
Abstract
A MgCl2-based Ziegler–Natta catalyst was characterized using X-ray diffraction (XRD) patterns, scanning electron microscopy (SEM) and transmission electron microscopy (TEM) and IR spectra. We focused on the XRD reflection at 2θ = 50° to determine the thickness of MgCl2 crystals, and validated these results with TEM pictures. SEM pictures were taken in order to measure the size of the nanoparticles formed by the MgCl2 crystals. Several compounds were synthesized for comparison and to aid interpretation of the infrared (IR) spectra. The catalysts were prepared by precipitating MgCl2, which was used as support material and subsequently treated with TiCl4. The thickness of the catalyst crystals was calculated from the XRD reflection at 2θ = 50°. Changing the precipitation temperature within a range from 40 to 90 °C altered the thickness of the MgCl2 crystal plates. The maximum thickness of 7 nm was achieved at a precipitation temperature of 60 °C. The SEM pictures showed that the nanoparticles had a diameter of ~200 nm. A crystal base unit had a volume that corresponded to that of a sphere of 3.5 nm radius. Thus, we estimated that a typical catalyst particle with a diameter of 20 μm contained about one million nanoparticles, each of which consisted of about 25,000 MgCl2 crystal units.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
In chemical industry, Ziegler–Natta catalysts are used to produce large volumes of polyethylene and polypropylene [1–3]. These catalysts produce a polymer material with a rather broad molecular weight distribution indicating that the catalysts have a structure of at least five different types of active sites [4–6]. The main components of these catalysts are: MgCl2 as support material, TiCl4 as the active transition metal, and triethylaluminum (AlEt3, TEA) as co-catalyst. The MgCl2 support material must be in a disordered, amorphous crystal state to enable the TiCl4 to link via coordination bonds to the edges of the MgCl2 crystal plates and thereby create active polymerization centers. The amorphous structure of the MgCl2 support material is created by chemical activation in the course of catalyst synthesis. AlEt3 acts as a co-catalyst and activates the titanium (Bahri-Laleh model) [7] by shifting an ethyl group to the titanium (Stukalov reaction) [8, 9]. Polymerization starts from the ethyl group that is bound to the titanium (Cossee and Trigger mechanisms) [10–12]. Coordination of the titanium to the amorphous MgCl2 results in high polymerization activity of the titanium.
Numerous studies have sought to determine the optimal structure of the MgCl2 support material of Ziegler–Natta catalysts and to describe the different types of active sites [13]. X-Ray diffraction (XRD) using a wavelength of 1.5406 Å has been employed to describe the activation of the catalyst components. Inactivated MgCl2 material has an XRD pattern with a strong dominant reflection at 15° 2θ, which gives the height of the crystal and indicates that inactivated MgCl2 has a needle-like structure [14]. At 30° 2θ and 35° 2θ, inactivated MgCl2 exhibits intense intermediate reflection peaks which result from the diagonal planes in the needle-like structure. The final significant reflection peak is at 50° 2θ; compared to the other peaks of inactivated MgCl2, it is usually much smaller and indicates the thickness of the MgCl2 crystal [15].
On activation of the MgCl2 support material, the most drastic changes in the crystal structure can be seen on the left half of the XRD pattern. The reflection peak at 15° 2θ is strongly reduced, which suggests that the needle-like structure of the MgCl2 crystals collapses almost completely, leaving only 5–10 crystal plates stacked on top of each other in the activated material. The intensities of the intermediate reflection peaks also decrease at 30° 2θ and 35° 2θ, and additionally the peaks merge and form an intermediate halo that indicates strong disorderliness in the MgCl2 material [15].
The only reflection peak that remains almost unchanged by the activation process of the MgCl2 material is 50° 2θ; suggesting that the thickness of MgCl2 crystal plates is almost the same as in inactivated MgCl2.
Since most of the changes in MgCl2 crystals during the activation process are thought to be related to the destruction of their needle-like structure, most X-ray studies in this area have focused on the degradation of the reflection at 15° 2θ and on the degradation and halo formation between 30° 2θ and 35° 2θ reflection peaks in order to explain the increasing disorder in the structure of the MgCl2 support material. Little or no attention has been paid to the reflection at 50° 2θ, probably since it usually remains largely unaffected by the activation process.
In this study, a typical MgCl2-based Ziegler–Natta polyethylene catalyst was characterized by interpreting the XRD patterns, SEM and TEM pictures and IR spectra of the catalyst and the MgCl2 support material. We focused on the XRD at 50° 2θ to determine whether additional information can be gained from the crystallographic data.
A typical Ziegler–Natta polyethylene catalyst was prepared for this investigation by first precipitating, and concurrently chemically activating the MgCl2 support material via a reaction between magnesium alcoholate [Mg(OR)2] and ethylaluminum dichloride (EtAlCl2, EADC). The final catalyst was obtained by treating this support material with titanium tetrachloride (TiCl4). In this treatment step, the titanium is linked to the support material via coordination bonds. We varied the synthetic conditions in terms of temperature and rate of addition of alcohol to determine whether this causes a significant change in the thickness of MgCl2 support material and thus of the final catalyst.
Further, TEM pictures were acquired to verify the results of the XRD measurements. SEM pictures were used to find out whether a correlation exists between the size of the nanoparticles [16] in the produced material and the crystal thickness measured by X-ray at 50° 2θ. With the help of IR spectroscopy the positions of the chlorine and the alchoholate groups on Mg, Al and Ti were determined. Compounds were prepared to interpret and compare the IR spectra of the support material and the catalysts. Our aim was to discover whether there was a clear change in the chemical composition of the support material due to the titanation process carried out in the last catalyst synthesis step. Finally, we used a fast polymerization method to demonstrate that the catalyst contained active titanium.
Experimental
Model compounds
Strictly inert conditions were used in all synthesis steps when preparing the model compounds, the support materials, and the catalysts. Nitrogen (5.0, Linde, Germany) was used to create an inert atmosphere and inert conditions for characterization of these materials. In handling of the KBr tablets for IR measurements, it was not possible to achieve totally inert conditions, as the tablets had to be transferred into the IR instrument after their preparations. Consequently, an absorption band between 3200 and 3500 cm−1 due to absorbed moisture could be seen in all IR spectra of the KBr tablets. This moisture absorption also shifted all metal–oxygen absorption peaks in the IR spectra to lower wavelengths. All reagents were of analytical grade.
Butyloctylmagnesium (Chemtura, USA), ethylaluminum dichloride (AcroSeal, USA), diethylaluminum chloride (Sigma Aldrich, USA), triethylaluminum (Aldrich, USA) and titanium tetrachloride (Aldrich, USA) were used as provided by the manufacturer. All monomers and solvents were purified in custom-made purification columns before polymerization process. Ethene (purity >99.9 %, obtained from Air Liquide) was purified by passing through three purification columns, first through a bed of oxidized BASF R3-16 catalyst (CO scavenging to CO2), second through reduced BASF R3-16 catalyst (absorption of O2), third through molecular sieves (3A, 4A, 13X, obtained from Sigma-Aldrich) to physically absorb CO2, H2O, and other impurities and finally passed through a 50/50-combination of Selexorb-types COS and CD (Alcoa) for removing COS, H2S, PH3 and oxygenates, e.g., ethers, alcohol, aldehydes, carbonyls, ketones, and peroxides. Hydrogen, nitrogen (purity >99.999 %, Linde), and heptane (purity >95 %, VWR) were purified over reduced BASF R3-11 catalyst and molecular sieves (13X, 4A, 3A). To achieve an optimum purification, all columns containing BASF catalysts were heated to 200 °C, except the columns for heptane, which were heated to 60 °C because of its vapor pressure. The purity of the compounds cleaned by this procedure was sufficiently high, so no additional treatment by Na or CaH2 was needed.
One of our aims was to use the IR spectra of the support material and the catalysts to localize the alcoholate, chlorine and ethyl groups in the product, more precisely and to determine in what quantities they were bound to Ti, Mg or Al. To be able to interpret the IR spectra of the support material and the catalysts, we prepared model compounds (Table 1). IR spectra of these model compounds were recorded, and their characteristic absorption peaks were identified.
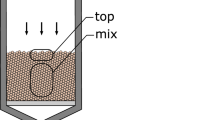
Magnesium dichloride was prepared using a 150 mL glass reactor (Fig. 1). The reactor was constructed such that it could be placed in a centrifuge, where precipitate and liquid phase could be separated efficiently. An amount of 4.3057 g (5.2465 mmol Mg) of a 20 % n-heptane solution of butyloctylmagnesium (BOMAG) was introduced into the glass reactor. The reactor was equipped with an outlet via an oil lock. An amount of 4.9816 g (26.2325 mmol) of TiCl4 was introduced into another glass reactor that was connected to the first reactor via a pipeline. The TiCl4 was diluted by adding 10 mL of n-heptane. Over a period of 30 min, 8.22 mL of 2-ethyl-hexanol was added dropwise at 90 °C into the reactor containing TiCl4. To improve the release of HCl, the temperature was increased to 90 °C. Reaction of the alcohol with TiCl4 yielded HCl gas, which passed through the pipeline and entered into the reactor containing the magnesium-alkyl, resulting in its chlorination and finally in precipitation of MgCl2 (0605-1). After the addition was completed, the MgCl2 precipitate was separated from the liquid by centrifugation. The MgCl2 was washed once at room temperature with 10 mL n-heptane and next with 10 mL pentane, and it was subsequently dried under a stream of nitrogen at 50–60 °C. A tablet consisting of 200 mg KBr and 2 mg of the MgCl2 precipitate was obtained and used for measuring the IR absorption spectrum.

