
Abstract
Purpose of Review
To highlight the added benefits of approved and upcoming, centrally-acting, anti-obesity drugs, focusing not only on the most common metabolic and cardiovascular effects but also on their less explored clinical benefits and drawbacks, in order to provide clinicians with a tool for more comprehensive, pharmacological management of obesity.
Recent Findings
Obesity is increasingly prevalent worldwide and has become a challenge for healthcare systems and societies. Reduced life expectancy and cardiometabolic complications are some of the consequences of this complex disease. Recent insights into the pathophysiology of obesity have led to the development of several promising pharmacologic targets, so that even more effective drugs are on the horizon. The perspective of having a wider range of treatments increases the chance to personalize therapy. This primarily has the potential to take advantage of the long-term use of anti-obesity medication for safe, effective and sustainable weight loss, and to concomitantly address obesity complications/comorbidities when already established.
Summary
The evolving scenario of the availability of anti-obesity drugs and the increasing knowledge of their added effects on obesity complications will allow clinicians to move into a new era of precision medicine.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Obesity is a chronic, progressive and relapsing disease, associated with decreased life expectancy and disability, depending on its duration, the magnitude of excess weight and the development of complications. It increases the incidence of type 2 diabetes (T2D) and cardiovascular disease (CVD) but also psychological, neurological, pulmonary and musculoskeletal diseases, resulting in a heavy financial burden on healthcare systems [1].
Despite major advances in diagnosis and an increased availability of anti-obesity treatment approaches, the prevalence of obesity continues to increase [2]. These data suggest that there is a lack of understanding as regards the management of obesity. Currently, medication used in Europe for obesity weight management is represented by orlistat, liraglutide and naltrexone hydrochloride (HCl)/bupropion HCl [3]. Orlistat is a pancreatic and gastric lipase inhibitor that generates an interference with lipase-catalysed breakdown and the subsequent systemic absorption of around 30% of dietary ingested fats [4, 5]. Liraglutide is a glucagon-like peptide-1 receptor agonist (GLP-1RA) and appetite suppressant, that acts centrally on the arcuate nucleus in the hypothalamus to suppress appetite and potentiate satiety [5,6,7,8]. Moreover, the main mechanism of action of naltrexone/bupropion relates to appetite suppression [9].
Safety and efficacy in relation to weight loss was also demonstrated in the case of new drugs, such as semaglutide, a GLP-1 RA [10], cagrilintide, a long-acting acylated amylin analogue [11••, 12••] and tirzepatide, a combination of the gastric inhibitory polypeptide (GIP) receptor with GLP-1RA [13].
Despite the availability of these drugs for the treatment of obesity, there are no guidelines specifying which therapeutic intervention should be used, based on patients’ comorbidities. Since obesity is a remarkably heterogeneous disease, it could also be expected that the heterogeneity among patients with obesity results in a different weight loss response to obesity interventions, such as diets, medication, devices and surgery.
Recently, a phenotype-guided approach for the treatment of obesity, based on measurable components of food intake and energy expenditure has been proven to be associated with greater weight loss; a higher proportion of patients lost more than 10% after one year, as compared with patients whose treatment was not phenotype-guided [14•].
Therefore, by moving a step closer towards a precision medicine and in order to optimize obesity treatment, we have questioned whether an obesity complications-guided approach, based on the evidence from preclinical and clinical studies, can be of help in individualizing obesity treatment.
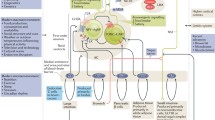
Thus, the aim of this manuscript is to review the current evidence on centrally-acting, anti-obesity drugs, both already approved and those which are upcoming, and to consider obesity-related, conventional comorbidities, as well as the added benefits, in order to provide tools for a more precise and patient-tailored treatment, potentially resulting in greater effectiveness (Fig. 1; Table 1).

Anti-obesity drugs and their effects on obesity complications or comorbidities, based on randomized clinical trials (RCT), cardiovascular outcome trials (CVOT) for CV disease, clinical and preclinical studies or indirect evidence. The data refer to dosages approved for the treatment of obesity, unless indicated otherwise (e.g., in light green). Should no data be available, the name of the drug is omitted
Approved and Upcoming, Centrally-Acting, Anti-Obesity Drugs
Naltrexone/Bupropion
is a drug, licensed as an adjunct to lifestyle changes, such as a low-calorie diet and regular physical activity and it is used for the management of weight in adults with obesity (Body Mass Index, BMI ≥ 30 kg/m2) or overweight (with a BMI ≥ 27 kg/m2) with at least one weight-related comorbidity [9].
Contained in each prolonged-release tablet is 8 mg of naltrexone hydrochloride and 90 mg of bupropion hydrochloride. The initial dose is one tablet daily, which should be increased over a four-week period to the recommended dose of two tablets twice daily (32 mg naltrexone and 360 mg bupropion per day). The suggestion is to assess weight loss after 16 weeks of treatment; the drug should be discontinued if at least 5% weight loss has not been achieved [9, 15].
Bupropion, originally developed as an antidepressant, acts by inhibiting dopamine and noradrenaline reuptake, while naltrexone is an opioid antagonist [16].
The main mechanism of action of naltrexone/bupropion relates to appetite suppression [9]. It has been suggested that naltrexone/bupropion exerts a synergistic effect on the hunger centre located in the hypothalamus, reducing appetite and the impact of food cues, and increasing activation of the regions of the brain responsible for self-control [17]. The most common side effect was nausea [18].
The efficacy of naltrexone/bupropion on obesity was mainly demonstrated in four randomized, double-blind, placebo-controlled, 56-week, phase-3 clinical trials, with 4536 adult subjects: COR-I, COR-II, COR-BMOD and COR-DM. COR-I and II trials were ad hoc designed trials, with weight change as the primary endpoint. In the COR-I study, 1742 subjects were enrolled and were randomized in a 1:1:1 ratio to naltrexone HCl 32 mg/bupropion HCl 360 mg, n = 583; naltrexone HCl 16 mg/bupropion HCl 360 mg, n = 578; placebo, n = 581. Half of the subjects (n = 870) completed 56 weeks of treatment (n = 296; n = 284; n = 290, respectively) and 83% (n = 1453) were included in the analysis [19]. The mean percentage change in body weight was -1.3% in the placebo group, while it was -6.1% in the naltrexone 32/bupropion 360 group (p < 0.0001 vs. placebo) and -5.0% in the naltrexone 16/bupropion 360 group (p < 0.0001 vs. placebo). Sixteen percent of subjects in the placebo group had a reduction in body weight of 5% or more, versus 48% in the naltrexone 32/bupropion 360 group and 39% in the naltrexone 16/ bupropion 360 group (p < 0.0001 vs. placebo) [19]. In the COR-II study, 1496 subjects were enrolled and were randomly divided 2:1 into naltrexone HCl 32 mg/bupropion HCl 360 mg (NB32) or placebo for up to 56 weeks. The primary end points were weight change and percentage of patients reaching ≥ 5% weight loss at week 28 [20]. Significantly greater weight loss was reported in the intervention group versus the placebo group at week 28 (-6.5 vs. -1.9%; p < 0.001) and week 56 (-6.4 vs. -1.2%; p < 0.001). A higher percentage of NB32-treated subjects achieved ≥ 5% weight loss versus those in the placebo group at week 28 (55.6 vs. 17.5%; p < 0.001) and week 56 (50.5 vs. 17.1%; p < 0.001) [20].
Liraglutide
is a glucagon-like peptide-1 (GLP-1) analogue with a 97% homology to human glucagon-like peptide-1 [5,6,7], due to molecular modifications [8]. GLP-1 is secreted by the L-cells of the gastrointestinal tract when nutrients arrive in the lumen. It produces glucose-dependent insulin secretion, a decrease in plasma glucagon concentrations, delayed gastric emptying and appetite suppression [8]. Since GLP-1 receptor agonists stimulate insulin release and inhibit glucagon secretion in a glucose-dependent fashion, there is a low risk of hypoglycaemia [3]. Endogenous GLP-1 has a half-life of 1.5–2 min, as it is degraded by dipeptidyl peptidase 4 (DPP-4) and neutral endopeptidases (NEP). Liraglutide, conversely, is relatively resistant to DPP- 4 degradation and has a plasma half-life of 11–13 h after subcutaneous administration, providing 24-h duration of action [21, 22]. Lastly, liraglutide does not interfere with the cytochrome P450 system and it is eliminated as metabolites in urine and faeces [22]. Saxenda®, liraglutide 3,0 mg, was approved in 2014 by the US Food and Drug Administration (FDA) [3] and in April 2015 by the European Medicines Agency (EMA) («Saxenda—EPAR summary for the public» 2015) for the treatment of obesity. The guidelines of the European Association for the Study of Obesity (EASO) recommend the use of pharmacologic therapy in adults who have a BMI ≥ 30 kg/m2 or ≥ 27 kg/m2 with at least one weight-related comorbid condition (e.g., hypertension, dyslipidaemia, insulin resistance, T2D) [23].
The most frequent adverse events are nausea and vomiting, which mainly occur during the first few weeks of treatment and are usually tolerable, not leading to the discontinuation of therapy; other adverse effects include diarrhoea, constipation, dyspepsia and abdominal pain [24,25,26,27]. The risk of acute pancreatitis has been reported in a small percentage of treated patients [6, 22].
