield: 69%; M.p. = 225–227 ºC; IR (KBr, ν/cm−1): 3305, 3213 (NH2), 3035 (CH-arom.), 2981, 2931 (CH-aliph.), 2218, 2191 (2C≡N), and 1647 (C = O amide); 1H NMR (400 MHz, DMSO-d6) δ/ppm 2.10, 2.32 (2 s, 6H, 2CH3), 6.66 (s, 2H, NH2 exchangeable by D2O), 7.16 (d, J = 6.8 Hz, 2H, Ar–H), 7.79–7.68 (m, 2H, Ar–H), 8.00 (d, J = 6.8 Hz, 2H, Ar–H), 8.19 (t, 1H, Ar–H); 13C NMR (101 MHz, DMSO-d6) δ/ppm 160.78 (C = O), 157.52 (C–NH2), 150.37 (C = C–Ar), 140.26, 139.18, 132.19, 131.07, 129.22, 125.97, 123.73, 121.67, 119.42, 115.08, 114.41 (2CN), 85.36, 79.13 (2C-CN), 24.68, 21.43 (2CH3); Anal. Calcd. for C20H15N5O (341.37): C, 70.37; H, 4.43; N, 20.52; Found: C, 70.21; H, 4.77, N, 20.78.
Ethyl 2-amino-5-cyano-1-(2,4-dichlorophenyl)-6-oxo-4-(4-methylphenyl)-1,6-dihydropyridine-3-carboxylate (5)
A mixture of acrylamide derivative 3b (0.01 mol) in absolute ethanol (20 mL) catalyzed with piperidine (3 drops) and ethyl cyanoacetate (0.01 mol) was heated under reflux for 6 h. The solid product obtained was collected by filtration and recrystallized from ethanol.
White crystals (EtOH); Yield 59%; M.p. = 200–202 ºC; IR (KBr, ν/cm−1): 3383, 3278 (NH2), 3066 (CH-arom.), 2993 (CH-aliph.), 2206 (C≡N) and 1639 (br C = O amide); 1H NMR (300 MHz DMSO-d6): δ/ppm 1.51 (t, 3H, CH3), 2.31 (s, 3H, CH3), 4.23 (q, 2H, J = 7.7 Hz, CH2), 7.22 (d, 2H, J = 7.5 Hz, Ar–H), 7.26 (s, 2H, NH2 exchangeable by D2O), 7.33 (d, 2H, J = 8.1 Hz, Ar–H), 7.49 (d, 1H, Ar–H), 7.55 (d, 1H, Ar–H), 7.64 (s, 1H, Ar–H); 13C NMR (75 MHz DMSO-d6): δ/ppm 167.61, 158.35 (C = O), 155.16 (C-NH2), 137.95, 136.35, 134.24, 130.70, 129.13, 128.82, 128.17, 128.26, 127.98, 127.76, 127.38, 125.15, 100.79, 69.58 (O-CH2CH3), 29.05 (CH3), 20.95 (O-CH2CH3); MS m/z (%): 442.86 (M+, 6.21), 286.38 (100%); Anal. Calcd. for C22H17Cl2N3O3 (442.30): C, 59.74; H, 3.87; N, 9.50 Found: C, 59.34; H, 3.33, N, 9.81.
2-Amino-5-cyano- N -(2,4-dichlorophenyl)-4-(4-methoxyphenyl)-6-oxo-1,6-dihydropyridine-3-carboxamide (9)
A solution of acrylamide derivatives 3a (0.01 mol) in absolute ethanol (20 mL) catalyzed with piperidine (3 drops) was heated under reflux condition with cyan acetamide (0.01 mol) for 7 h. The solid product obtained was collected by filtration and recrystallized from ethanol.
Pale orange (EtOH); Yield 71%; M.p. = 198–200 ºC; IR (KBr, ν/cm−1): 3448, 3303, 3174 (NH2, NH), 3043 (CH-arom.), 2961, 2908 (CH-aliph.), 2206 (C≡N) and 1697 (C = O amide); 1H NMR (300 MHz DMSO-d6): δ/ppm 3.84 (s, 3H, OCH3), 7.13 (d, 2H, Ar–H), 7.32 (d, 1H, Ar–H), 7.51 (d, 2H, Ar–H), 7.77 (s, 2H, NH2 exchangeable by D2O), 7.83 (s, 1H, NH exchangeable by D2O), 7.88 (d, 1H, Ar–H), 7.96 (s, 1H, NH exchangeable by D2O), 8.11 (s, 1H, Ar–H); 13C NMR (76 MHz DMSO-d6): δ/ppm 163.13, 162.56 (2C = O), 150.16 (C-NH2), 144.65 (C = C), 142.95, 132.45, 128.64, 126.34, 124.40, 120.47, 117.09, 114.88, 112.54, 102.90, 55.60 (OCH3); MS m/z (%): 428.95 (M+, 25.52). Anal. Calcd. for C20H14Cl2N4O3 (428.04): C, 55.69; H, 3.29, N, 13.05. Found: C, 55.84; H, 3.53, N, 12.84.