The experimental setup used for preparation of MgCl2
Our procedure for synthesizing the Ziegler–Natta PE catalyst followed a typical industrial procedure that can be found in patented literature [17], and it also resembles procedures described in other publications [18, 19] and to some extent the recipe provided by Wang et al. [20]. The carrier synthesis described by Nakayama et al. [21] is an example of a support material preparation that resembles the recipe used in this study. The carrier material produced for the immobilization of homogeneous catalysts, described by Severn et al. also has a composition corresponding to the support material produced in our study [22].
Preparation of magnesium alcoholate
A magnesium alcoholate solution was prepared according the following procedure: under inert conditions, 10.0 g (18.1 mmol) of a 20.0 % toluene solution of butylethyl magnesium (BEM) was introduced into a glass reactor. Alternatively, 14.9 g (18.1 mmol) of a 20.3 % n-heptane solution of BOMAG was introduced. A volume of 10 mL n-heptane was added to reduce viscosity. Over a period of 20 min, a mass of 4.34 g (33.2 mmol) of 2-ethyl-hexanol (ROH) was added dropwise into the reactor while mixing. The temperature was kept between 20 and 25 °C at all times. The ROH/Mg molar ratio was 1.83. After complete addition, the reaction solution was allowed to stabilize for an hour at room temperature. The main part of the reaction was:
Precipitation of MgCl2 carrier material
The reactor system described above was used to prepare both the MgCl2 support material and the catalyst. 9.34 g (18.1 mmol) of a 1.80 M toluene solution of EADC was introduced into this reactor under inert conditions. Alternatively, 14.9 g (18.1 mmol) of a 0.90 M n-heptane solution of EADC was introduced. The aim of our study was to adjust MgCl2 crystal thickness by varying the precipitation temperature or the EADC/Mg(OR)2 ratio, because small catalyst particle size, and thus often low crystal thickness can be achieved by precipitation. It is assumed that small MgCl2 crystals have more defects where Ti can form active centers, which in turn leads to higher activity. The precipitation conditions were varied by which the MgCl2 support material was created in order to adjust catalyst crystal size and thereby catalyst performance. In the first test series, the temperature in this precipitation step was selected between 40 and 100 °C, and in the second test series the EADC/Mg(OR)2 ratio was varied within the range 0.5–2. The magnesium alcoholate solution was then added dropwise into the reaction solution while mixing. A white precipitate started to form after about 25 % of the magnesium alcoholate solution had been added into the reactor. The total addition time was 20 min. The precipitation reaction was according to the following scheme:
The precipitated amount of MgCl2 was dependent on the reaction conditions. According to Chien et al. [23]. the “x” in the precipitate of MgCl2(EtAl(OR)2)x can be any value between 0 and 1, as it is not a stoichiometric compound like TiCl3(AlCl3)0.33 or MgCl2(H2O)6. The composition of the neutral complex between MgCl2 and EtAl(OR)2 would produce a soluble product MgCl2(EtAl(OR)2)3.
After complete addition of the Mg(OR)2, the reactants were allowed to react with each other for 30 min at 60 °C. When precipitation of the MgCl2 carrier had finished, the precipitate was separated from the liquid phase in a centrifuge for 5 min at 2000 rpm. Subsequently, the precipitate was washed twice with 20 mL portions of n-heptane at 60 °C for 20 min. Support material intended for composition investigation was washed at room temperature with 20 mL of hexane and then dried under a stream of nitrogen at 40–60 °C.
Synthesis of the Ziegler–Natta catalyst
Catalyst preparation started by producing the support material in a slurry form in 7 mL of n-heptane. Next, 1.72 g (9.07 mmol) TiCl4 diluted with 10 mL of n-heptane were added. The vial that had contained the TiCl4 was washed with 3 mL of n-heptane, which was then added into the reactor to ensure complete transfer of the TiCl4. The reaction components were then allowed to react for 30 min at 60 °C. In this reaction, the EtAl(OR)2 was partially washed out from the precipitate and exchanged with TiCl4. In addition, some of the alcoholate groups shifted to the titanium according to:
According to Chien et al. [23] the presence of EtAl(OR)2 in the precipitate also causes part of the titanium to reduce from Ti(IV) to Ti(III) via the reactions:
After completion of the reaction, the catalyst was separated from the solution by centrifugation for 5 min at 2000 rpm. Subsequently, the catalyst was washed once with 20 mL of n-heptane for 20 min at 60 °C and then with 20 mL of hexane at room temperature. Finally, the catalyst was dried under a stream of nitrogen at 40–50 °C. The yield was about 2 g of catalyst per synthesized batch. XRD of the catalyst was then measured, and IR spectra, SEM and—in some cases—also TEM pictures of the catalyst and the carrier were recorded. The catalysts were also tested for active titanium in a polymerization quick test [24]. The synthetic content and catalyst codes are given in Table 2.
As previously mentioned, we sought to investigate the effects of small changes in crystal structures in relation to the synthetic conditions of the catalyst. First, the temperature was varied between 40 and 90 °C, and then the addition rate of the Mg(OR)2 solution into the reactor was varied between 1 min and 300 min. A few tests were performed using a temperature of 120 °C in the titanation step. Further, we increased the amount of EtAlCl2 in the precipitation step, and finally treated the catalyst with TiCl4 a second time at TiCl4/MgCl2 molar ratio of 12.
IR spectra
IR Analyses were performed on a Thermo Fisher Scientific Nicolet iN10MX (Thermo Scientific, USA) microscope with an iN10Z unit using attenuated total reflection (ATR) mode or transmission mode with CaF2 windows or KBr tablets. Spectra were recorded in the range of 400–4000 cm−1 at a resolution of 4 cm−1. KBr tablets were prepared from a blend of 200 mg KBr and 2 mg sample. When CaF2 windows were used, a drop of the sample solution (in n-heptane) was placed between the two windows.
X-ray diffraction (XRD) patterns
XRD measurements were performed using a Philips Pro X’Pert (PANalytical, Netherlands) system with CuKα radiation and a wavelength λ = 154.056 pm. The X-ray tube was powered using an acceleration voltage of 45 kV and a current of 40 mA. The catalyst powders were placed in a specially designed holder that allowed covering the powder with a Mylar film, which was necessary to reduce the exposure to moisture during the measurement and thus to hinder the formation of hydrated or hydroxide phases. All diffractograms were acquired in Bragg–Brentano geometry, with a step size of 0.0021° and 40 s per step. The crystallite grain size was determined based on the peak broadening using the single-line method and the Scherrer formula:
where Γ is the full width at half maximum (FWHM) of a diffraction line, K denotes the Scherrer constant, which was considered to be 0.9 (assuming spherical particles), 2θ is the diffraction angle, and D is the grain diameter D [25]. In general, when determining the grain size based on peak broadening, also the broadening related to the XRD equipment and that caused by the microstrain must be taken into consideration [26]. Equation (6) is a good approximation in the case of extensive broadening (small crystallite size) when no large microstrain is present. The broadening related to the equipment was determined using a highly crystalline silicon powder and evaluated based on the peak at 2θ = 28.46°. The peak broadening value obtained for silicon (0.0854°) was deducted from the peak broadening values obtained for the catalysts powders.
SEM pictures
SEM pictures allow the size of the nanoparticles in a Ziegler–Natta catalyst to be estimated [14, 16, 27]. For example, Busico et al. [28]. estimated that the size of the nanoparticles of their PP catalyst, which was ball-milled for 20 h, was about 100 nm. Additionally, SEM images can be used to estimate the pore structure of catalyst. For instance, Nejad et al. [26] stated on the basis of their SEM investigations that a catalyst with a large pore structure of 50–100 nm produces good replica in PE polymerization, whereas for pore structures between 20 and 50 nm fine porosity, and for catalyst pore structures as small as 10–20 nm, polymerization yields only fine particles.
The SEM images were acquired using a ZEISS 1540XB CrossBeam instrument (Carl Zeiss Microscopy, Germany) with a field emission filament, using an acceleration voltage of 2 kV with a SE2-Detector. The catalyst samples were applied to the sample holder and covered with a Mylar® film in a glove box to prevent hydration from the atmosphere prior to the SEM investigations.
TEM pictures
The catalyst samples were suspended in n-heptane and then further diluted with n-heptane to cause disintegration of the catalyst particles into nanoparticles and basic MgCl2 crystalline units. A drop of this diluted suspension was placed on a membrane covering a copper grid. The sample was then put in a desiccator to dry. All sample preparation steps were carried out under strictly inert conditions. TEM images were acquired on a Jeol-2011 TEM with a LaB6-filament with an acceleration voltage of 200 kV (Jeol, Japan) [29].
Polymerization quick test
The quick test was performed in a micro-reactor equipped with a video-microscope [24, 30]. The quick test was started by pre-contacting catalyst and co-catalyst (TEA) for 30 min at an Al/Ti molar ratio of 25. 10 µL of concentrated TEA was placed next to the polymerization area for scavenging. The activated catalyst was placed on the polymerization table by means of a micropipette, and catalyst particles without contact to each other were chosen for evaluation. After flushing the feed pipe five times with vacuum and ethylene, ethylene was injected to start the polymerization. A constant ethylene pressure of 20 bar (2 MPa) was applied, and the polymerization temperature was kept at 70 °C.
Results and discussion
IR results
IR absorption bands in the model compounds
For model compounds with more than one ethyl ligand group, inert conditions were created by enclosing the sample between CaF2 windows during measurement. The progress of the reaction between 2-ethyl-hexanol and the ethyl-aluminum group could be tracked by observing the disappearance of the broad absorption band between 3200 and 3500 cm−1 (with a maximum at 3420 cm−1) [23, 31, 32] originating from the –OH group in the added 2-ethyl-hexanol. The situation was the same for tracking the alcohol reaction with TiCl4 and with butyl-ethyl-magnesium. The results indicated quantitative reactions in all cases in which TEA, EADC and BEM were involved. No remaining absorption of an –OH group between 3200 and 3500 cm−1 was observed for these reaction products. In contrast, all reaction products originating from DEAC and the alcohol showed a flat and broad absorption starting from 3000 cm−1 and continuing up to 3500 cm−1. However, since the same flat absorption band was also observed for the DEAC reagent itself, the –OH absorption seen in the reaction product most probably originated from the Et2AlCl reagent and did not indicate an incomplete reaction. The only case in which an incomplete alcohol reaction was observed was that involving TiCl4. In both instances in which TiCl4 was used, a remaining absorption between 3200 and 3500 cm−1 could be seen, which indicated that unreacted alcohol was present in the reaction mixture. Generally only two of the chlorine atoms of TiCl4 tended to react, even in the presence of excess alcohol. Hence, the results indicated that an Al–Et group reacted more readily with alcohol than a Ti–Cl group in TiCl4.
The positions of the Al–OR, Al–Et, Al–Cl and Ti–OR groups were determined using the IR spectra and ignoring all the absorption peaks originating from 2-ethyl-hexanol and the solvent. The Al–OR group showed strong absorption at 635 and 1037 cm−1, and weaker absorption at 906 and 1120 cm−1 (Fig. 2). However, the strong absorption at 635 cm−1 was relatively close to the limits of accurate measurement of the instrument, so the position of this absorption peak must be regarded as the only confirmation. The Al–Et group showed moderately weak absorptions at 987, 1055 and 1100 cm−1 and the Al–Cl group showed absorption only at 872 cm−1.