It was demonstrated that liraglutide causes thyroid C-cell tumours in mice, however, there is no evidence to support the fact that liraglutide causes such tumours in humans [24]. Liraglutide does not interfere with other medication and does not affect the absorption of oral medication. There are currently no dosage adjustments provided for the use of liraglutide in the presence of renal or hepatic dysfunction, however, caution should be used. Pregnant patients should avoid the use of liraglutide [22].
The dose of liraglutide should be titrated by 0.6 mg weekly, up to a 3.0 mg daily dose. If the dose escalation is not tolerated due to adverse gastrointestinal effects, delaying the titration by one week may be considered. Patients should be evaluated after 16 weeks: if the patient has not lost at least 4% of his or her baseline body weight, liraglutide should be discontinued [22].
The SCALE (Satiety and Clinical Adiposity – Liraglutide Evidence) Obesity study was a 56-week, double-blind, randomized controlled trial (RCT) involving 3731 patients without T2D and with a body-mass index of ≥ 30 kg/m2 or ≥ 27 kg/m2 and with dyslipidaemia or hypertension [6]. The coprimary endpoints were weight change from the baseline and, in particular, a loss ≥ 5% and ≥ 10% of their baseline body weight at week 56. After 56 weeks, patients in the liraglutide group had lost a mean of 8.4 ± 7.3 kg of body weight, and those in the placebo group had lost a mean of 2.8 ± 6.5 kg. Patients in the liraglutide group lost at least 5% of their body weight, compared to the placebo group (63.2% vs. 27.1%), more than 10% of their body weight (33.1% vs. 10.6%), and more than 15% of their body weight (14.4% vs. 3.5%) [6]. There were no differences in the treatment effects or safety profile of 3.0 mg of liraglutide for individuals with a BMI of 27 to < 35 or ≥ 35 kg/m2, and its effects and safety profile were consistent across racial subgroups [28]. The efficacy of liraglutide in terms of causing weight loss among patients with T2D was also demonstrated [26]. Recent clinical trials and metanalyses investigated the use of liraglutide in adolescents (> 12 years old) suffering from obesity, as it has demonstrated a significantly greater reduction in the BMI standard-deviation score than the placebo plus lifestyle therapy [29•, 30]). The existing literature offers some case reports of successful treatment with liraglutide in subjects with compulsive, food-related behaviour, associated with autism [31] and hypothalamic obesity [32]; some encouraging results have been noted in patients with obesity and severe mental illness [33].
Semaglutide
is a GLP-1RA, with 94% homology with native human GLP-1 and structural modifications that allow for reversible albumin binding, reduced renal clearance and degradation by DPP-4, resulting in a longer half-life (155 to 184 h) [10, 34]. As with other GLP-1RAs, the most common side effects of semaglutide are gastrointestinal issues, mostly mild-to-moderate in severity, transient and occurring during dose escalation [35]. Although semaglutide increases the risk of biliary disease (primarily cholelithiasis), no unexpected safety issues have arisen to date, therefore, the established safety profile is similar to that of other GLP-1RAs.
Semaglutide was approved in 2021 by the FDA and EMA as an adjunct to reduced calorie intake and increased physical activity for chronic weight management in adults with obesity (BMI ≥ 30 kg/m2) or overweight (BMI ≥ 27 kg/m2) with at least one weight-related comorbidity [36, 37]. The semaglutide-weight loss was obtained by limiting food intake with minimal effects on energy expenditure [38]. Despite preclinical studies reported that semaglutide had limited efficacy in crossing the blood–brain barrier, semaglutide was proven to access the brainstem, septal nucleus and hypothalamus directly [39]. Semaglutide also induces central c-Fos activation in secondary brain areas without direct GLP-1R interaction, such as the lateral parabrachial nucleus, thus having direct and indirect effects on neutral pathways involved in homeostatic (appetite, hunger, satiety) and hedonic (food preference, cravings, control of eating) aspects of food intake and reward-related behaviours pertaining to food [39]. Conversely, only a very small percentage of weight loss is explained by delayed gastric emptying and gastrointestinal side effects (nausea or vomiting) [40].
The dose of semaglutide for reaching an efficient weight loss is 2.4 mg, injected subcutaneously once-weekly [41] and is reached by increasing the initial dose of 0.25 mg at four-week intervals.
The Semaglutide Treatment Effect in People with obesity (STEP) was a pivotal, global, phase-3, clinical development programme that evaluated 2.4 mg of semaglutide once-weekly for weight management [42]. As a whole, the first four STEP trials reported reductions of approximately 15% of initial body weight after 68 weeks of treatment, associated with improvements in cardiovascular (CV) risk factors and physical functioning [42]. The STEP 5 trial assessed the durability of the drug effect and examined the trajectory of weight loss over 104 weeks, showing that the initial reduction in weight, which plateaued after approximately week 60, was maintained for the remainder of the study [43].
A placebo-controlled, phase-3 trial (NCT05035095, OASIS 1) is ongoing, to address the effects of 50 mg of oral semaglutide once-daily over a period of 68 weeks in subjects who are either overweight or with obesity [44].
Semaglutide/Cagrilintide
Cagrilintide is a long-acting, acylated amylin analogue, with agonistic effects on both native amylin and calcitonin receptors and has been investigated for weight management. Amylin is a glucoregulatory hormone, co-secreted with insulin by pancreatic β-cells that regulate glucose homeostasis by delaying gastric emptying, suppressing postprandial glucagon release and inducing meal‐ending satiety [45]. Native amylin is involved in the regulation of appetite and satiation through the activation of receptors in the area postrema and nucleus of the solitary tract of the hindbrain [46]. In addition, the hypothalamus, ventral tegmental area and laterodorsal tegmental nucleus are targeted by amylin to influence the hedonic aspects of food intake, such as reward-guided behaviours that influence food choices and preferences [47]. Thus, although both semaglutide and cagrilintide induce satiety, while operating in different regions of the brain, their combination has the potential to act additively to improve weight control. The key point with regard to developing combination therapies is the need to be more effective than monotherapy but to maintain a reasonable price in terms of adverse events and cost.
Based on a phase-2 trial that reported dose-dependent reductions in body weight of up to 10.8% after a 26-week treatment with sc cagrilintide once-weekly [12••], a phase-1-b study tested the safety and tolerability of six multiple-ascending doses of sc cagrilintide (with doses ranging from 0.16 to 4.5 mg), administered once-weekly in combination with 2.4 mg of semaglutide to subjects, who are either overweight or with obesity. Changes in body weight, assessed as the exploratory endpoint, were as high as 17.1% (15.9 kg; with 2.4 mg of cagrilintide plus 2.4 mg of semaglutide) after 20 weeks of treatment [11••]. A RCT currently ongoing, aims to test the efficacy and safety of cagrilintide sc 2.4 mg in combination with semaglutide sc 2.4 mg (cagrisema sc 2.4 mg/2.4 mg) once-weekly in participants with overweight or obesity (REDEFINE 1, NCT05567796) [48].
Tirzepatide
Further to the aforementioned GLP-1Ras, such as liraglutide and semaglutide, a new anti-obesity drug, administered once a week and combining the GIP receptor with GLP-1R agonism into a single novel molecule has been developed and named tirzepatide [49]. It seems that GIP is a hormone that potentiates the effects of GLP-1RAs.
The adverse drug effect of tirzepatide is similar to that seen in the GLP-1RAs [50], whereby the most common side effects associated with tirzepatide are related to gastrointestinal symptoms, i.e., diarrhoea, nausea and vomiting [13].
A large, controlled trial [51] was recently conducted and enrolled 2539 adults with overweight or obesity, who were randomly divided into four groups and received 5 mg, 10 mg or 15 mg of sc tirzepatide once-weekly or a placebo for 72 weeks. The mean weight loss percentage (WL%) at week 72 was − 15.0% with 5 mg, − 19.5% with 10-mg and − 20.9% with 15-mg weekly doses of tirzepatide, and − 3.1% with the placebo. The proportion of patients who had a WL% of 5% or more was 85%, 89%, 91% and 35% with 5 mg, 10 mg and 15 mg of tirzepatide and placebo, respectively. Moreover, more than 50% of patients in the 10 mg and 15 mg groups experienced a WL% ≥ 20% as compared with 3% in the placebo group [51]. In addition, a recent systematic review and meta-analysis confirmed that the WL% with tirzepatide is dose-dependent and this drug appears to induce a greater WL when compared to GLP-1 RAs, (up to 7.16 kg more with 15 mg of tirzepatide) [52].
Food Cravings
Food cravings are strongly associated with increased food intake and are a significant predictor of greater weight gain over time and a lifetime high BMI [53]. Although several types of food cravings have been defined, they are not always completely dissociable. However, the type of craving may be less important than its severity in terms of contributing to increased eating. While the obesogenic food environment negatively impacts on everyone to some extent, it has been recognized that some individuals may be more sensitive to food-related cues or may experience more cravings.