6-Hydroxy-6'-methyl-2-oxo-4-(4-methylphenyl)-2H-[1,2'-bipyridine]-5-carbonitrile (13).
A mixture of acrylamide derivatives 3d (0.01 mol) in absolute ethanol (20 mL) catalyzed with piperidine (3 drops) and ethyl acetoacetate (0.01 mol) was heated under reflux for 6 h. The solid product obtained was collected by filtration and recrystallized from ethanol.
Pale orange (EtOH); Yield 73%; M.p. = 165–167 ºC; IR (KBr, ν/cm−1): 3402 (OH), 3008 (CH-arom.), 2966 (CH-aliph.), 2206 (C≡N), and 1693 (C = O amide); 1H NMR (300 MHz DMSO-d6): δ/ppm 2.37, 2.43 (2 s, 6H, 2CH3), 7.03 (d, 1H, J = 7.5 Hz, Ar–H), 7.38 (d, 2H, J = 7.8 Hz, Ar–H), 7.68 (s, 1H, Ar–H), 7.73 (d, 1H, J = 7.8 Hz, Ar–H), 7.89 (d, 2H, J = 8.1 Hz, Ar–H), 8.35 (s, 1H, pyridine-H), 10.62 (s, 1H, OH; exchangeable by D2O); 13C NMR (76 MHz DMSO-d6): δ/ ppm 161.22 (C–OH), 156.73 (C = O), 151.14 (C = N), 150.74 (C = C-Ar), 143.25, 138.58, 130.31, 129.82, 129.14, 119.46, 116.35, 111.22, 105.49, 23.51 (CH3), 21.25 (CH3); MS m/z (%): 317.72 (M+, 20.61), 137.39 (100%); Anal. Calcd. For C19H15N3O2 (317.35): C, 71.91; H, 4.76, N, 13.24. Found: C, 71.62; H, 4.32; N, 12.95.
Synthesis of 5-(Aryl)-3 H -pyrazol-3-one (14a,b)
To a solution of acrylamide derivatives 3a–d (0.01 mol) in absolute ethanol (20 mL), hydrazine hydrate (0.01 mol) was added and the reaction mixture was heated under reflux for 3–5 h. The solid products obtained were collected by filtration and recrystallized from ethanol.
5-(4-Methoxyphenyl)-3 H -pyrazol-3-one (14a)
Yellow powder (EtOH); Yield 66%; M.p. = 165–167 ºC; IR (KBr, ν/cm−1): 3062 (CH-arom.), 2970 (CH-aliph.), and 1660 (C = O amide) 1H NMR (300 MHz DMSO-d6): δ/ppm 3.82 (s, 3H, OCH3), 7.55 (d, 2H, Ar–H), 7.82 (d, 2H, Ar–H), 8.62 (s, 1H, Pyrazole-H); 13C NMR (76 MHz DMSO-d6): δ/ppm 161.66 (C = O), 160.50 (C-OMe), 129.96, 126.55, 114.39, 55.36 (C-OMe); MS m/z (%): 188.04 (M+, 38.72), 185.86 (100%); Anal. Calcd. For C10H8 N2O2 (188.19): C, 63.83; H, 4.29, N; 14.89. Found: C, 63.52; H, 3.687; N, 14.65.
5-(4-Methylphenyl)-3 H -pyrazol-3-one (14b)
Brown powder (EtOH); Yield 69%; M.p. = 150–152 ºC; IR (KBr, ν/cm−1): 3062 (CH-arom.), 2970 (CH-aliph.), and 1655 (C = O amide); 1H NMR (300 MHz DMSO-d6): δ/ppm 2.36 (s, 3H, CH3), 7.32 (d, 2H, J = 8.1 Hz, Ar–H), 7.77 (d, 2H, J = 7.8 Hz, Ar–H), 8.55 (s, 1H, pyrazole-H); 13C NMR (76 MHz DMSO-d6): δ/ppm 161.24 (C = O), 141.32, 131.19, 129.51, 128.31, 21.51 (CH3); MS m/z (%): 172.02 (M+, 42.54), 69.09 (100%); Anal. Calcd. For C10H8 N2O (172.19): C, 69.77; H, 4.68, N; 16.27. Found: C,70.23; H, 4.22; N, 16.54.
5-Amino-1-carbamothioyl- N -(2,4-dichlorophenyl)-3-(4-methoxyphenyl)-1 H -pyrazole-4-carboxamide (16).
A mixture of acrylamide derivative 3b (0.01 mol) in absolute ethanol (20 mL) and thiosemicarbazide (0.01 mol) was heated under reflux for 6 h. The solid product obtained was collected by filtration and recrystallized from ethanol.