IR spectrum of EtAl(OR)2 showing strong absorption at 635 and 1037 cm−1. The 635 cm−1 absorption peak is relatively close to the measurement limit of the instrument, so its position should be regarded only as indicative. Additionally, the normal hydrocarbon peaks can be seen, but no sign of moisture has been detected
The Ti–OR group showed strong absorption at 845 and 718 cm−1 and weaker absorption at 1006 cm−1. The stretching vibration of a single 2-ethyl-hexanolate group on titanium chloride showed up at 1050 cm−1 according to Gupta et al. [33] and between 1110 and 1130 cm−1 according to Kissin et al. [11], so in several cases there seemed to be disagreement not only between our results and published results, but also between the values published in different articles. According to Thongdonjui et al. Ti–Cl exhibits absorption at 466 and 618 cm−1 [19]. For the model compounds presented in this work, these absorptions peaks were outside the measuring range of the instrument used.
The results obtained for the model compounds indicated that the IR spectrum was a valid basis for determining whether the alcoholate group was attached to Al or to Ti because it exhibited strong absorption peaks in totally different locations in these cases. However, it turned out that forming a complex with MgCl2 caused a change in the absorption of the model compound. Table 3 lists the absorption peaks identified for the model compounds.
Interference of moisture with the IR results of Mg complexes
The reaction between the magnesium-alkyl and 2-ethyl-hexanol was strongly exothermic, thus causing a temperature rise. Towards the end of the alcohol addition, the reaction mixture turned into a gel with higher viscosity. The IR spectrum of this complex is shown in Fig. 3. The spectrum of the Mg(OR)2 complex shows a strong absorption peak at 1095 cm−1. This absorption peak originates most probably from the Mg–O–CH2– bond. An absorption peak at the right end of the spectrum at 589 cm−1 is very close to the measuring limits of the instruments and must therefore be regarded as only confirmation with respect to its position. According to Ault [34], the main absorption of Mg–C bonds should appear between 530 and 550 cm−1, which indicates that the absorption peaks at the right side of our Mg(OR)2 spectrum originates from the unreacted part of the added MgR2. The absorption band of the free –OH group in the added alcohol between 3200 and 3500 cm−1 disappeared completely, suggesting a quantitative reaction between the Mg-alkyl and the alcohol. According to Thongdonjui et al. and Zohuri et al. the Mg–O–Et group shows strong absorption at 1449 cm−1 and weaker absorption at 1632 cm−1 [19, 35]. This could not be confirmed in our investigation. The n-heptane solvent exhibited regular hydrocarbon absorption between 2800 and 3000 cm−1 and two individual peaks at 1378 and 1462 cm−1 and the absorbed carbon dioxide showed an absorption peak at 2300 cm−1.