Naltrexone/Bupropion
Naltrexone antagonism of the opioid receptors has been reported as decreasing the subjective delightfulness or liking of particular foods; in particular, naltrexone plays a role in modulating the reward aspects of eating and the consumption of energy dense sugar and fat-laden foods [54,55,56]. The blockade of the l-opioid receptor with naltrexone, in order to antagonize the auto-inhibitory actions of the bupropion-stimulated release of endogenous opioids, is the basis of the combination treatment with bupropion and naltrexone for obesity management. Indeed, the injection of naltrexone and bupropion into the reward system of mice resulted in a reduction of food intake, and the effect was potentiated when these were administered together. A phase-3 trial demonstrated that the synergic treatment of naltrexone and bupropion significantly reduces food reward aspects, measured by the Control of Eating Questionnaire (CoEQ) [19, 20, 57]. Thus, patients displayed reduced frequency and intensity of food cravings. A recent randomized double-blind placebo-controlled trial showed that behavioral weight loss therapy (BT) and naltrexone-bupropion were efficient for binge-eating disorder [58], with BT being superior to the absence of BT.
Liraglutide
acts on the central nervous system increasing satiety and hence reducing ad libitum meal intake [59]. In response to highly palatable foods, liraglutide decreases neural activation in the brain areas associated with appetite and reward [60]. To regulate food intake, our brain integrates external cues with internal state signals, such as the feeling of hunger [61,62,63]. Incentive motivation is defined as the process that translates an expected reward into the effort spent to obtain it and this is regulated by the dopaminergic midbrain and its mesoaccumbens dopaminergic projections [64]. GLP-1 has a modulatory role in the midbrain dopamine function, blunting the interaction effect of hunger on motivation, depending on insulin sensitivity, meaning that it can restore dysregulated motivational behaviour in insulin-resistant humans [65]. Liraglutide, combined with intensive behavioural therapy (IBT), is associated with greater short-term results in dietary disinhibition, eating disorder psychopathology and shape concern than IBT alone [59, 66].
The Binge Eating Liraglutide Intervention (BELIEVE), a phase-3 RCT, confirmed the efficacy of liraglutide compared to placebo in reducing the number of binge episodes per week, ameliorating dietary disinhibition, perceived hunger, quality of life and a depressed mood [67].
Another RCT showed a significant difference in hunger, fullness and food preoccupation up until week 24 between liraglutide + IBT and placebo + IBT: these differences were not maintained at week 52, despite the greater weight loss in the liraglutide-treated participants [68].
Semaglutide
Clinically, 2.4 mg of semaglutide once-weekly has been shown to decrease energy intake by reducing appetite, food cravings and a preference for fatty, energy-dense foods, as well as improving control of eating and meal portion size management [38, 40].
The severity and type of food cravings were assessed in a subgroup of participants from Canada/USA in a STEP 5 trial [69] by using the CoEQ, comprising 19 items grouped into four domains: craving control, craving for savoury food, craving for sweet food or positive mood. Among the participants receiving treatment with semaglutide (n = 88) vs. placebo (n = 86), all four domain scores significantly improved at week 20 and 52. The domains of craving control and craving for savoury food maintained their improvements up to week 104 and the individual, craving-related items which were significantly reduced were: desire to eat salty and spicy food, craving for dairy food, craving for starchy food, difficulty in resisting cravings and difficulty in controlling eating. By contrast, the scores for hunger and fullness improved significantly at week 20 only [70].
In an exploratory phase-1 trial investigating the effects of 14 mg of oral semaglutide once-daily for 12 weeks (four-week dose escalation) on the control of eating among 15 subjects with T2D, fewer and less strong food cravings, as well as better eating control (assessed by CoEQ) were reported compared to the placebo [71].
A clinical trial is underway to assess whether 50 mg of semaglutide once-daily affects food intake, hunger, satiety and food cravings among those with obesity [72].
Type 2 Diabetes
In most cases, obesity precedes diabetes and is the most important risk factor in relation to the worldwide increase of T2D prevalence [73]; consequently, it has been increasingly recognized that many patients with T2D would benefit from having a primary weight-centric approach to diabetes treatment [74]. As such, weight loss ≥ 15% can have a disease-modifying effect among people with T2D. From this perspective, we are revising the impact of centrally-acting, anti-obesity drugs on T2D.
Naltrexone/Bupropion
The efficacy of naltrexone/bupropion in T2D was investigated in the COR-DM trial. In this study, 505 subjects with T2D with haemoglobin A1c (HbA1c) between 7 and 10% and fasting blood glucose < 270 mg/dl (15.0 mmol/dl) [75] and taking any combination of oral diabetes medication (metformin, sulfonylureas and thiazolidinediones) were enrolled. These subjects were randomized in a 2:1 ratio to NB32 or placebo for 56 weeks and stratified by HbA1c ≤ 8% or > 8% and sulfonylurea use. Subjects in both groups followed a hypocaloric diet (a 500 kcal per day deficit, based on estimated metabolic rate) and participated in moderate physical activity (30 min of walking, five days per week). Co-primary end points were represented by the percentage weight loss from the baseline and the percentage of patients achieving ≥ 5% weight loss. Secondary end points were represented by the percentage of patients achieving ≥ 10% weight loss and changes in waist circumference, triglycerides, HDL- and LDL- cholesterol (HDL-C and LDL-C) and hs-CRP. The metabolic parameters that were followed up were HbA1c, fasting blood glucose, fasting insulin, HOMA-IR and the percentage of patients achieving HbA1c < 7.0% and < 6.5%, in addition to changes in oral antidiabetic agents.
Based on the modified intent to treat (mITT) analysis, the average weight loss was significantly higher in the intervention group versus the placebo group (-5.0 ± 0.3% vs. -1.8 ± 0.4%; p < 0.001). HbA1c reduction was higher in the intervention group versus the placebo group (-0.6 vs. -0.1%; p < 0.001) at the end of 56 weeks. In addition, a greater percentage of patients in the intervention versus the placebo group achieved HbA1c < 7.0% (44.1 vs. 26.3%) or < 6.5% HbA1c (20.7 vs. 10.2%). Statistical analysis showed that the HbA1c change was significantly correlated with weight loss in both the intervention and the placebo groups.
Changes in fasting blood glucose and fasting insulin did not significantly differ between the intervention and placebo groups. In addition, over the course of the study, fewer patients in the intervention versus the placebo group needed to increase oral antidiabetic agents, due to worsening HbA1c values [75].
A post-hoc analysis of naltrexone/bupropion versus the placebo was carried out among subjects with stable T2D on an incretin agent prior to randomization in a double-blind, placebo-controlled CV outcome trial (CVOT) [76]. After one year, the mean weight loss was significantly greater among naltrexone/bupropion patients versus placebo patients, particularly those taking DPP-4i (mean absolute difference 4.6% [p < 0.0001]) and those taking GLP-1RAs (mean absolute difference 5.2%, p < 0.0001). The rates of serious adverse effects in all areas of this analysis were lower than have been reported in other CVOTs in T2D [76].
In summary, naltrexone/bupropion has been demonstrated to be effective in reducing HbA1c and is safe among subjects with T2D, who are taking oral antidiabetic agents.
Liraglutide
Beta-cell dysfunction is the main defect in patients with T2D, whose beta-cells number and function reduce by 50% by the time of their diagnosis [77]. Among the many ways in which the β-cell fails, there is the loss of incretin effect, which is defined as the increase in glucose-stimulated insulin secretion by intestinal insulinotropic peptides such as GIP and GLP-1. These two hormones can stimulate insulin secretion in a glucose-dependent way [78]. Liraglutide is currently approved for once-daily sc treatment of T2D at a dose of 1.8 mg [79].
The Liraglutide Effect and Action in Diabetes (LEAD™), completed in 2007, is a phase-3 development programme, divided into six clinical trials, each studying the effect of liraglutide in combination with other commonly used antidiabetic medication (LEAD 1, 2, 4, 5, 6) and in monotherapy (LEAD 3). This programme included 6500 people in 41 countries worldwide [80] and demonstrated the superiority of liraglutide compared with other antidiabetic drugs (including glimepiride, rosiglitazone, metformin, insulin glargine and exenatide) in lowering HbA1c, fasting plasma glucose and body weight [80]. A recent RCT (NCT01541215) was able to confirm that 1.8 mg of liraglutide per day (added to metformin, with or without basal insulin) improves glycaemic control in children and adolescents with T2DM, with only an increased frequency of adverse gastrointestinal events, mainly nausea [81]. Finally, and rather interestingly, liraglutide as an adjunct to insulin has been shown to improve glycaemic control, induce body weight loss and decrease exogenous insulin requirements and severe hypoglycaemia in patients with type 1 diabetes mellitus [82].
Semaglutide
is currently approved for the once-weekly sc and once-daily oral treatment of T2D. Semaglutide acts through the incretin pathway, stimulating insulin and inhibiting glucagon secretion from the pancreatic islets in a glucose-dependent manner [83]. Subcutaneous semaglutide was evaluated in the Semaglutide Unabated Sustainability in Treatment of Type 2 Diabetes (SUSTAIN) clinical trial programme, consisting of seven randomized, controlled phase-3 trials and involving more than 8000 patients with T2D. Six efficacy trials (SUSTAIN 1–5 and SUSTAIN 7) were designed to evaluate the efficacy and safety of semaglutide vs. comparators, across the broad spectrum of T2D pharmacotherapy [84]. Semaglutide consistently reduced HbA1c and fasting plasma glucose, and self-monitored blood glucose profiles and body weight, with a lower risk of hypoglycaemia (excluding placebo and sitagliptin), demonstrating superior glycaemic control vs. a dipeptidyl peptidase-4 inhibitor (sitagliptin), other once-weekly GLP-1RAs (exenatide ER and dulaglutide) and basal insulin (IGlar) [85,86,87,88,89,90].