Pale brown crystals (EtOH); Yield 78%; M.p. = 150–152 ºC; IR (KBr, ν/cm−1): 3406, 3290, 3151 (NH, NH2), 3041 (CH-arom.), 2970 (CH-aliph.), and 1651 (C = O amide); 1H NMR (300 MHz DMSO-d6): δ/ppm 3.82 (s, 3H, OCH3), 6.94 (d, 2H, J = 6.9 Hz, Ar–H), 7.01 (d, 2H, J = 8.1 Hz, Ar–H), 7.60 (s, 2H, NH2 exchangeable by D2O), 7.73 (d, 1H, J = 6.9 Hz, Ar–H), 7.89 (s, 2H, NH2 exchangeable by D2O), 8.03 (d, 1H, Ar–H), 8.11 (s, 1H, Ar–H), 11.33 (s, 1H, NH exchangeable by D2O); 13C NMR (76 MHz DMSO-d6): δ/ppm 177.66 (C = S), 160.72 (C-OMe), 152.47 (C = O), 144.63 (C = N), 142.40 (C = C), 134.87, 132.95, 130.03, 128.94, 128.13, 127.72, 127.63, 127.44, 126.75, 55.26 (C-OMe); Anal. Calcd. For C18H15Cl2N5O2S (435.03): C, 49.55; H, 3.47, N; 16.05. Found: C, 49.78; H, 3.84; N, 16.37.
Antimicrobial activity
The in vitro antibacterial activity against two gram-positive strains as S. aureus (RCMB010010) and B. subtilis (RCMB 015), and two gram-negative strains as E. coli (RCMB010052) and P. vulgaris (RCMB004) by agar well diffusion method according to the previously reported methods [54, 55, 69].
DFT calculation
All the theoretical calculation were performed using DFT/B3LYP method with a basic set 6-31 g (d) and Gaussian 9.0 package and Gaussian view 6.0.16 software. Additionally, the energy descriptors involving E HOMO, ELUMO, energy bandgap, ionization potential, electron affinity, electronegativity, chemical hardness, chemical softness, chemical potential, and electrophilic index were calculated as described previously [70].
Molecular docking study
The docking process was performed using Molecular operating Environmental (MOE) 10.2008, Chemical computing Group Inc, Montreal, Quench, Canada [71, 72] according to the previously reported method. The most active derivatives 3a, 4a, 4b, 9, and 16 were docked inside the active site of topoisomerase IV (PDB:3FV5). The crystal structure of E. coli topoisomerase IV (PDB:3FV5) was retractive and downloaded from the protein data bank (https://www.rcsb.org/structure/3fv5). Subsequently, the protein structure in pdb format was exported to MOE 10.2008 software. The active site was generated according to standard protocol and according to previously reported method [64], where only one chain was sellected. The missed hydrogen was added, the protein was minimized, refined, and redocked using the original ligand that exerted binding energy S = -23.60 kcal/mol with RMSD = 0.828 Å. The most active derivatives 3a, 4a, 4b, 9, and 16 were drawn and exported to MOE involving added hydrogen, energy minimized using forcefield MMFF94X, and finally saved as a new database. The docking process performed using the trigonal matcher placement and refinement forcefield.
References
R. Wang, X. Yin, Y. Zhang, W. Yan, Design, synthesis and antimicrobial evaluation of propylene-tethered ciprofloxacin-isatin hybrids. Eur. J. Med. Chem. 156, 580–586 (2018). https://doi.org/10.1016/j.ejmech.2018.07.025
Y. Hu, S. Zhang, Z. Xu, Z. Lv, M. Liu, 4-Quinolone hybrids and their antibacterial activities. Eur. J. Med. Chem. 141, 335–345 (2017). https://doi.org/10.1016/j.ejmech.2017.09.050
R. Hasan, M. Acharjee, R. Noor, Prevalence of vancomycin resistant Staphylococcus aureus (VRSA) in methicillin resistant S. aureus (MRSA) strains isolated from burn wound infections. Tzu Chi Med. J. 28, 49–53 (2016). https://doi.org/10.1016/j.tcmj.2016.03.002
Y.A. Ammar, S.Y. Abbas, M.M. Ghorab, M.S. Al Said, Transmonocyanoacetylation of phenylenediamines: a simple and efficient synthesis of novel N-(aminophenyl)-2-cyanoacetamides and their derivatives. Tetrahedron Lett. 57, 275–277 (2016). https://doi.org/10.1016/j.tetlet.2015.11.098
A.A. Fadda, S. Bondock, R. Rabie, H.A. Etman, Cyanoacetamide derivatives as synthons in heterocyclic synthesis. Turkish J. Chem. 32, 259–286 (2008)
P.P. Pandey, Pyridine, BoD–Books on Demand, 2018.