IR spectrum of Mg(OR)2 directly after preparation, showing no sign of moisture
After recording the IR spectrum of the Mg(OR)2 complex, the sample was left in the instrument for a few minutes, and then a second IR spectrum of the complex was recorded, which is shown in Fig. 4.

IR spectrum of Mg(OR)2 after exposure to air. Absorption between 3200 and 3500 cm−1 and a new peak at 1636 cm−1 are clear signs of the presence of moisture
For a short time the Mg(OR)2 sample was exposed to ambient air and strongly affected its IR absorption spectrum. A broad absorption band of free water or –OH groups between 3200 and 3500 cm−1 and a single additional absorption peak at 1636 cm−1 can be seen. The hydrocarbon peaks between 2800 and 3000 cm−1 and at 1381 and 1466 cm−1 were not affected, but the Mg–O–CH2– peak position shifted significantly to the right (lower wavelength) to 1047 cm−1. This indicated that the adsorbed moisture had weakened the Mg–O–CH2– bond and caused the shift to a lower energy level. These results show that magnesium compounds easily adsorb moisture from ambient air, which changes the nature of the absorbing Mg compound considerably. The new peak at 1636 cm−1 might originate from H2O that is in complexation with magnesium. This absorption peak at 1636 cm−1 corresponded to the absorption peak at 1632 cm−1 that was reported in the investigations by Thongdonjui et al. and Zohuri et al. [19, 35] describing the IR-absorption of Mg(OEt)2. Our results can indicate that the Mg(OEt)2 investigated by Thongdonjui et al. and Zohuri et al. could have been contaminated by moisture.
IR results for precipitated MgCl2
The KBr tablet containing the MgCl2 precipitate exhibited a strong absorption peak at 1633 cm−1 and peaks due to moisture adsorption and of the remaining hydrocarbons. Additional absorption peaks were found at 1039 and 609 cm−1 in the spectrum of the neat MgCl2 (Fig. 5). Two publications support the assumption that the Mg–Cl group is the cause of the absorption peak at 1633 cm−1. According to Sinthusai et al. [18] and to Thongdonjui et al. [19], Mg–Cl stretching results in a weak absorption at 2253 and 1852 cm−1 and a strong absorption at 1633 cm−1 in a ZN catalyst system that very much resembles the system we investigated in this work. According to Zohuri et al. [36], strong absorption of the Mg–Cl group that depends on the nature of the catalyst can be seen in the area between 1590 and 1614 cm−1 in a silica-based Ziegler–Natta catalyst, which indicates that the Mg–Cl bond is weakened due to a complex forming between the MgCl2 and the SiO2 carrier. They suggested that the absorption peak at 1633 cm−1 was caused by the Mg–Cl group, but—as can be seen from the experimental results described above—this peak can also be caused by adsorbed moisture.

IR spectrum of pure MgCl2, showing strong absorption at 1633 cm−1 and absorption peaks at 1039 and 609 cm−1. The peaks between 3200 and 3500 cm−1 indicate moisture adsorption
IR results of the MgCl2-EtAl(OR)2 and the MgCl2-TiCl3(OR) model compounds
MgCl2-EtAl(OR)2 also showed a strong absorption peak at 1635 cm−1, which indicated the presence of a Mg–Cl/moisture bond. Two additional absorption peaks appeared at 1051 and at 630 cm−1 (Fig. 6). The absorption at 630 cm−1 can be related to the free Al–O–CH2– groups, as they exhibited absorption at 635 cm−1 in all model compounds. The absorption at 1051 cm−1 may have originated from the same group in a strong complex with magnesium forming a Mg–O(Al)–CH2 complex and may even include a shift of a chlorine group from Mg to Al. Figure 7 shows the assumed locations of the absorption peaks. The MgCl2-TiCl3(OR) complex also shows a strong Mg–Cl/moisture absorption at 1635 cm−1, and additional peaks at 1073 and 603 cm−1. Logically, the 603 cm−1 absorption must originate from Mg–O–CH2– group, since for the model compound, the Mg–O–CH2– group showed an absorption at 595 cm−1. If this is the case, then a part of the –OR groups from the titanium compound must have shifted to magnesium according to:

IR spectrum of MgCl2-EtAl(OR)2, showing Mg–Cl/moisture absorption at 1635 cm−1, the Al–O–CH2– group at 630 cm−1, and a peak at 1051 cm−1. In addition, the normal hydrocarbon and –OH/H2O absorption peaks can be seen in the spectrum

Schematic drawing showing the possible locations of the bonds responsible for the absorption peaks at 630–635, 1045–1051 and 1633–1635 cm−1
However, since the absorption peak for the Mg–Cl group remained strong, only few of the –OR groups must have shifted toward magnesium. The third absorption peak at 1073 cm−1 resulted most likely from the Ti–OR group coordinated to MgCl2.
IR Results for the MgCl2 support material
IR Spectra of several MgCl2 support materials were taken. In the measurements, we used KBr tablets. All support materials exhibited strong absorption at 1635 cm−1, which originated from the Mg–Cl bond. This is in good agreement with what we observed for the MgCl2 and MgCl2-EtAl(OR)2 model compounds which also accords with reports in the literature [18, 19]. Another distinct absorption peak was detected at 1045 cm−1. This may correspond to the absorption of the Mg–O–Al–CH2– group seen in the MgCl2-EtAl(OR)2 model compound. In addition, there was a clear but somewhat broader peak between 630 and 670 cm−1, which fitted with the strong absorption at 635 cm−1 of the free Al–O–CH2– group seen in the MgCl2–EtAl(OR)2 model compound and in the EtAl(OR)2 and Al(OR)3 model compounds. Hence, it can be concluded that the support material produced closely resembles the MgCl2–EtAl(OR)2 model compound in Fig. 6.
The n-heptane solution remaining in the catalyst showed absorption between 2900 and 2967 cm−1 and two separate peaks at 1465 and 1380 cm−1, which is in agreement with reports by Blitz et al., Deslauriers et al. and Thushara et al. [34, 37, 38].
The spectrum of the support material featured a large absorption band between 3200 and 3500 cm−1 with a maximum at 3446 cm−1. This absorption was in accordance with the results of Chien et al. [39], who reported for moisture-contaminated Ziegler–Natta catalysts a broad absorption band between 3350 and 3320 cm−1 originating from –OH groups and an absorption peak at 3570 cm−1 resulting from free water. In our case, the absorption originated from moisture, adsorbed by the support material, when the IR tablet was moved from the press to the IR instrument. Additional water may have originated from an internal equilibrium reaction as it is illustrated in Fig. 8.