Oral semaglutide represents the first available oral GLP-1RA for the management of T2D. Its approval was based on the results of the randomized, phase-3a clinical trial (Peptide InnOvatioN for Early diabEtes tReatment, PIONEER) that tested oral semaglutide among patients with T2D in relation to the placebo group and active glucose-lowering medication [91]. Over periods of treatment of up to 78 weeks, oral semaglutide (7 and 14 mg once-daily) reduced HbA1c, fasting plasma glucose and body weight, thereby showing significantly greater efficacy than sitagliptin, empagliflozin, liraglutide (at week 26) and dulaglutide [91].
Semaglutide/Cagrilintide
A phase-3 study (REDEFINE 2, NCT04982575) has been testing the efficacy of combination treatment with 2.4 mg of semaglutide plus 2.4 mg of cagrilintide in glycaemic control among subjects with overweight/obesity and T2D [92].
Tirzepatide
The impact of tirzepatide on the glycaemic profile has been reported as fasting glycaemia, post-prandial glycaemia, HbA1c and insulin sensitivity as well as hypoglycaemia. When compared to the placebo, tirzepatide seems to exhibit a significantly higher reduction of both fasting and post-prandial glycaemia (nearly − 60 mg/dL, and nearly − 2% as HbA1c) [13, 50, 93] and improves insulin resistance by reducing the HOMA-IR index by − 0.70 [50, 94].
However, more interestingly, tirzepatide action appears to exceed other treatments, such as GLP-1 RAs and basal insulin, in lowering fasting and post-prandial glycemia by 15 to 20 mg/dL and HbA1c by nearly 1% [94,95,96]. Moreover, patients receiving tirzepatide had significantly lower insulin resistance, expressed as HOMA-IR, compared to GLP-1 RAs by − 0.44 [50, 94].
Finally, the occurrence of hypoglycaemia with tirzepatide did not differ from that seen in the placebo group and had a lower incidence by comparison with basal insulin [97]. However, the difference in hypoglycaemia incidences between tirzepatide and GLP-1RAs is still unclear, due to the variability in definitions of hypoglycaemia among studies [52].
Cardiovascular Disease
Obesity is associated with an increased incidence of CV events, and weight loss achieved through lifestyle changes lowers risk factors but has no clear effect on CV outcomes. In contrast, some classes of drugs used to treat obesity exert cardioprotective effects, as shown by CVOTs in T2D patients or by indirect clinical evidence on inflammation and other CV risk factors.
Naltrexone/Bupropion
Evidence from a randomized clinical trial, the LIGHT trial, poses the question whether naltrexone/bupropion is safe for the CV system or even decreases CV risk [98]. Indeed, this trial was prematurely interrupted by the FDA because of serious confidentiality breaches, that led to the disclosure of preliminary findings which indicated a potential positive effect on CV risk factors. The findings of the LIGHT trial, including those collected at the time, showed that 25 and 50% of the planned major CV events (MACE) (death from CV causes, nonfatal myocardial infarction, or nonfatal stroke) occurred, and when the study was interrupted, these data were published in 2016 [98]. The results at 25% MACE highlight some CV benefit from this drug, while those at 50% of MACE only demonstrated non-inferiority vs. placebo at the limit of 2.0 but not at 1.4, as hypothesized. Data at 64% MACE, when the study was closed, suggested a worse safety profile, with a higher prevalence of nonfatal stroke in the naltrexone/bupropion. However, no definite conclusion can be drawn on CV safety regarding this drug combination, since the trial was not completed and provided different results of the analyses performed at different time points.
Taken together, bupropion and naltrexone are expected to have both beneficial and detrimental CV effects. Indeed, bupropion, while possibly increasing heart rate and blood pressure by potentiating catecholaminergic neurotransmission, it exerts anti-inflammatory effects [99]. Naltrexone could blunt cardiac adaptation to ischemia, although the beneficial protective effects mediated by endogenous opioid in the endothelium, could have a positive effect on strokes and reduce toll-like, receptor4-dependent inflammation [99].
Liraglutide
The impact of liraglutide on CV outcomes was studied in a large RCT, The Liraglutide Effect and Action in Diabetes: Evaluation of Cardiovascular Outcome Results (LEADER) trial (ClinicalTrials.gov NCT01179048) [100] including 9340 patients with advanced T2DM and high baseline CV risk. After a median follow-up of 3.8 years, patients with liraglutide exhibited a significant reduction in the first occurrence of death from CV causes, non-fatal myocardial infarction or non-fatal stroke primary outcomes, compared to patients in the placebo group, with a hazard ratio (HR) of 0.87; 95% CI 0.78–0.97, p = 0.01. Furthermore, nephropathy events were significantly lower after liraglutide therapy than placebo (HR 0.78; 95% CI 0.67–0.92), but there was no significant difference in retinopathy events [101]. Another study suggested that liraglutide did not have an impact on left ventricular systolic function compared with placebo in stable, chronic heart failure patients with and without T2D (LIVE) [102]. On the other hand, it was demonstrated that liraglutide use in subjects with heart failure and reduced left ventricular ejection fraction was implicated with an increased adverse risk of heart failure-related outcomes [103]. Since it is challenging to elucidate whether GLP-1 analogues reduce the risk of atherosclerosis independent of glycaemic control, in a mouse model of arterial hypertension, GLP-1Ras, including liraglutide, reduce blood pressure and protect endothelial function independent of glucose control. These protective effects by GLP-1Ras require the endothelial but not myeloid cells [104]. Recently, a RCT was performed to evaluate the effects of 3.0 mg of injectable liraglutide daily, plus lifestyle intervention on body fat distribution in overweight or adults with obesity with and without T2D and at high CVD risk over 40 weeks of treatment [105]. Liraglutide showed consistent effects on visceral adipose tissue (VAT), measured with MRI, across all subgroups of age, sex, race–ethnicity, BMI classification and prediabetes status at baseline, with no significant interactions observed. Therefore, it was suggested that VAT reduction may be one mechanism capable of explaining the benefits seen in CV outcomes in previous trials with liraglutide, among patients with T2D, given its different effects on glucose homeostasis, atherogenic lipids, neuromediated appetite suppression and inflammation.
Semaglutide
The results of the SUSTAIN-6, a multicentre CVOT with a median observation time of 2.1 years, were significant for the noninferiority and superiority (although the latter was not prespecified) of semaglutide vs. placebo, reporting a 26% lower risk of the primary composite CV outcome (CV death, nonfatal myocardial infarction or nonfatal stroke) in patients treated once-weekly with 0.5 mg or 1.0 mg sc semaglutide, compared to those receiving placebo [106]. CV and all-cause mortality were also significantly reduced in the PIONEER 6 trial, which aimed to evaluate the CV safety of once-daily, oral formulations of semaglutide [107].
CV outcomes are currently being investigated among those with obesity and with established CVD (but not T2D) over 31 to 59 months of treatment in the Semaglutide Effects on Heart Disease and Stroke in Patients with Overweight or Obesity (SELECT) trial (NCT03574597) [108]. The SELECT-LIFE (NCT04972721), an observational, non-interventional survey study, will examine the long-term effects (up to 10 years) on SELECT trial participants.
Tirzepatide
In a clinical trial that included 475 adults randomized in a 1:1:1:1 ratio to receive 5 mg (n = 116), 10 mg (n = 119) or 15 mg (n = 120) of sc injections of tirzepatide or placebo once-weekly (n = 120) over a 40-week period, one of the outcomes was changes in blood pressure [97]. At week 40, the mean reduction in systolic and diastolic blood pressure was − 6.1 to − 12.6 mm Hg and − 2.0 to − 4.5 mm Hg for the tirzepatide groups and − 1.7 mm Hg and − 2.1 mm Hg for the placebo group [97].
On the other hand, a recent meta-analysis examined the adverse CV events of tirzepatide, including seven RCTs [109]. The primary aim was to compare the time to the first confirmed occurrence of four-component, major, adverse CV events (CV death, myocardial infarction, stroke and hospitalized unstable angina) between pooled tirzepatide groups and control groups [109]. The data of 4,887 participants treated with tirzepatide and those of the 2,328 control participants were analysed. Tirzepatide did not increase the risk of major CV events in participants with T2D versus controls, revealing its safety [109].
Few studies detected changes in lipid profile, expressed as LDL-c, HDL-c and triglycerides due to tirzepatide, as compared with the placebo and GLP-1RAs. Firstly, patients receiving tirzepatide displayed significantly reduced triglycerides by nearly − 30 mg/dL and LDL‑c − 5 mg/dL, and higher HDL c blood levels by + 0.5 mg/dL, as compared to the placebo [50]. On the other hand, patients receiving tirzepatide did not have significantly different triglycerides and LDL‑c compared with GLP-1RAs (i.e., dulaglutide/semaglutide) [94], but had significantly higher HDL-c compared to dulaglutide/semaglutide + 0.72 mg/dL [50].