H. Ashihara, I.A. Ludwig, R. Katahira, T. Yokota, T. Fujimura, A. Crozier, Trigonelline and related nicotinic acid metabolites: occurrence, biosynthesis, taxonomic considerations, and their roles in planta and in human health. Phytochem. Rev. 14, 765–798 (2015). https://doi.org/10.1007/s11101-014-9375-z
Y. Hamada, H.D. Tagad, Y. Nishimura, S. Ishiura, Y. Kiso, Tripeptidic BACE1 inhibitors devised by in-silico conformational structure-based design. Bioorg. Med. Chem. Lett. 22, 1130–1135 (2012). https://doi.org/10.1016/j.bmcl.2011.11.102
A. Malani, A. Makwana, J. Monapara, I. Ahmad, H. Patel, N. Desai, Synthesis, molecular docking, DFT study, and in vitro antimicrobial activity of some 4-(biphenyl-4-yl)-1,4-dihydropyridine and 4-(biphenyl-4-yl)pyridine derivatives. J. Biochem. Mol. Toxicol. (2021). https://doi.org/10.1002/jbt.22903
N.C. Desai, H. Somani, A. Trivedi, K. Bhatt, L. Nawale, V.M. Khedkar, P.C. Jha, D. Sarkar, Synthesis, biological evaluation and molecular docking study of some novel indole and pyridine based 1,3,4-oxadiazole derivatives as potential antitubercular agents. Bioorg. Med. Chem. Lett. 26, 1776–1783 (2016). https://doi.org/10.1016/j.bmcl.2016.02.043
F.A. Bassyouni, H.A. Tawfik, A.M. Soliman, M.A. Rehim, Synthesis and anticancer activity of some new pyridine derivatives. Res. Chem. Intermed. 38, 1291–1310 (2012). https://doi.org/10.1007/s11164-011-0413-9
B. Martinez-Gualda, S.-Y. Pu, M. Froeyen, P. Herdewijn, S. Einav, S. De Jonghe, Structure-activity relationship study of the pyridine moiety of isothiazolo[4,3-b]pyridines as antiviral agents targeting cyclin G-associated kinase. Bioorg. Med. Chem. 28, 115188 (2020). https://doi.org/10.1016/j.bmc.2019.115188
A. Ragab, S.A. Fouad, O.A.A. Ali, E.M. Ahmed, A.M. Ali, A.A. Askar, Y.A. Ammar, Sulfaguanidine Hybrid with Some New Pyridine-2-One Derivatives: Design Synthesis, and Antimicrobial Activity against Multidrug-Resistant Bacteria as Dual DNA Gyrase and DHFR Inhibitors. Antibiotics 10, 162 (2021). https://doi.org/10.3390/antibiotics10020162
A. Chaubey, S.N. Pandeya, Pyridine" a versatile nucleuse in pharmaceutical field. Asian J. Pharm. Clin. Res. 4, 5–8 (2011)
A. Kamboj, B. Sihag, D.S. Brar, A. Kaur, D.B. Salunke, Structure activity relationship in β-carboline derived anti-malarial agents. Eur. J. Med. Chem. 221, 113536 (2021). https://doi.org/10.1016/j.ejmech.2021.113536
A. Mehanna, Antidiabetic agents: past, present and future. Future Med. Chem. 5, 411–430 (2013). https://doi.org/10.4155/fmc.13.13
S.M. Gomha, Z.A. Muhammad, M.R. Abdel-aziz, H.M. Abdel-aziz, H.M. Gaber, M.M. Elaasser, One-pot synthesis of new thiadiazolyl-pyridines as anticancer and antioxidant agents. J. Heterocycl. Chem. 55, 530–536 (2018). https://doi.org/10.1002/jhet.3088
J.-F. Renard, F. Lecomte, P. Hubert, X. de Leval, B. Pirotte, N-(3-Arylaminopyridin-4-yl)alkanesulfonamides as pyridine analogs of nimesulide: Cyclooxygenases inhibition, anti-inflammatory studies and insight on metabolism. Eur. J. Med. Chem. 74, 12–22 (2014). https://doi.org/10.1016/j.ejmech.2013.12.033
A.A. Bekhit, A.M. Baraka, Novel milrinone analogs of pyridine-3-carbonitrile derivatives as promising cardiotonic agents. Eur. J. Med. Chem. 40, 1405–1413 (2005). https://doi.org/10.1016/j.ejmech.2005.06.005
N. Bharti, M.R. Maurya, F. Naqvi, A. Azam, Synthesis and antiamoebic activity of new cyclooctadiene ruthenium(II) complexes with 2-acetylpyridine and benzimidazole derivatives. Bioorg. Med. Chem. Lett. 10, 2243–2245 (2000). https://doi.org/10.1016/S0960-894X(00)00446-7
B. Wu, V. Oesker, J. Wiese, R. Schmaljohann, J.F. Imhoff, Two new antibiotic pyridones produced by a marine fungus, Trichoderma sp. strain MF106. Mar. Drugs. 12, 1208–1219 (2014). https://doi.org/10.3390/md12031208