Possible equilibrium reaction at the position of the –OR group on an Mg–Al adduct
IR Results for the catalyst
In order to prepare KBr tablets for the IR measurements, we ground 2 mg of the catalyst together with 200 mg of KBr in a glove box and placed the powder between the clams of a tablet press. The clams were quickly taken to the laboratory, where force was applied to the press to obtain the KBr tablet, which was immediately transferred to the measuring instrument to minimize adsorption of moisture from ambient air. Nonetheless, the spectra (Fig. 9) show a clear absorption band between 3200 and 3500 cm−1, which indicates the presence of moisture. It is possible to increase the height of the Mg–Cl absorption peak at 1633–1635 cm−1 due to moisture adsorption. A separate peak to the right of the main moisture peak is detected at 3241 cm−1 which may originate from the moisture and form a complex with MgCl2, because, according to Rönkkö et al. the peaks of free –OH groups and moisture peaks tend to shift to the right when a complex is formed with MgCl2 [40]. As the catalyst had been a slurry in n-heptane, the spectra also showed absorption due to remaining n-heptane between 2873 and 2961 cm−1, and two separate peaks at 1381 and 1464 cm−1. However, the n-heptane absorption did not dominate the spectra. The catalysts showed strong absorption at 1634–1635 cm−1 corresponding to the Mg–Cl bond. These bands were in accordance with the findings by Sinthusai et al. and by Thongdonjui et al. [18, 19]. One of the Ti–Cl absorption peaks may have appeared in some of the catalyst IR spectra that showed absorption between 603 and 617 cm−1. This would correspond to the Ti–Cl absorption at 618 cm−1 reported by Thongdonjui et al., though these results should not be regarded as conclusive. The main absorption peak of Ti–Cl at 466 cm−1 was too close to the measurement limit of the instrument, so it could not be identified. Additionally, there was a strong absorption peak at 1045 cm−1 [19] which corresponded to that seen in the support material and was assumed to originate from the complex between the EtAl(OR)2 and the MgCl2 carrier material. The only indication of the presence of titanium in the catalyst was the absorption peak at 617 cm−1.

Example IR spectrum of catalyst 0105-1, showing the distinct absorption peak of Mg–Cl at 1635 cm−1 and the absorption peak at 1045 cm−1 originating from the complex between EtAl(OR)2 and MgCl2
On the basis of these results, it can be concluded that there is a strong resemblance between the IR spectra of the support material and that of catalyst. Both showing the same structure which suggests that the base material in the catalyst consists of MgCl2 to which EtAl(OR)2 is coordinated. There is no clear sign of the presence of titanium compounds, as the main absorption peaks of Ti–Cl are outside the measuring limits of the IR instrument used. Nor is there any sign of a Cl3Ti–OR compound, which should, according to the model compound 1206-2, exhibit a strong absorption peak at 1073 cm−1. This suggests that there is no significant shift of –OR groups from the EtAl(OR)2 compound to TiCl4. The fact that most or all of the titanium was in the form of TiCl4 in this catalyst, and thereby outside the range of the measuring instrument, can explain why no titanium could be detected in the IR-spectra of the catalysts even if the catalyst contained 4 w% of titanium. It also indicates that there was no reaction between adsorbed H2O and TiCl4 in these catalysts as this would have created Ti–OH groups or even Ti–O–Ti groups that should have shown up in the IR-spectra. This has a practical consequence, since, according to Salajka et al. RO-groups on titanium cause often the catalyst to yield a polymer with lower molecular weight and a somewhat broader molecular weight distribution (MWD) than TiCl4 [41–43]. According to Wolf et al., –OR groups on titanium can also result in R-Cl splitting off and formation of either Ti–O–Ti or Ti=O groups in the catalyst [29, 44]. The large absorption band around 3500 cm−1 that is seen in all catalyst IR-spectra is typical for MgCl2 based Ziegler–Natta catalysts. This absorption band originates from adsorbed H2O on the active MgCl2 carrier. The reason why this adsorbed moisture is not reacting with the TiCl4 in the catalyst creating Ti–OH groups or Ti–O–Ti bonds that could immediately appear in a reaction between free TiCl4 and H2O is not known.
X-ray results
In our study, XRD of the prepared catalysts generally showed a small reflection peak at 2θ = 15°, further—partly merged—reflection peaks at 2θ = 30° and 2θ = 35°, and a peak at 2θ = 50°. This is in agreement with results described in the literature about the small reflection peak at 2θ = 15° [1, 15] and the partly merged—reflection peaks at 2θ = 30° and 2θ = 35° [45–47].
Some publications have described MgCl2 crystal thickness: Nakayama et al. [21]. prepared support material with a thickness of 28 Å, Chien et al. [48] produced a ball-milled catalyst with a crystal thickness of 36 Å, Busico et al. [28] prepared a PP catalyst with a crystal thickness of 130 Å, Singh et al. [49] synthesized a similar kind of catalyst with a crystal thickness of 31 Å, and Kojoh et al. [50] described their use of TiCl4 to hinder MgCl2 from forming larger crystals in precipitation reactions.
Reproducibility of the synthesis in terms of crystal thickness
A so-called standard synthesis was repeated in order to determine to what degree the thickness of the catalyst crystals was reproducible in parallel catalyst syntheses. In the standard synthesis, the Mg(OR)2 was added within 20 min at 60 °C, and the EtAlCl2/Mg(OR)2 molar ratio was 1:1. The titanation temperature was 60 °C, and there was no second titanation. The results from these two standard syntheses are listed in Table 4. The thickness of the MgCl2 crystal was calculated from the reflection peak at 2θ = 50°, on which our study concentrated. Given the peak position, the most probable crystalline phase is rhombohedral MgCl2. However, due to the relatively small difference (approximately 0.3°) in the 2θ positions between the rhombohedral and hexagonal phases and the significant peak broadening caused by the small crystallite size, the presence of a hexagonal phase cannot be completely excluded. Thus, most diffractograms were measured only between 2θ ranging from 45° to 55° (Fig. 10). The results show that both measurements yielded a crystal thickness close to 70 Å, which corresponds to an acceptable maximum error of 5 %.

XRD measurement between 45° 2θ and 55° 2θ of standard catalyst 0206-1 from a synthesis in which Mg(OR)2 was added over 20 min at 60 °C and the EtAlCl2/Mg(OR)2 molar ratio was 1:1. The titanation temperature was 60 °C, and there was no second titanation
Change in MgCl2 crystal thickness due to titanation
To determine to what extent the TiCl4 treatment of the support material influenced MgCl2 crystal thickness, XRD measurements of the support material (0206-1s) and of the final catalyst (0206-2) after TiCl4 treatment were taken. The results listed in Table 5 show that the TiCl4 treatment of the support material caused an increase in MgCl2 crystal thickness from 60 Å to almost 68 Å.
Influence of the temperature during synthesis on crystal thickness
In order to observe the effect of the temperature during synthesis on the crystal size of the MgCl2 carrier material, we prepared a series of catalysts at different temperatures within the range from 40 to 120 °C. At 120 °C, the synthesis had to be carried out in a nonane solution to prevent the reaction solution from boiling. After preparation, the thickness of the catalyst crystals was measured by XRD. The results listed in Table 6 show that the largest crystals (around 68 Å) were formed at 60 °C. At lower temperatures, crystal size decreased dramatically, dropping to 44 Å at 40 °C. The decrease in crystal size when using higher synthesis temperatures can be explained by the normal tendency of creating smaller crystal size when the precipitation reaction is increased due to higher and faster reaction rate leading to a faster over saturation of the precipitating product giving smaller crystal size. The decrease in crystal size when using lower synthesis temperature can only be explained by a slower chlorination reaction of Mg(OR)2 by EADC leading to an insufficient crystal growth.
A gradual decrease in crystal size was also observed at higher temperatures: at about 90 °C, the crystals had a thickness of about 59 Å (Fig. 11; Table 6). At even higher temperatures, a partial change in the crystal structure occurred, as indicated by the development of another reflection peak in the XRD pattern (Fig. 12). The second peak found at approximately 2θ = 52.3° might be related either to the development of hexagonal MgCl2 in addition to the rhombohedral one or to an increase in crystals with different orientations compared to the rhombohedral phase.