A recent study assessed the effect of once-weekly tirzepatide (1, 5, 10 or 15 mg), GLP-1RAs (i.e., dulaglutide) (1.5 mg) and placebo on inflammation-related biomarkers, measured at baseline, 4, 12 and 26 weeks [110]. At 26 weeks, 10 and 15 mg of tirzepatide decreased YKL-40 (also known as chitinase-3 like-protein-1), intercellular adhesion molecule 1 (ICAM-1), leptin and growth differentiation factor 15 levels vs. the baseline, as well as YKL-40 and leptin levels vs. both the placebo and GLP-1RAs. Similarly, 15 mg of tirzepatide also decreased ICAM-1 levels vs. the placebo and GLP-1RAs, as well as high-sensitivity C-reactive protein (hsCRP) levels vs. the baseline and the placebo, but not vs. GLP-1RAs. YKL-40, hsCRP and ICAM-1 levels rapidly decreased within four weeks of treatment with 10 and 15 mg of tirzepatide, whereas the decrease in leptin levels was more gradual and did not plateau by 26 weeks [110].
Non-Alcoholic Fatty Liver Disease
Obesity is not associated only with atherogenic dyslipidaemia, hypertension and CVD, but also with metabolic complications linked to insulin resistance, such as non-alcoholic fatty liver disease (NAFLD) [111].
This term refers to a spectrum of conditions defined by the presence of approximately 5% liver fat accumulation, which progresses over time to (non-alcoholic) steatohepatitis, that might occur with or without the presence of liver fibrosis, cirrhosis and hepatocellular carcinoma [112]. A growing body of evidence supports the notion that NAFLD is a multisystem disease, representing a strong independent predictor of CV events [113], chronic kidney disease and some types of extrahepatic cancers [114]. The magnitude of this risk parallels the severity of NAFLD (especially the liver fibrosis stage) [114].
Naltrexone/Bupropion
Naltrexone/bupropion has not been evaluated in-depth to date among patients with obesity and NAFLD, and only limited data demonstrating the hepatic effect of these medications are available. In a post-hoc analysis of four RCTs, the combination of naltrexone/bupropion for one year improved the fibrosis-4 index (FIB-4), regarded as a non-invasive index of hepatic fibrosis and independent of potential cofounders such as weight loss [115]; no change in ALT was reported [115]. Although there is scarce evidence relating to the efficacy of naltrexone/bupropion in relation to NAFLD, it could be hypothesized that the magnitude of weight loss may potentially lead to an improvement in hepatic steatosis and possibly inflammation.
Liraglutide
Although there are no licensed treatments for NAFLD, GLP-1RAs are promising [116, 117]. Indeed, this class of drugs is beneficial for patients with NAFLD, not only by reducing the long-term risk of CVD, which is the leading cause of death in patients with NAFLD, but also by exerting hepato-protective effects [116]. Indeed, although the presence of GLP-1R in hepatocytes is still controversial [118], GLP-1RAs have been shown to improve hepatic mitochondrial function and insulin sensitivity and to inhibit endoplasmic reticulum stress. They also promote autophagy to limit free fatty acid accumulation and lipotoxicity [119].
Certain studies have demonstrated that liraglutide was able to improve liver steatosis, measured as a reduction in liver fat content in RCT [120] or in open label studies [121].
Among 52 overweight patients with biopsy-proven NASH and treated either with 1.8 mg of liraglutide daily or the placebo for 48 weeks [122], the resolution of NASH occurred mostly in patients treated with liraglutide compared with those given the placebo. Fibrosis was also less in patients with liraglutide (9% versus 36%, p = 0.04).
In a 26-week RCT, investigating the effect of antidiabetic agents on NAFLD in T2M with an inadequate glycaemic control by metformin, 75 patients were randomized (1:1:1) to receive add-on liraglutide, sitagliptin or insulin glargine. Intrahepatic lipid (by MRI-estimated proton density fat fraction, MRI-PDFF), VAT and weight decreased significantly with liraglutide (p < 0.001; p = 0.003; p = 0.005, respectively) and sitagliptin (p = 0.001; p = 0.027; p = 0.005, respectively). No significant change in MRI-PDFF, VAT or body weight was observed with insulin glargine. Subcutaneous AT decreased significantly in the liraglutide group (p = 0.020) compared to other groups. Changes in MRI-PDFF, VAT and body weight were significantly greater with liraglutide treatment than insulin glargine but did not differ significantly between liraglutide and sitagliptin (J. Yan et al. 2019). In conclusion, the improvement of NAFLD by GLP-1RA involve different pathways including, on the one hand, an adaptation of plasma insulin and glucagon concentrations with a higher insulin sensitivity and, on the other hand, a decrease in AT lipotoxicity with weight-independent mechanisms [123]. However, hepatocytes lack GLP-1 receptors [124] thus it does not seem to be a direct effect of liraglutide and its precise role has yet to be elucidated.
Semaglutide
The effects of semaglutide, which had been reported to reduce levels of alanine aminotransferase and inflammatory markers in subjects with T2D and/or obesity in a dose-dependent manner [125], have been investigated in 67 subjects with NAFLD, using non-invasive MRI methods. After 72 weeks of treatment with 0.4 mg of semaglutide once-daily, a greater reduction in liver steatosis was achieved compared to the placebo, however, no significant changes in liver stiffness, a surrogate marker of fibrosis, were observed [126].
In patients with NASH and liver fibrosis at stage F1, F2 or F3, treatment with sc semaglutide (0.1, 0.2, 0.4 mg once-daily) for 72 weeks resulted in a higher prevalence of NASH resolution, with no worsening of fibrosis compared with the placebo [127]. However, there was no improvement in fibrosis [127], possibly due to the short trial duration, a small study population, disease heterogeneity and the intrinsic limitations of liver biopsy (e.g., sample variability and reading across pathologists) [128].
As proof of concept, the phase-2 trial (NCT03987074) has been exploring the effectiveness of 2.4 mg of semaglutide administered alone or co-administered with firsocostat and cilofexor over a 24-week period in patients with NASH and mild to moderate fibrosis. Improvements in hepatic steatosis (measured by magnetic resonance imaging proton density fat fraction), liver injury (measured by serum alanine aminotransferase) and fibrosis (assessed and measured by vibration-controlled transient elastography and enhanced liver fibrosis score) were observed with semaglutide monotherapy, with the combination regimen further improving hepatic steatosis [129].
Another placebo-controlled, phase-2 trial (NCT03987451) has been investigating the effectiveness of 2.4 mg of semaglutide in patients with NAFLD and cirrhosis for 48 weeks [130].
Lastly, a phase-3 clinical trial (NCT04822181), aiming to study the effect of sc semaglutide monotherapy once-weekly over a period of five years in patients with NASH is currently in progress. Progression to cirrhosis, improvement of inflammation and histology of ballooned hepatocytes are some of the endpoints [131].
Semaglutide/Cagrilintide
A placebo-controlled phase-2 study (NCT05016882) has been investigating the efficacy and safety of sc semaglutide (2.4 mg once-weekly), co-administered with cagrilintide with three different doses (7.5 mg, 15 mg and 30 mg once-weekly) in subjects with NASH and fibrosis stage 2 − 4 (F2 − F4). The main endpoints are the improvement in liver fibrosis and the improvement/resolution of NASH [132].
Tirzepatide
Few studies have examined the impact of tirzepatide on NAFLDs. A post-hoc analysis of a 26-week, phase 2 study in which T2D patients were randomized to receive tirzepatide, basal insulin or placebo, tirzepatide (especially at higher doses of 10 and 15 mg/week) reduced transaminases and non-invasive indices of hepatic fibrosis (including keratin-18 and procollagen III) [133]. In line, one specific recent study [134] assessed the changes in liver fat content (LFC), measured by magnetic resonance imaging-proton density fat fraction, volume of VAT and abdominal subcutaneous adipose tissue (SAT) in response to tirzepatide or basal insulin. Exactly 296 patients were randomly assigned to treatment (tirzepatide 5 mg, n = 71; tirzepatide 10 mg, n = 79; tirzepatide 15 mg, n = 72 and basal insulin, n = 74). Baseline demographics and clinical characteristics were similar across all treatment groups. The absolute reduction in LFC at week 52 was significantly greater for the pooled tirzepatide groups with a dosage of 10 mg and 15 mg vs. the basal insulin group. Tirzepatide showed a significant reduction in LFC, VAT and SAT volumes compared with basal insulin.
A 52-week, multicenter, double-blind, placebo-controlled, phase 2 RCT investigating the efficacy and safety of tirzepatide (at 5, 10, 15 mg/week) on histological endpoints in overweight/patients with obesity and NASH is ongoing (SYNERGY-NASH; NCT04166773) [135].
Obstructive Sleep Apnoea
Obstructive sleep apnoea (OSA) is a common, chronic, sleep-related breathing disorder, characterized by repetitive upper airway collapse during sleep, causing sleep fragmentation, oxygen desaturation and excessive daytime sleepiness. OSA is common especially in older male subjects with obesity [136]. In addition, to increased all-cause mortality, untreated OSA is associated with other adverse outcomes including CVD, cerebrovascular events, T2D and cognitive impairment [137]. OSA may also be considered as a potential influent factor of weight management outcomes, since it was found to be associated with impaired muscle energy metabolism [138], reduced levels of physical activity and exercise performance [139], as well as higher levels of ghrelin, known to be an appetite stimulant [140].