Y. Yan, X. Li, C. Zhang, L. Lv, B. Gao, M. Li, Research progress on antibacterial activities and mechanisms of natural alkaloids: a review. Antibiot. (2021). https://doi.org/10.3390/antibiotics10030318
N.C. Desai, B.Y. Patel, B.P. Dave, Synthesis and antimicrobial activity of novel quinoline derivatives bearing pyrazoline and pyridine analogues. Med. Chem. Res. 26, 109–119 (2017). https://doi.org/10.1007/s00044-016-1732-6
L.J. Piddock, R.N. Walters, J.M. Diver, Correlation of quinolone MIC and inhibition of DNA, RNA, and protein synthesis and induction of the SOS response in Escherichia coli. Antimicrob. Agents Chemother. 34, 2331–2336 (1990). https://doi.org/10.1128/AAC.34.12.2331
A. Titi, M. Messali, B.A. Alqurashy, R. Touzani, T. Shiga, H. Oshio, M. Fettouhi, M. Rajabi, F.A. Almalki, T. Ben Hadda, Synthesis, characterization, X-Ray crystal study and bioctivities of pyrazole derivatives: Identification of antitumor, antifungal and antibacterial pharmacophore sites. J. Mol. Struct. 1205, 127625 (2020). https://doi.org/10.1016/j.molstruc.2019.127625
S.O. Ojwach, J. Darkwa, Pyrazole and (pyrazol-1-yl)metal complexes as carbon–carbon coupling catalysts, Inorganica Chim. Acta. 363, 1947–1964 (2010). https://doi.org/10.1016/j.ica.2010.02.014
A. Jamwal, A. Javed, V. Bhardwaj, A review on pyrazole derivatives of pharmacological potential. J. Pharm. BioSci. 3, 114–123 (2013)
S.T. Heller, S.R. Natarajan, Rapid access to pyrazolo[3,4-c]pyridines via alkyne annulation: limitations of steric control in nickel-catalyzed alkyne insertions. Org. Lett. 9, 4947–4950 (2007). https://doi.org/10.1021/ol701784w
J. Zhang, Y. Zhang, W.F.K. Schnatter, J.W. Herndon, Coupling of N-Heterocycle-Fused Enyne Aldehydes with γ, δ-Unsaturated Fischer Carbene Complexes. Organometallics 25, 1279–1284 (2006). https://doi.org/10.1021/om051008q
S.F. Vasilevsky, E.V. Mshvidobadze, V.I. Mamatyuk, G.V. Romanenko, J. Elguero, Unexpected results in the heterocyclization of 5-acetylenylpyrazole-4-carboxylic acid hydrazides under the influence of CuCl: formation of a diazepinone and dehydrodimerization into the corresponding bis(pyrazolo[4,3-d][1,2]diazepinone). Tetrahedron Lett. 46, 4457–4459 (2005). https://doi.org/10.1016/j.tetlet.2005.04.127
A.R. Katritzky, M. Wang, S. Zhang, M.V. Voronkov, P.J. Steel, Regioselective synthesis of polysubstituted pyrazoles and isoxazoles. J. Org. Chem. 66, 6787–6791 (2001). https://doi.org/10.1021/jo0101407
S.M.M.R. In, Comprehensive Heterocyclic Chemistry II Vol. 3: Katritzky AR. Rees CW. Scriven EFV, (1996).
A.S. Hassan, A.A. Askar, A.M. Naglah, A.M. Naglah, A.A. Almehizia, A.A. Almehizia, A. Ragab, Discovery of new schiff bases tethered pyrazole moiety: design, synthesis, biological evaluation, and molecular docking study as dual targeting DHFR/DNA gyrase inhibitors with immunomodulatory activity. Molecules 25, 2593 (2020). https://doi.org/10.3390/molecules25112593
A. Masih, A.K. Agnihotri, J.K. Srivastava, N. Pandey, H.R. Bhat, U.P. Singh, Discovery of novel pyrazole derivatives as a potent anti-inflammatory agent in RAW264.7 cells via inhibition of NF-ĸB for possible benefit against SARS-CoV-2. J. Biochem. Mol. Toxicol. 35, e22656 (2021). https://doi.org/10.1002/jbt.22656
S.S. Abd El-Karim, H.S. Mohamed, M.F. Abdelhameed, A. El-Galil, E. Amr, A.A. Almehizia, E.S. Nossier, Design, synthesis and molecular docking of new pyrazole-thiazolidinones as potent anti-inflammatory and analgesic agents with TNF-α inhibitory activity. Bioorg. Chem. 111, 104827 (2021). https://doi.org/10.1016/j.bioorg.2021.104827
M. Abdel-Aziz, G.E.-D.A. Abuo-Rahma, A.A. Hassan, Synthesis of novel pyrazole derivatives and evaluation of their antidepressant and anticonvulsant activities. Eur. J. Med. Chem. 44, 3480–3487 (2009). https://doi.org/10.1016/j.ejmech.2009.01.032