The MgCl2 crystal thickness measured at 2θ = 50° is correlated to the synthesis temperature during preparation of the MgCl2 support material

X-ray diffraction of the 0406-1 catalyst prepared at 120 °C, showing changes in the crystalline phase and/or orientation indicated by the presence of another reflection at approximately 2θ = 52.3°
Influence of the addition time of Mg(OR)2 on crystal thickness
In order to investigate to what extent it is possible to influence MgCl2 crystal thickness by changing the Mg(OR)2 addition time, we carried out a series of syntheses where the Mg(OR)2 addition time was changed from 1 to 300 min. Otherwise, standard conditions were used (synthesis at 60 °C with an EtAlCl2/Mg(OR)2 molar ratio of 1:1 and no second titanation). The results (Table 7) show that the crystal size of the MgCl2 support increases minimally with increasing Mg(OR)2 addition time. Extending the addition time from 20 min to 2 h resulted in a crystal thickness that increased from approximately 71–74 Å. For even longer addition times the crystals became narrower. The same was the case when the addition times were shorter than 20 min.
Influence of EtAlCl2/Mg(OR)2 molar ratio or a second titanation on crystal thickness
Next we sought to assess whether it is possible to modify the thickness of the MgCl2 crystals through stronger chlorination. We carried out two syntheses at 60 °C with a Mg(OR)2 addition time of 20 min: In the first, the EtAlCl2/Mg(OR)2 molar ratio was increased from 1:1 to 2:1, and in the second a second titanation with a TiCl4/MgCl2 molar ratio of 12:1 was introduced. Neither resulted in a significant increase in crystal thickness (Table 8). In the case in which the EtAlCl2/Mg(OR)2 molar ratio was doubled, the carrier had nearly the same crystal thickness (69 Å) as observed in the standard synthesis of 0206-2 catalyst (68 Å). In the case of a second TiCl4 treatment of the catalyst, a slightly smaller crystal size of 63 Å was achieved.
SEM results
In a typical Ziegler–Natta catalyst, the particles usually have a size of 20–30 μm. The particles are formed by clusters of nanoparticles of about 100 nm. The SEM images in our study were used to describe the physical appearance of the catalyst and to investigate whether the changes in the catalysts synthesis conditions caused any clear changes in the inner structure of the catalyst.
First, we compared the SEM pictures of the catalysts synthesized with different addition times of magnesium alcoholate to the solution of EADC. We found that increasing the addition time resulted in larger catalyst nanoparticles of up to 200 nm. This trend was observed for 1, 20 min and 2 h addition time. For even longer addition times, no further increase in nanoparticle size could be identified.
Our comparison of the SEM pictures of the catalysts prepared at different temperatures showed that the largest nanoparticles, in the range of 200–300 nm (Fig. 13), were obtained at 60 °C. For higher or lower temperatures, nanoparticle size decreased to approximately 100 nm (Fig. 14). Nanoparticle size of catalysts prepared at a temperature of 40 °C seemed larger than that of particles prepared at 120 °C (Table 9.).

SEM picture of the catalyst produced at 60 °C with a Mg(OR)2 addition time of 20 min showing a nanoparticle size of around 200 nm

SEM picture of the catalyst produced at 120 °C with an addition time of 20 min showing a nanoparticle size of around 100 nm
Further, the SEM pictures showed that the EtAlCl2/Mg(OR)2 molar ratio had no impact on nanoparticle size.
The relationships between nanoparticle size, addition time and synthesis temperature as identified from SEM pictures correlate with those determined by XRD.
TEM results
We recorded TEM images for selected catalyst samples. Figure 15 shows a typical example image from our study, where the crystalline MgCl2 discs forming the base units of the crystalline material can be seen. The size of these base units varied between 4 and 7 nm. The TEM image of the 0206-2 catalysts (Fig. 15) showed crystallite sizes in the range of 5–6 nm. These results are therefore in good agreement with those calculated from the XRD patterns.

TEM picture showing the size of the individual MgCl2 crystals in a Ziegler-Natta catalyst
Estimating the physical composition of the catalyst
XRD measurements showed that the thickness of the MgCl2 crystals was around 7 nm, which gave a plate radius of 3.5 nm. Should five crystal plates stacked on top of each other form a base unit, it has a volume that roughly corresponds to that of a sphere with a radius of 3.5 nm. The volume therefore amounts to 3π/4 × (3.5 nm)3. Our SEM pictures showed that the nanoparticles had a diameter of 200 nm, which corresponded to a volume of 3π/4 × (100 nm)3. Dividing this volume by the volume of the base unit, we find that around 25 000 base units can be fitted into one nanoparticle, leading to about 125 000 MgCl2 crystal plates per nanoparticle. As the yield of the catalyst synthesis was about constant it could be assumed that the smaller the crystal size was, the greater number of crystals was needed to make up the same yield.
Catalyst particles commonly have a size of 20 μm, which corresponds to a volume of 3π/4 × (10 000 nm)3. Dividing this volume by the volume of the nanoparticle yields one million. The calculation thus shows that a 20 μm Ziegler–Natta catalyst particle contains about one million nanoparticles, each of which consists of about 105 of MgCl2 crystal plates (Fig. 16). The absolute particle size of the catalyst could only visually be estimated in our investigation. This was done from the microscope pictures. The catalyst particles did not have a regular shape

The physical composition of a 20 μm Ziegler-Natta catalyst particle. The catalyst particle contains about one million nanoparticles, each of which consists of about 25 000 of MgCl2 crystal units that contain five MgCl2 crystal plates
and many times it was difficult to judge if a particle was consisting of a cluster or of a single particle. This is a common feature for the precipitated Ziegler–Natta catalysts where there has been no directing force in connection to the preparation of the MgCl2 support material. Therefore, only an indicative estimation of the relation between the MgCl2 crystal size and the catalyst particle size can be made. This estimation indicated that if higher temperatures had been used in the synthesis, the catalysts tended to be somewhat bigger. The catalyst showed then also a more regular morphology having a higher bulk density. The catalyst prepared at 40 °C showed clearly a smaller particle size and also a smaller MgCl2 crystal broadness.
In general it can be said that the better morphology a catalyst possesses, the better is the physical strength of the catalyst particle which in turn gives a better morphology to the produced polymer. A correlation between the crystal size and strength of the catalyst particles could be seen only when using lower synthesis temperatures. Then a smaller crystal size was created and at the same time a worse morphology of the catalyst which in turn led to a weaker catalyst particle that in turn created a worse morphology to the produced polymer. No physical tests were however done to measure the catalyst particle strength in connection to this investigation.
Polymerization quick test
Figure 17 illustrates an example of this polymerization quick test where catalyst 1205-1 was polymerized. The test clearly shows that the size of the studied particle increases from the initial size of 53–284 µm after 30 min of polymerization and thus that the catalyst contains active titanium. All catalysts produced were tested with this procedure and showed reasonable growth, which indicated that they all contained active titanium. In our polymerization quick tests we could not see any difference in catalyst activity that would correspond to the MgCl2 crystal thickness of the catalyst. In literature no systematic study focusing on the correlation between the catalyst MgCl2 crystal thickness and the activity of the catalyst could be found.