Naltrexone/Bupropion
Currently there is no evidence relating to the efficacy and safety of naltrexone/bupropion in subjects with obesity and OSA. One crossover RCT in 12 OSA patients reported a reduction in the Apnoea-Hypopnea Index (AHI) following a single dose of naltrexone [141]. Although this is a promising result, further trials are needed in order to use naltrexone/bupropion in a safe manner in subjects with obesity and OSA.
Liraglutide
OSA was found to be associated with a lower GLP-1 response to a glucose challenge after adjusting for gender, BMI and glycaemic status [142], whereas in severe OSA, higher fasting GLP-1 levels were found and interpreted as a compensatory mechanism [143].
The SCALE Sleep Apnoea RCT aimed to investigate whether 3.0 mg of liraglutide reduced OSA severity compared with the placebo, using the primary endpoint of change in AHI after 32 weeks, both in combination with lifestyle intervention, in participants with obesity and moderate or severe OSA, who are unable or unwilling to use CPAP treatment [144]. At week 32, the mean weight loss was significantly greater with liraglutide than with the placebo (p < 0.0001). Consistent with the greater weight loss effect, significantly greater reductions in waist and neck circumference were also observed with liraglutide compared with the placebo. In both treatment groups, most of the reduction in mean AHI occurred by week 12. At week 32, the mean reduction in AHI with liraglutide was statistically significant compared with the placebo (estimated treatment difference: − 6.1 events h − 1 (95% CI, − 11.0 to − 1.2). The treatment effect on AHI did not depend on the participants’ gender, baseline BMI or OSA severity category. However, participants with severe OSA at baseline experienced a greater mean reduction in AHI in both treatment groups. Furthermore, there was a trend which showed that more liraglutide-treated patients, rather than placebo-treated participants, tended not to meet the diagnostic criteria for OSA. At week 32, weight loss with liraglutide had not plateaued. Thus, it is possible that further weight loss-related improvement in AHI would have been possible with a longer duration of liraglutide treatment [144].
Tirzepatide
To the best of our knowledge, there are no published results to date regarding the effect of tirzepatide on OSA, but actions have been already taken in this direction. Currently, recruitment to certain on-going clinical trials have started. For instance, the SURMOUNT-OSA study [145], initiated a few months ago, aims to evaluate the effect and safety of tirzepatide in 412 adults of both genders with OSA and obesity, randomly selected to use/not use the Positive Airway Pressure therapy, the primary outcome being the change in AHI from the baseline to the one year follow up.
Neurodegenerative Diseases
Epidemiological evidence suggest an increased risk of developing cognitive impairment and neurodegenerative diseases among individuals with obesity or T2D [146, 147]. Insulin is an important neuroprotective growth factor and neuronal insulin resistance may underlie the link between the presence of metabolic abnormalities and both Alzheimer’s disease and Parkinson’s disease [148, 149]. In addition, obesity might be associated with neurodegeneration, by causing brain inflammation, oxidative stress, protein deposition and morphological changes in brain cells [150].
Alzheimer’s Disease
Alzheimer's disease is the most common form of neurodegenerative dementia and is characterized by impaired memory and progressive changes in cognition, behaviour and the motor system. The main neuropathological features of Alzheimer's disease include cortical atrophy, amyloid plaques formed by aggregated amyloid β (Aβ) peptides and intracellular neurofibrillary tangles, made of hyperphosphorylated tau protein, causing a reduction in synaptic strength, synaptic loss and neurodegeneration [151]. However, the current treatments for Alzheimer's disease are symptomatic and they target neurotransmission rather than neurodegeneration or neuronal metabolism [152].
Liraglutide
In experimental mice models, caloric restriction has been proven to reduce Aβ and neurofibrillary tangle loads. However, the neurotrophic/neuroprotective actions of GLP-1 RAs can be considered direct actions, rather than secondary actions, due to reduced food intake [153]. Indeed, preclinical models have shown that GLP-1 can promote synaptic formation, neurogenesis and neuroprotection, and may reduce neuroinflammation [154,155,156]. Liraglutide was shown to attenuate impaired learning and memory, as well as synaptic plasticity, decrease hippocampal neuronal loss, reduce Aβ oligomer levels and tau hyperphosphorylation, and promote neurogenesis and homeostatic autophagy in preclinical studies [156]. The use of 1.8 mg of liraglutide, used as a treatment in Alzheimer's disease patients with a long-standing disease, prevents the expected decline of cerebral glucose metabolism that reflects disease progression, but studies up until now have not shown any differences between the groups treated with liraglutide and the placebo regarding amyloid deposition or cognition [152]. The Evaluating Liraglutide in Alzheimer's Disease (ELAD, NCT01843075) is an ongoing multi-centre, phase-2b RCT using liraglutide in patients with mild Alzheimer's dementia, with the aim of assessing the change in the cerebral glucose metabolic rate as its primary outcome [157].
Semaglutide
GLP-1 not only enhances insulin signalling in the brain but, since GLP-1Rs are expressed in the brain [158], the former also seems to influence the control of synaptic plasticity, thus exerting neuroprotective effects [159, 160].
In human neuroblastoma (SH-SY5Y) cell lines, semaglutide exerted neuroprotective effects against extracellular amyloid-β plaques, by enhancing autophagy and inhibiting apoptosis [161]. Preclinical models, real-world evidence studies and the post-hoc analysis of data from large CV outcome trials prompted the testing of oral semaglutide in Alzheimer’s patients [162]. Indeed, two placebo-controlled phase-3 trials (EVOKE NCT04777396 and EVOKE Plus NCT04777409) have been investigating the efficacy of oral semaglutide (once-daily 14 mg) in patients with early Alzheimer’s disease. Change in the clinical dementia rating, time of progression to dementia and the change in the Alzheimer's Disease Composite Score are some of the outcomes that will be assessed [163].
Parkinson’s Disease
Parkinson’s disease is a progressive nervous system disorder the aetiology of which remains unclear, although genetic and environmental factors seem to be involved. The clinical hallmark of Parkinson’s disease is a motor syndrome characterized by bradykinesia, rest tremor, rigidity postural instability and gait difficulties, due to nigral degeneration and striatal dopamine depletion. Parkinson’s disease is also associated with a variety of non-motor symptoms (e.g., constipation, urinary dysfunction, orthostatic hypotension, memory loss, depression and sleep disturbances) which are likely to be related to the neurodegeneration of other structures, including the peripheral autonomic nervous system [164]. Loss of dopaminergic neurons by apoptosis or autophagy and intracellular accumulation of misfolded α-synuclein (protein involved in dopamine metabolism) in abnormal aggregates, termed Lewy bodies, are the main pathological features of the disease [165].
Liraglutide
GLP1-RAs not only have a protective role in relation to toxic insults, but are also able to elevate endogenous TH levels in dopaminergic neurons [166, 167]. GLP1-RAs are able to change the microglial phenotype from a pro-inflammatory to a quiescent state; moreover, microglial cells generate additional GLP-1 in their anti-inflammatory state both as a potential trophic and as an anti-inflammatory factor [149, 167]. Preclinical studies showed that liraglutide reverses nigral neuronal loss, decreases pro-inflammatory cytokines and increases striatal dopamine, nigral glial cell line-derived neurotrophic factors and TH + cells. As a result, the pro-apoptotic environment in nigrostriatal tissues reduces significantly [168]. Long-term treatment with liraglutide can protect against motor function decay and dopaminergic neuron loss [169].
An ongoing phase-2, single centre, double-blind, placebo-controlled clinical trial is testing the efficacy and safety of liraglutide in the treatment of patients with idiopathic Parkinson’s disease [170].
Semaglutide
In the methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) mouse model of Parkinson’s disease, semaglutide (25 nmol/kg once-daily) improved most of the neuropathological features of Parkinson’s disease, reducing motor impairment, oxidative damage, inflammation and apoptosis in the substantia nigra [171]. It also increased dopamine levels by supplementing tyrosine hydroxylase levels in the brain and protected dopaminergic neurons, by reducing the aggregation of α-synuclein and enhancing the expression of “glial cell line-derived neurotrophic factor” (GDNP) [172].
A phase-2 placebo-controlled clinical trial aiming to explore the effects of 1.0 mg of sc semaglutide once-weekly on Parkinson’s disease was registered by Oslo University Hospital in 2018, but recruitment has yet to start [173]. This study was designed to assess the neuroprotective and anti-inflammatory effects of semaglutide on both motor and non-motor symptoms, nigro-striatal degeneration, cognitive function and quality of life.
Depression
Depression is a mood disorder with a high prevalence of coexistence with many other diseases including metabolic disorders, such as obesity and T2D. Obesity increases the risk of depression and, conversely, depressive disorders are predictive of the development of obesity [174], therefore, a bidirectional relationship with a weight-related gradient has been described [175]. In addition to modulating eating behaviour, centrally-acting, anti-obesity drugs are also likely to alter emotional behaviour, due to the high expression of their receptors within the fronto-striatal and limbic circuitry.