M.M.S. Wassel, W.M.G. El-din, A. Ragab, G.A.M.E. Ali, Y.A. Ammar, Antiviral activity of adamantane-pyrazole derivatives against foot and mouth disease virus infection in vivo and in vitro with molecular docking study. J. Appl. Vet. Sci. 5, 37–46 (2020). https://doi.org/10.21608/JAVS.2020.118001
M.M.S. Wassel, A. Ragab, G.A.M. Elhag Ali, A.B.M. Mehany, Y.A. Ammar, Novel adamantane-pyrazole and hydrazone hybridized: design, synthesis, cytotoxic evaluation, SAR study and molecular docking simulation as carbonic anhydrase inhibitors. J. Mol. Struct. 1223, 128966 (2020). https://doi.org/10.1016/j.molstruc.2020.128966
K.Y. Lee, J.M. Kim, J.N. Kim, Regioselective synthesis of 1,3,4,5-tetrasubstituted pyrazoles from Baylis-Hillman adducts. Tetrahedron Lett. 44, 6737–6740 (2003). https://doi.org/10.1016/S0040-4039(03)01648-4
H. Ali Mohamed, Y.A. Ammar, G.A.M. Elhagali, H.A. Eyada, D.S. Aboul-Magd, A. Ragab, In vitro antimicrobial evaluation, single-point resistance study, and radiosterilization of novel pyrazole incorporating thiazol-4-one/thiophene derivatives as dual DNA gyrase and DHFR inhibitors against MDR pathogens. ACS Omega (2022). https://doi.org/10.1021/acsomega.1c05801
H.F. Rizk, M.A. El-Borai, A. Ragab, S.A. Ibrahim, M.E. Sadek, A novel of azo-thiazole moiety alternative for benzidine-based pigments: design, synthesis, characterization, biological evaluation, and molecular docking study. Polycycl. Aromat. Compd. (2021). https://doi.org/10.1080/10406638.2021.2015402
A. Ezzat, M.B.I. Mohamed, A.M. Mahmoud, R.S. Farag, A.S. El-Tabl, A. Ragab, Synthesis, spectral characterization, antimicrobial evaluation and molecular docking studies of new Cu (II), Zn (II) thiosemicarbazone based on sulfonyl isatin. J. Mol. Struct. 1251, 132004 (2022). https://doi.org/10.1016/j.molstruc.2021.132004
E.S.A.E.H. Khattab, A. Ragab, M.A. Abol-Ftouh, A.A. Elhenawy, Therapeutic strategies for Covid-19 based on molecular docking and dynamic studies to the ACE-2 receptors, Furin, and viral spike proteins. J. Biomol. Struct. Dyn. 40, 1–19 (2022). https://doi.org/10.1080/07391102.2021.1989036
S.A. Ibrahim, A. Ragab, H.A. El-Ghamry, Coordination compounds of pyrazolone-based ligand: design, characterization, biological evaluation, antitumor efficiency, and DNA binding evaluation supported by in silico studies. Appl. Organomet. Chem. 36, 1–17 (2022). https://doi.org/10.1002/aoc.6508
Y.A. Ammar, M.M. Aly, A.A.G. Al-Sehemi, M.A. Salem, M.S.A. El-Gaby, Cyanoacetanilides intermediates in heterocyclic synthesis. Part 5: preparation of hitherto unknown 5-aminopyrazole and pyrazolo [1, 5-a] pyrimidine derivatives containing sulfamoyl moiety. J. Chin. Chem. Soc. 56, 1064–1071 (2009)
Y.A. Ammar, M.S.A. El-Gaby, M.A. Salem, Cyanoacetanilides intermediates in heterocyclic synthesis. Part 6: Preparation of some hitherto unknown 2-oxopyridine, bipyridine, isoquinoline and chromeno[3,4-c]pyridine containing sulfonamide moiety. Arab. J. Chem. 7, 615–622 (2014). https://doi.org/10.1016/j.arabjc.2013.11.026
P. Wang, S.M. Batt, B. Wang, L. Fu, R. Qin, Y. Lu, G. Li, G.S. Besra, H. Huang, Discovery of novel thiophene-arylamide derivatives as DprE1 inhibitors with potent antimycobacterial activities. J. Med. Chem. 64, 6241–6261 (2021). https://doi.org/10.1021/acs.jmedchem.1c00263
S.T. Cheung, M.S. Miller, R. Pacoma, J. Roland, J. Liu, A.M. Schumacher, L.C. Hsieh-Wilson, Discovery of a small-molecule modulator of glycosaminoglycan sulfation. ACS Chem. Biol. 12, 3126–3133 (2017). https://doi.org/10.1021/acschembio.7b00885
M. Hajjami, F. Ghorbani, S. Rahimipanah, S. Roshani, Efficient preparation of Zr(IV)-salen grafted mesoporous MCM-41 catalyst for chemoselective oxidation of sulfides to sulfoxides and Knoevenagel condensation reactions. Chin. J. Catal. 36, 1852–1860 (2015). https://doi.org/10.1016/S1872-2067(15)60968-8