Example of polyethylene formation in the polymerization quick test. The tested catalyst 1205-1 was prepared at 60 °C with a Mg(OR)2 addition time of 120 min
Conclusion
This study investigated how changes in the synthesis conditions alter the crystal thickness of a typical MgCl2-based PE Ziegler–Natta catalyst. The MgCl2 support material was created by adding Mg(OR)2 to EtAlCl2. TiCl4 was added after the support material had been washed. The thickness of the catalysts was measured by X-ray diffraction using the reflection at 2θ = 50°. The results were confirmed by means of TEM pictures. To determine whether a correlation exists between MgCl2 crystal thickness and nanoparticle size, we took SEM pictures of the catalysts, based on which the nanoparticle size could be estimated. IR spectroscopy was employed to determine the chemical composition of the catalysts. In summary, the results of these investigations were:
-
Overcoming the settling problems in the synthesis process by using a catalyst synthesis reactor that could be transferred to a centrifuge both accelerated production significantly and introduced a high degree of precision to the process.
-
The IR results for the MgCl2-EtAl(OR)2 model compound indicated that an exchange of Cl and –OR groups between Mg and Al may occur.
-
The IR results for the MgCl2–Cl3Ti–OR model compound indicated that an exchange of Cl and –OR groups between Mg and Ti was possible.
-
The IR spectra also revealed that there is a strong resemblance between the chemical composition of the support material and that of the catalyst, which suggests that the base material in the catalyst consists of MgCl2 in coordination equilibrium with Mg(OR)2 and EtAl(OR)2. The IR results gave no clear indication of the presence of titanium compounds in the catalyst.
-
According to the XRD results, there was an increase from 60 Å to almost 68 Å in MgCl2 crystal thickness due to the TiCl4 treatment of the support material.
-
The XRD results showed that the largest crystals (70 Å) were created at a synthesis temperature of 60 °C. For lower temperatures, the crystal size decreased dramatically to only about 44 Å at 40 °C. A gradual decrease in crystal size was also observed for higher temperatures: at 90 °C a crystal thickness of about 59 Å was obtained. For even higher temperatures, an additional peak appeared in the diffraction pattern.
-
The XRD results showed that increasing the addition time of Mg(OR)2 results in only a minor increase in crystal size of the MgCl2 support material.
-
The XRD results further revealed that increased chlorination by higher amounts of EtAlCl2 or TiCl4 did not cause a significant change in the thickness of MgCl2 crystals.
-
The TEM pictures confirmed that the size of the MgCl2 crystals was between 40 and 70 Å.
-
Our SEM pictures showed that there was a slight correlation between MgCl2 crystal thickness and nanoparticle size. Nanoparticles of 200 nm and about 100 nm were obtained at synthesis temperatures of 60 and 100 °C, respectively.
-
Our calculation showed that a 20 μm Ziegler–Natta catalyst particle contained about one million nanoparticles, each of which consisted of about 105 MgCl2 crystal plates.
-
All catalysts showed chemical activity in a polymerization quick test.
This investigation was planned as the first of a series of studies in which we have sought to reveal correlations between the thickness of the MgCl2 support material in a typical polyethylene Ziegler–Natta catalyst and its co-polymerization performance. As part of future work, we will evaluate the effect of MgCl2 crystal thickness on polymerization activity, molecular weight capability, width of molecular weight distribution, and amount of soluble polymer material in co-polymers, especially when higher amounts of co-monomer are incorporated.
References
Kashiwa N (2004) The discovery and progress of MgCl2-supported TiCl4 catalysts. J Polym Sci Part A Polym Chem 42:1–8
Galli P, Vecellio G (2001) Technology: driving force behind innovation and growth of polyolefins. Prog Polym Sci 26:1287–1336
Vaughan A, Davis DS, Hagadorn JR (2012) Industrial catalysts for alkene polymerization. Polym Sci A Compr Ref 3:657–672
Kissin YV (2012) Active centers in Ziegler-Natta catalysis: formation kinetics and structure. J Catal 292:180–200
Kebritchi A, Nekoomanesh M, Mohammadi F (2015) The interrelationship between microstructure and melting, crystallization and thermal degradation behaviours of fractionated ethylene/1-butene copolymer. Iran Polym J 24:267–277
Nouri M, Parvazinia M, Arabi H (2015) Effect of fragment size distribution on reaction rate and molecular weight distribution in heterogeneous olefin polymerization. Iran Polym J 24:437–448
Bahri-Laleh N, Correa A, Mehdipour-Ataei S, Arabi H, Haghighi MN, Zohuri G, Cavallo L (2011) Moving up and down the titanium oxidation state in Ziegler-Natta Catalysts. Macromolecules 44:778–783
Stukalov DV, Zakharov VA (2009) Active site formation in MgCl2-supported Ziegler-Natta catalyst. A density functional theory study. J Phys Chem C 113:21376–21382
Mikenas TB, Koshevoy EI, Zakharov VA, Nikolaeva MI (2014) Formation of isolated titanium(III) ions as active sites of supported titanium-magnesium catalysts for polymerization of olefins. Macromol Chem Phys 215:1707–1720
Soares JBP, McKenna TFL (2012) Polyolefin Reaction Engineering. Wiley-VCH, Weinheim
Kissin YV, Mink RI, Brandolini AJ, Nowlin TE (2009) AlR2Cl/MgR2 combinations as universal co-catalysts for Ziegler-Natta, metallocene, and post-metallocene catalysts. J Polym Sci A Polym Chem 47:3271–3285
Boero M, Parrinello M, Hüffer S, Weiss H (2000) First principles study of propene polymerization in Ziegler-Natta heterogeneous catalysis. J Am Chem Soc 122:501–509
Cipullo R, Mellino S, Busico V (2014) Identification and count of the active sites in olefin polymerization catalysis by oxygen quench. Macromol Chem Phys 215:1728–1734
Liu Z, Zhang X, Huang H, Yi J, Liu W, Liu W, Zhen H, Huang O, Gao K, Zhang M, Yang W (2012) Synthesis of (co-)polyethylene with broad molecular weight distribution by the heterogeneous Ziegler-Natta catalysts via one-pot strategy. J Ind and Eng Chem 18:2217–2224
Di Noto V, Zanetti R, Viviani M, Marega C, Marigo A, Bresadola S (1992) MgCl2-supported Ziegler-Natta catalysts: a structural investigation by X-ray diffraction and Fourier-transform IR spectroscopy on the chemical activation process through MgCl2-ethanol adducts. Makromol Chem 193:1653–1663
Dwivedi S, Taniike T, Terano M (2014) Understanding the chemical and physical transformations of a Ziegler-Natta catalyst at the initial stage of polymerization kinetics: the key role of alkylaluminum in the catalyst activation process. Macromol Chem Phys 215:1698–1706