Naltrexone/Bupropion
may be a particularly appealing option for weight management among those with depression, as one of its components, bupropion, is an agent which has been used as an antidepressant indicator for almost 40 years. A post-hoc analysis of five randomized clinical trials (duration range, 24–56 weeks) evaluating adverse effects on mood related to the use of naltrexone/bupropion in subjects with overweight or obesity as well as with or without clinical depression at baseline found a lower incidence of depression among naltrexone/bupropion-treated patients, compared to the placebo [176]. In a small 24-week pilot study that evaluated naltrexone/bupropion effects in 25 female patients affected by a major depressive disorder, the treatment was associated with a significant reduction of Montgomery–Åsberg Depression Rating Scale (MADRS) scores, improving depressive symptoms and a reduction of body weight in these patients [177]. Moreover, in patients with obesity who are on antidepressants, naltrexone/bupropion is well tolerated and is effective in promoting weight loss regardless of antidepressant use [178].
Liraglutide
Although GLP-1RAs seem to have a beneficial influence in terms of reversing many maladaptive brain changes relevant to depression, at the same time, they are reported to have certain unfavourable effects, such as activation of the hypothalamic–pituitary–adrenal axis and the intensification of anxiety-like behaviour in some animal studies [179]. Liraglutide has been studied in relation to depression and mood disorders, mainly for safety reasons, but with some important outcomes: not only is there no cause for concern regarding the neuropsychiatric safety of treatment with 3.0 mg of liraglutide, but phase-3 trials have shown an identical rate of suicidal ideation between the placebo and treatment groups [180]. Regarding its possible use as an antidepressant, there are some interesting behavioural studies showing that liraglutide attenuates depressive- and anxiety-like behaviours in the corticosterone induced depression mouse model via improving hippocampal neural plasticity [181, 182]. A recent study involving adults with mood disorders using the Trail Making Test-B to measure executive function and MRI for a morphometry assessment demonstrated a cognitive improvement following liraglutide administration, associated with changes in brain volumes, including caudate, putamen, middle frontal cortex, superior frontal cortex and lateral orbitofrontal cortex. These findings underscore the relationship between weight, brain structure and function [183].
Osteoarthritis
Osteoarthritis is the most common degenerative joint condition and is the leading cause of disability. Obesity, together with ageing, remains the most important risk factor as regards the occurrence and progression of osteoarthritis (OA) not only by overloading the joints with the consequent destruction of articular cartilage, but also by alterations in innate and adaptive immune responses that could lead to inflammatory cell recruitment into the synovial joints [184].
Liraglutide
Considering the overlap between OA risk factors and metabolic derangements, as well as the anti-inflammatory properties of GLP-1 and the expression of GLP-1R in joint tissues [185], GLP-1RAs may be considered as therapeutic candidates for managing OA. A double-blind, placebo-controlled RCT (LOSEIT) assessed that in patients with overweight or obesity and knee OA, liraglutide in adjunct to an eight-week diet induced significant weight loss but did not reduce knee pain compared to the placebo [186]. Treatment with a GLP-1RA was shown to increase bone formation and prevent bone loss after weight loss [187]. An in-vitro study on human chondrocytes, exposed to liraglutide, demonstrated that this may exert a powerful antioxidant effect in the context of OA, reducing proinflammatory cytokines and mediating cartilage degradation [188]. Some animal studies showed that an intra-articular injection of liraglutide alleviated pain-related behaviour in an in-vivo OA mouse model [189].
Semaglutide
A placebo-controlled phase-3 trial (NCT05064735) has started to investigate the efficacy of sc semaglutide (2.4 mg, once-weekly) over a 68-week period in patients with obesity and a clinical diagnosis of knee OA in terms of change in WOMAC pain, physical function and stiffness score [190].
Conclusions
Obesity still represents a growing worldwide health challenge. Shortened life expectancy, increased healthcare costs and cardiometabolic complications are some of the consequences of this complex disease.
Over the last few years, substantial advances in the understanding of the determinants, pathophysiology, assessment and treatment of obesity have been made. Although bariatric surgery still represents the most effective therapeutical approach, with well-established benefits, pharmacotherapy has been enriched with new and more effective, centrally-acting drugs, showing promise for even greater weight loss efficacy that could revolutionize obesity management in the years to come. The perspective of having a wider range of treatments increases the interest in their putative, pleiotropic effects and the chance to tailor therapy. In this review, we highlight the added benefits of these approved and upcoming, centrally-acting, anti-obesity drugs, focusing not only on the most common metabolic and CV effects but also on their less explored clinical benefits and drawbacks. The evolving scenario of the availability of anti-obesity drugs and the increasing knowledge of their added effects on obesity complications will allow clinicians to move into a new era of precision medicine.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Bray GA, Kim KK, Wilding JPH. Obesity: a chronic relapsing progressive disease process. A position statement of the World Obesity Federation. Obes Rev. 2017;18:715–23.
Powell-Wiley TM, Poirier P, Burke LE, Després J-P, Gordon-Larsen P, Lavie CJ, et al. Obesity and Cardiovascular Disease: A Scientific Statement From the American Heart Association. Circulation. 2021;143:E984-1010.
Apovian CM, Aronne LJ, Bessesen DH, McDonnell ME, Murad MH, Pagotto U, et al. Pharmacological management of obesity: an endocrine Society clinical practice guideline. J Clin Endocrinol Metab. 2015;100:342–62.
Apovian CM, Garvey WT, Ryan DH. Challenging obesity: Patient, provider, and expert perspectives on the roles of available and emerging nonsurgical therapies. Obesity (Silver Spring). 2015;23 Suppl 2:S1–26.
Kakkar AK, Dahiya N. Drug treatment of obesity: Current status and future prospects. Eur J Intern Med. 2015;26(2):89–94. https://doi.org/10.1016/j.ejim.2015.01.005.
Pi-Sunyer X, Astrup A, Fujioka K, Greenway F, Halpern A, Krempf M, et al. A Randomized, Controlled Trial of 3.0 mg of Liraglutide in Weight Management. N Engl J Med. 2015;373:11–22.
Malm-Erjefält M, Bjørnsdottir I, Vanggaard J, Helleberg H, Larsen U, Oosterhuis B, et al. Metabolism and excretion of the once-daily human glucagon-like peptide-1 analog liraglutide in healthy male subjects and its in vitro degradation by dipeptidyl peptidase IV and neutral endopeptidase. Drug Metab Dispos. 2010;38:1944–53.
Mehta A, Marso SP, Neeland IJ. Liraglutide for weight management: a critical review of the evidence. Obes Sci Pract. 2017;3:3–14.
Mysimba. Summary of product characteristics, EU. Orexigen Therapeutics Ireland Limited, March 2015. https://www.bing.com/search?pc=CBHS&ptag=N3110D122919A9DFA1A1FF2&form=CONMHP&conlogo=CT3210127&q=1)+Mysimba.+Summary+of+product+characteristics%2C+EU.+Orexigen+Therapeutics+Ireland+Limited%2C+March+2015.
Lau J, Bloch P, Schäffer L, Pettersson I, Spetzler J, Kofoed J, et al. Discovery of the Once-Weekly Glucagon-Like Peptide-1 (GLP-1) Analogue Semaglutide. J Med Chem. 2015;58:7370–80.
•• Enebo LB, Berthelsen KK, Kankam M, Lund MT, Rubino DM, Satylganova A, et al. Safety, tolerability, pharmacokinetics, and pharmacodynamics of concomitant administration of multiple doses of cagrilintide with semaglutide 2·4 mg for weight management: a randomised, controlled, phase 1b trial. Lancet. 2021;397:1736–48. Conmbined treatment with cagrilintide and semaglutide 2·4 mg was well tolerated with an acceptable safety profile.
•• Lau DCW, Erichsen L, Francisco AM, Satylganova A, le Roux CW, McGowan B, et al. Once-weekly cagrilintide for weight management in people with overweight and obesity: a multicentre, randomised, double-blind, placebo-controlled and active-controlled, dose-finding phase 2 trial. Lancet. 2021;398:2160–72. https://doi.org/10.1016/S0140-6736(21)01751-7. Treatment with cagrilintide in subjects with overweight and obesity led to significant reductions in bodyweight and was well tolerated.
Frias JP, Nauck MA, Van J, Benson C, Bray R, Cui X, et al. Efficacy and tolerability of tirzepatide, a dual glucose-dependent insulinotropic peptide and glucagon-like peptide-1 receptor agonist in patients with type 2 diabetes: A 12-week, randomized, double-blind, placebo-controlled study to evaluate different do. Diabetes Obes Metab. 2020;22:938–46.
• Acosta A, Camilleri M, Abu Dayyeh B, Calderon G, Gonzalez D, McRae A, et al. Selection of Antiobesity Medications Based on Phenotypes Enhances Weight Loss: A Pragmatic Trial in an Obesity Clinic. Obesity. 2021;29:662–71. The phenotype-guided (hungry brain (abnormal satiation), emotional hunger (hedonic eating), hungry gut (abnormal satiety), and slow burn (decreased metabolic rate) approach was associated with 1.75-fold greater weight loss after 12 months with mean weight loss of 15.9% compared with 9.0% in the non-phenotype-guided group.
Vaxchora | European Medicines Agency. https://www.ema.europa.eu/en/medicines/human/EPAR/mysimba.
Booth K, Clements JN. Role of bupropion plus naltrexone for the management of obesity. J Pharm Technol. 2016;32:125–32.