A.S. Hassan, N.M. Morsy, H.M. Awad, A. Ragab, Synthesis, molecular docking, and in silico ADME prediction of some fused pyrazolo[1,5-a]pyrimidine and pyrazole derivatives as potential antimicrobial agents. J. Iran. Chem. Soc. 19, 521–545 (2022). https://doi.org/10.1007/s13738-021-02319-4
H.F. Rizk, M.A. El-Borai, A. Ragab, S.A. Ibrahim, Design, synthesis, biological evaluation and molecular docking study based on novel fused pyrazolothiazole scaffold. J. Iran. Chem. Soc. 17, 2493–2505 (2020). https://doi.org/10.1007/s13738-020-01944-9
D. Secci, A. Bolasco, P. Chimenti, S. Carradori, The state of the art of pyrazole derivatives as monoamine oxidase inhibitors and antidepressant/anticonvulsant agents. Curr. Med. Chem. 18, 5114–5144 (2011)
S. Singh Jadav, B. Nayan Sinha, B. Pastorino, X. de Lamballerie, R. Hilgenfeld, V. Jayaprakash, Identification of pyrazole derivative as an antiviral agent against Chikungunya through HTVS. Lett. Drug Des. Discov. 12, 292–301 (2015)
Y.A. Ammar, S.M.A.A. El-Hafez, S.A. Hessein, A.M. Ali, A.A. Askar, A. Ragab, One-pot strategy for thiazole tethered 7-ethoxy quinoline hybrids: synthesis and potential antimicrobial agents as dihydrofolate reductase (DHFR) inhibitors with molecular docking study. J. Mol. Struct. 1242, 130748 (2021). https://doi.org/10.1016/j.molstruc.2021.130748
D.M. Elsisi, A. Ragab, A.A. Elhenawy, A.A. Farag, A.M. Ali, Y.A. Ammar, Experimental and theoretical investigation for 6-Morpholinosulfonylquinoxalin-2(1H)-one and its haydrazone derivate: synthesis, characterization, tautomerization and antimicrobial evaluation. J. Mol. Struct. 1247, 131314 (2022). https://doi.org/10.1016/j.molstruc.2021.131314
A. Ragab, D.M. Elsisi, O.A. Abu Ali, M.S. Abusaif, A.A. Askar, A.A. Farag, Y.A. Ammar, Design, synthesis of new novel quinoxalin-2(1H)-one derivatives incorporating hydrazone, hydrazine, and pyrazole moieties as antimicrobial potential with in-silico ADME and molecular docking simulation. Arab. J. Chem. 15, 103497 (2022). https://doi.org/10.1016/j.arabjc.2021.103497
M. Erol, I. Celik, E. Uzunhisarcikli, G. Kuyucuklu, Synthesis, molecular docking, and DFT studies of some new 2,5-disubstituted benzoxazoles as potential antimicrobial and cytotoxic agents. Polycycl. Aromat. Compd. (2020). https://doi.org/10.1080/10406638.2020.1802305
A.Y. Alzahrani, Y.A. Ammar, M. Abu-Elghait, M.A. Salem, M.A. Assiri, T.E. Ali, A. Ragab, Development of novel indolin-2-one derivative incorporating thiazole moiety as DHFR and quorum sensing inhibitors: synthesis, antimicrobial, and antibiofilm activities with molecular modelling study. Bioorg. Chem. 119, 105571 (2022). https://doi.org/10.1016/j.bioorg.2021.105571
S.A. Khan, A.M. Asiri, H.M. Basisi, M. Asad, M.E.M. Zayed, K. Sharma, M.Y. Wani, Synthesis and evaluation of quinoline-3-carbonitrile derivatives as potential antibacterial agents. Bioorg. Chem. 88, 102968 (2019). https://doi.org/10.1016/j.bioorg.2019.102968
I. Celik, M. Erol, O. Temiz Arpaci, F. Sezer Senol, I. Erdogan Orhan, Evaluation of activity of some 2,5-disubstituted benzoxazole derivatives against acetylcholinesterase, butyrylcholinesterase and tyrosinase: ADME prediction, DFT and comparative molecular docking studies. Polycycl. Aromat. Compd. 42, 412–423 (2022). https://doi.org/10.1080/10406638.2020.1737827
M.M.S. Wassel, Y.A. Ammar, G.A.M. Elhag Ali, A. Belal, A.B.M. Mehany, A. Ragab, Development of adamantane scaffold containing 1,3,4-thiadiazole derivatives: design, synthesis, anti-proliferative activity and molecular docking study targeting EGFR. Bioorg. Chem. 110, 104794 (2021). https://doi.org/10.1016/j.bioorg.2021.104794
S.A. Ibrahim, H.F. Rizk, D.S. Aboul-Magd, A. Ragab, Design, synthesis of new magenta dyestuffs based on thiazole azomethine disperse reactive dyes with antibacterial potential on both dyes and gamma-irradiated dyed fabric. Dye. Pigment. 193, 109504 (2021). https://doi.org/10.1016/j.dyepig.2021.109504