Garoff T, Waldvogel P, Pesonen K (2004) Method for the preparation of olefin polymerization catalyst support and an olefin polymerization catalyst. U.S. Patent 7,432,220
Sinthusai L, Trakarnpruk W, Strauss R (2009) Ziegler-Natta catalyst with high activity and good hydrogen response. J Met Mater Miner 19:27–32
Thongdonjui A, Trakarnpruk W, Strauss RH (2009) Effect of electron donor on PE polymerization. J Met Mater Miner 19:17–23
Wang J, Cheng R, He X, Liu Z, Tian Z, Liu B (2015) A novel (SiO2/MgO/MgCl2)·TiClx Ziegler-Natta catalyst for ethylene and ethylene/1-hexene polymerization. Macromol Chem Phys 216:1472–1482
Nakayama Y, Bando H, Sonobe Y, Fujita T (2004) Development of single-site new olefin polymerization catalyst system using MgCl2-based activators: MAO-free MgCl2-supported FI catalyst systems. Bull Chem Soc Jpn 77:617–625
Severn JR, Chadwick JC (2004) MAO-free activation of metallocenes and other single-site catalysts for ethylene polymerization using spherical supports based on MgCl2. Macromol Rapid Commun 25:1024–1028
Chien JCW, Wu JC, Kuo CI (1982) Magnesium chloride supported high-mileage catalysts for olefin polymerization. I. Chemical composition and oxidation states of titanium. J Polym Sci Polym Chem Ed 20:2019–2032
McKenna TFL, Tioni E, Ranieri MM, Alizadeh A, Boisson C, Monteil V (2013) Catalytic olefin polymerization at short times: studies using specially adapted reactors. Can J Chem Eng 91:669–686
Gusev AI, Rempel AA (2004) Nanocrystalline materials. Cambridge International Science Publishing, Cambridge
Nejad MH, Ferrari P, Pennini G, Cecchin G (2008) Ethylene homo and copolymerization over MgCl2-TiCl4 catalysts: polymerization kinetics and polymer particle morphology. J Appl Polym Sci 108:3388–3402
Pirinen S, Jayaratne K, Denifl P, Pakkanen TT (2014) Ziegler-Natta catalysts supported on crystalline and amorphous MgCl2/THF complexes. J Mol Catal A Chem 395:434–439
Busico V, Causà M, Cipullo R, Credendino R, Cutillo F, Friederichs N, Lamanna R, Segre A, Castelli VVA (2008) Periodic DFT and high-resolution magic-angle-spinning (HR-MAS) 1H NMR investigation of the active surfaces of MgCl2-supported Ziegler-Natta catalysts. The MgCl2 matrix. J Phys Chem C 112:1081–1089
Zijlstra HS, Stuart MCA, Harder S (2015) Structural investigation of methylalumoxane using transmission electron microscopy. Macromolecules 48:5116–5119
Abboud M, Kallio K, Reichert KH (2004) Video microscopy for fast screening of polymerization catalysts. Chem Eng Technol 27:694–698
Natter H, Schmelzer M, Löffler MS, Krill CE, Fitch A, Hempelmann R (2000) Grain-growth kinetics of nanocrystalline iron studied in situ by synchrotron real-time X-ray diffraction. J Phys Chem B 104:2467–2476
Andoni A (2009) A flat model approach to Ziegler-Natta olefin polymerization catalysts. Doctor Thesis, Technische Universiteit Eindhoven
Gupta VK, Satish S, Bhardwaj IS (1993) MgCl2·6H2O-based titanium catalysts for propene polymerization. Die Angew Makrom Chem 213:113–125
Ault BS (1980) Infrared matrix isolation study of magnesium metal atom reactions. Spectra of an unsolvated Grignard species. J Am Chem Soc 102:3480–3484
Deslauriers PJ, Rohlfing DC, Hsieh ET (2002) Quantifying short chain branching microstructure in ethylene 1-olefin copolymers using size exclusion chromatography and Fourier transform infrared spectroscopy (SEC-FTIR). Polymer 43:159–170
Zohuri GH, Ahmadjo S, Jamjah R, Nekoomanesh M (2001) Structural study of mono- and bi- supported Ziegler-Natta catalysts of MgCl2/SiO2/TiCl4/donor systems. Iran Polym J 10:149–155
Blitz JP, Meverden CC, Diebel RE (1998) Reactions of dibutyl magnesium with modified silica gel surfaces. Langmuir 14:1122–1129
Thushara KS, Gnanskumar ES, Mathew R, Jha RK, Ajithkumar TG, Rajamohanan PR, Sarma K, Padmanabhan S, Bhaduri S, Gopinath CS (2011) Toward an understanding of the molecular level properties of Ziegler-Natta catalyst support with and without the internal electron donor. J Phys Chem C 115:1952–1960
Chien JCW, Wu JC, Kuo CI (1983) Magnesium chloride supported high-mileage catalysts for olefin polymerization. IV. FTIR and quantitative analysis of modifiers in the catalysts. J Polym Sci Polym Chem Ed 21:725–736
Rönkkö HL, Knuuttila H, Linnolahti M, Haukka M, Pakkanen TT, Denifl P, Leinonen T (2011) Complex formation and characterization of MgCl2/2-(2-ethylhexyloxy)ethanol adduct. Inorg Chimica Acta 371:124–129
Salajka Z, Kratochvíla J, Hudec P, Vĕčorek P (1993) One-phase supported titanium-based catalysts for polymerization of ethylene. II. Effect of hydrogen. J Polym Sci A Polym Chem 31:1493–1498
Kong Y, Yi J, Dou X, Liu W, Huang O, Gao K, Yang W (2010) With different structure ligands heterogeneous Ziegler-Natta catalysts for the preparation of copolymer of ethylene and 1-octene with high comonomer incorporation. Polymer 51:3859–3866
Lemos C, Franceschini F, Radtke C, Santos JHZ, Wolf CR (2012) The effect of partial replacement of TiCl4 by Ti(OR)4 on the performance of MgCl2-supported Ziegler-Natta catalysts. Appl Catal A General 423–424:69–77
Wolf CR, de Camargo Forte MM, dos Santos JHZ (2005) Characterization of the nature of chemical species of heterogeneous Ziegler-Natta catalysts for the production of HDPE. Catal Today 107–108:451–457
Kashiwa N (1980) Super active catalyst for olefin polymerization. Polym J 12:603–608
Di Noto V, Bresadola S (1996) New synthesis of a highly active δ-MgCl2 for MgCl2/TiCl4/AlEt3 catalytic systems. Macromol Chem Phys 197:3827–3835
Rojanotaikul P, Ayudhya SKN, Charoenchaidet S, Faungnawakij K, Soottitantawat A (2012) Preparation of porous anhydrous MgCl2 particles by spray drying process. Eng J 16:109–114
Chien JCW, Wu JC, Kuo CI (1983) Magnesium chloride supported high-mileage catalysts for olefin polymerization. V. BET, porosimetry, and X-ray diffraction studies. J Polym Sci Polym Chem Ed 21:737–750
Singh G, Kaur S, Makwana U, Patankar RB, Gupta VK (2009) Influence of internal donors on the performance and structure of MgCl2 supported titanium catalysts for propylene polymerization. Macrom Chem Phys 210:69–76
Kojoh SI, Fujita T, Kashiwa N (2001) Recent advances in olefin polymerization catalysts based on group 4 transition metals. Recent Res Develop Polym Sci 5:43–58
Acknowledgments
Open access funding provided by [Johannes Kepler University Linz]. The authors are Grateful to Borealis Polyolefine GmbH for financial support and to Dr. Ingrid Abfalter for her intellectual support.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Redzic, E., Garoff, T., Mardare, C.C. et al. Heterogeneous Ziegler–Natta catalysts with various sizes of MgCl2 crystallites: synthesis and characterization. Iran Polym J 25, 321–337 (2016). https://doi.org/10.1007/s13726-016-0424-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13726-016-0424-x