E.A. Fayed, Y.A. Ammar, M.A. Saleh, A.H. Bayoumi, A. Belal, A.B.M. Mehany, A. Ragab, Design, synthesis, antiproliferative evaluation, and molecular docking study of new quinoxaline derivatives as apoptotic inducers and EGFR inhibitors. J. Mol. Struct. 1236, 130317 (2021). https://doi.org/10.1016/j.molstruc.2021.130317
M.O. Puskullu, I. Celik, M. Erol, H. Fatullayev, E. Uzunhisarcikli, G. Kuyucuklu, Antimicrobial and antiproliferative activity studies of some new quinoline-3-carbaldehyde hydrazone derivatives. Bioorg. Chem. 101, 104014 (2020). https://doi.org/10.1016/j.bioorg.2020.104014
M.A. Salem, A. Ragab, A.A. Askar, A. El-Khalafawy, A.H. Makhlouf, One-pot synthesis and molecular docking of some new spiropyranindol-2-one derivatives as immunomodulatory agents and in vitro antimicrobial potential with DNA gyrase inhibitor. Eur. J. Med. Chem. 188, 111977 (2020). https://doi.org/10.1016/j.ejmech.2019.111977
A.R.Y.A. Ammar, A.MSh. El-Sharief, A. Belal, S.Y. Abbas, Y.A. Mohamed, A.B.M. Mehany, Design, synthesis, antiproliferative activity, molecular docking and cell cycle analysis of some novel (morpholinosulfonyl) isatins with potential EGFR inhibitory activity. Eur. J. Med. Chem. 156, 918–932 (2018). https://doi.org/10.1016/j.ejmech.2018.06.061
E.A. Fayed, A. Ragab, R.R. Ezz Eldin, A.H. Bayoumi, Y.A. Ammar, In vivo screening and toxicity studies of indolinone incorporated thiosemicarbazone, thiazole and piperidinosulfonyl moieties as anticonvulsant agents. Bioorg. Chem. (2021). https://doi.org/10.1016/j.bioorg.2021.105300
S.A. Ibrahim, E.A. Fayed, H.F. Rizk, S.E. Desouky, A. Ragab, Hydrazonoyl bromide precursors as DHFR inhibitors for the synthesis of bis-thiazolyl pyrazole derivatives; antimicrobial activities, antibiofilm, and drug combination studies against MRSA. Bioorg. Chem. 116, 105339 (2021). https://doi.org/10.1016/j.bioorg.2021.105339
A. Ragab, Y.A. Ammar, A. Ezzat, A.M.M.M.B.I. Mohamed, A.S. El-Tabl, R.S. Farag, Synthesis, characterization, thermal properties, antimicrobial evaluation, ADMET study, and molecular docking simulation of new mono Cu (II) and Zn (II) complexes with 2-oxoindole derivatives. Comput. Biol. Med. 145, 105473 (2022). https://doi.org/10.1016/j.compbiomed.2022.105473
A.Y. Alzahrani, Y.A. Ammar, M.A. Salem, M. Abu-Elghait, A. Ragab, Design, synthesis, molecular modeling, and antimicrobial potential of novel 3-[(1H-pyrazol-3-yl)imino]indolin-2-one derivatives as DNA gyrase inhibitors. Arch. Pharm. (Weinheim) 355, e2100266 (2022). https://doi.org/10.1002/ardp.202100266
M. Eldeeb, E.F. Sanad, A. Ragab, Y.A. Ammar, K. Mahmoud, M.M. Ali, N.M. Hamdy, Anticancer effects with molecular docking confirmation of newly synthesized Isatin sulfonamide molecular hybrid derivatives against hepatic cancer cell lines. Biomedicines (2022). https://doi.org/10.3390/biomedicines10030722
E.A. El-Kalyoubi, S.A.; Ragab, A.; Abu Ali, O.A.; Ammar, Y.A.; Seadawy, M.G.; Ahmed, A.; Fayed, One-Pot Synthesis and Molecular Modeling Studies of New Bioactive Spiro-Oxindoles Based on Uracil Derivatives as SARS-CoV-2 Inhibitors Targeting RNA Polymerase and Spike Glycoprotein, Pharm. Artic. 15 (2022). https://doi.org/10.3390/ph15030376 Academic.
Funding
Open access funding provided by The Science, Technology & Innovation Funding Authority (STDF) in cooperation with The Egyptian Knowledge Bank (EKB).
Author information
Authors and Affiliations
Corresponding authors
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Saadon, K.E., Taha, N.M.H., Mahmoud, N.A. et al. Synthesis, characterization, and in vitro antibacterial activity of some new pyridinone and pyrazole derivatives with some in silico ADME and molecular modeling study. J IRAN CHEM SOC 19, 3899–3917 (2022). https://doi.org/10.1007/s13738-022-02575-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13738-022-02575-y