Caixàs A, Albert L, Capel I, Rigla M. Naltrexone sustained-release/bupropion sustained-release for the management of obesity: Review of the data to date. Drug Des Devel Ther. 2014;8:1419–27. https://doi.org/10.2147/DDDT.S55587.
Apovian CM. Naltrexone/bupropion for the treatment of obesity and obesity with Type 2 diabetes. Future Cardiol. 2016;12:129–38.
Greenway FL, Fujioka K, Plodkowski RA, Mudaliar S, Guttadauria M, Erickson J, et al. Effect of naltrexone plus bupropion on weight loss in overweight and obese adults (COR-I): A multicentre, randomised, double-blind, placebo-controlled, phase 3 trial. Lancet. 2010;376:595–605.
Apovian CM, Aronne L, Rubino D, Still C, Wyatt H, Burns C, et al. A Randomized, Phase 3 Trial of Naltrexone SR/Bupropion SR on Weight and Obesity-related Risk Factors (COR-II). Obesity. 2013;21:935.
Agersø H, Jensen LB, Elbrønd B, Rolan P, Zdravkovic M. The pharmacokinetics, pharmacodynamics, safety and tolerability of NN2211, a new long-acting GLP-1 derivative, in healthy men. Diabetologia. 2002;45(2):195–202. https://doi.org/10.1007/s00125-001-0719-z.
Fda, Cder. SAXENDA (liraglutide [rDNA origin] injection). www.fda.gov/medwatch.
Yumuk V, Tsigos C, Fried M, Schindler K, Busetto L, Micic D, et al. European Guidelines for Obesity Management in Adults. Obes Facts. 2015;8:402–24.
Manigault KR, Thurston MM. Liraglutide: A glucagon-like peptide-1 agonist for chronic weight management. Consult Pharm. 2016;31(12):685–97. https://doi.org/10.4140/TCP.n.2016.685.
Wadden TA, Hollander P, Klein S, Niswender K, Woo V, Hale PM, et al. Weight maintenance and additional weight loss with liraglutide after low-calorie-diet-induced weight loss: the SCALE Maintenance randomized study. Int J Obes. 2013;37:1443–51.
Davies MJ, Bergenstal R, Bode B, Kushner RF, Lewin A, Skjøth TV, et al. Efficacy of Liraglutide for Weight Loss Among Patients With Type 2 Diabetes. JAMA. 2015;314:687.
Astrup A, Carraro R, Finer N, Harper A, Kunesova M, Lean MEJ, et al. Safety, tolerability and sustained weight loss over 2 years with the once-daily human GLP-1 analog, liraglutide. Int J Obes. 2012;36:843–54.
Ard J, Cannon A, Lewis CE, Lofton H, Vang Skjøth T, Stevenin B, et al. Efficacy and safety of liraglutide 3.0 mg for weight management are similar across races: subgroup analysis across the SCALE and phase II randomized trials. Diabetes Obes Metab. 2016;18:430–5.
• Kelly AS, Auerbach P, Barrientos-Perez M, Gies I, Hale PM, Marcus C, et al. A Randomized, Controlled Trial of Liraglutide for Adolescents with Obesity. N Engl J Med. 2020;382:2117–28. In adolescents with obesity, the use of liraglutide (3.0 mg) plus lifestyle therapy led to a significantly greater reduction in the BMI standard-deviation score than placebo plus lifestyle therapy.
Ryan PM, Seltzer S, Hayward NE, Rodriguez DA, Sless RT, Hawkes CP. Safety and Efficacy of Glucagon-Like Peptide-1 Receptor Agonists in Children and Adolescents with Obesity: A Meta-Analysis. J Pediatr. 2021;236:137-147.e13.
Järvinen A, Laine MK, Tikkanen R, Castrén ML. Beneficial effects of GLP-1 agonist in a Male with compulsive food-related behavior associated with autism. Front Psychiatry. 2019;10:97. https://doi.org/10.3389/fpsyt.2019.00097.
Bretault M, Carette C, Zaharia R, Vychnevskaia K, Bouillot JL, Czernichow S, et al. Liraglutide 3 mg as a weight-loss strategy after failed bariatric surgery in a patient with hypothalamic obesity following craniopharyngioma. Diabetes Metab. 2020. https://doi.org/10.1016/j.diabet.2019.07.004.
Barnard-Kelly K, Whicher CA, Price HC, Phiri P, Rathod S, Asher C, et al. Liraglutide and the management of overweight and obesity in people with severe mental illness: qualitative sub-study. BMC Psychiatry. 2022;22(1):21. https://doi.org/10.1186/s12888-021-03666-5.
Knudsen LB, Lau J. The discovery and development of liraglutide and semaglutide. Front Endocrinol. (Lausanne); 2019;10:155. https://doi.org/10.3389/fendo.2019.00155.
Chao AM, Tronieri JS, Amaro A, Wadden TA. Semaglutide for the treatment of obesity. Trends Cardiovasc Med. 2021;S1050–1738(21):00158–64. https://doi.org/10.1016/j.tcm.2021.12.008.
U.S Food and Drug Administration. FDA Approves New Drug Treatment for Chronic Weight Management. FDA Drug Saf. Commun. 2022. https://www.fda.gov/news-events/press-announcements/fda-approves-new-drug-treatment-chronic-weight-management-first-2014.
Wegovy | European Medicines Agency. https://www.ema.europa.eu/en/medicines/human/EPAR/wegovy.
Blundell J, Finlayson G, Axelsen M, Flint A, Gibbons C, Kvist T, et al. Effects of once-weekly semaglutide on appetite, energy intake, control of eating, food preference and body weight in subjects with obesity. Diabetes Obes Metab. 2017;19:1242–51.
Gabery S, Salinas CG, Paulsen SJ, Ahnfelt-Rønne J, Alanentalo T, Baquero AF, et al. Semaglutide lowers body weight in rodents via distributed neural pathways. JCI Insight. 2020;5(6):e133429. https://doi.org/10.1172/jci.insight.133429.
Friedrichsen M, Breitschaft A, Tadayon S, Wizert A, Skovgaard D. The effect of semaglutide 2.4 mg once weekly on energy intake, appetite, control of eating, and gastric emptying in adults with obesity. Diabetes Obes Metab. 2021;23:754–62.
O’Neil PM, Birkenfeld AL, McGowan B, Mosenzon O, Pedersen SD, Wharton S, et al. Efficacy and safety of semaglutide compared with liraglutide and placebo for weight loss in patients with obesity: a randomised, double-blind, placebo and active controlled, dose-ranging, phase 2 trial. Lancet. 2018;392:637–49.
Kushner RF, Calanna S, Davies M, Dicker D, Garvey WT, Goldman B, et al. Semaglutide 2.4 mg for the Treatment of Obesity: Key Elements of the STEP Trials 1 to 5. Obesity. 2020;28:1050–61.
Garvey WT, Batterham RL, Bhatta M, Buscemi S, Christensen LN, Frias JP, et al. Two-year effects of semaglutide in adults with overweight or obesity: the STEP 5 trial. Nat Med. 2022;28:2083–91.
Research Study to Investigate How Well Semaglutide Tablets Taken Once Daily Work in People Who Are Overweight or Living With Obesity (OASIS 1) - Full Text View - ClinicalTrials.gov. https://clinicaltrials.gov/ct2/show/NCT05035095.
Hay DL, Chen S, Lutz TA, Parkes DG, Roth JD. Amylin: Pharmacology, physiology, and clinical potential. Pharmacol. Rev. 2015. p. 564–600.
Potes CS, Lutz TA. Brainstem mechanisms of amylin-induced anorexia. Physiol Behav. 2010;100:511–8.
Boyle CN, Lutz TA, Le Foll C. Amylin – Its role in the homeostatic and hedonic control of eating and recent developments of amylin analogs to treat obesity. Mol Metab. 2018;8:203–10. https://doi.org/10.1016/j.molmet.2017.11.009.
A Research Study to See How Well CagriSema Helps People With Excess Body Weight Lose Weight - Full Text View - ClinicalTrials.gov. https://clinicaltrials.gov/ct2/show/NCT05567796.
Chavda VP, Ajabiya J, Teli D, Bojarska J, Apostolopoulos V. Tirzepatide, a New Era of Dual-Targeted Treatment for Diabetes and Obesity: A Mini-Review. Molecules. 2022;27(13):4315. https://doi.org/10.3390/molecules27134315.
Frias JP, Nauck MA, Van J, Kutner ME, Cui X, Benson C, et al. Efficacy and safety of LY3298176, a novel dual GIP and GLP-1 receptor agonist, in patients with type 2 diabetes: a randomised, placebo-controlled and active comparator-controlled phase 2 trial. Lancet. 2018;392:2180–93. https://doi.org/10.1016/S0140-6736(18)32260-8.
Jastreboff AM, Aronne LJ, Ahmad NN, Wharton S, Connery L, Alves B, et al. Tirzepatide Once Weekly for the Treatment of Obesity. N Engl J Med. 2022;387:205–16.
Karagiannis T, Avgerinos I, Liakos A, Del Prato S, Matthews DR, Tsapas A, et al. Management of type 2 diabetes with the dual GIP/GLP-1 receptor agonist tirzepatide: a systematic review and meta-analysis. Diabetologia. 2022;65:1251–61.
Boswell RG, Kober H. Food cue reactivity and craving predict eating and weight gain: A meta-analytic review. Obes Rev. 2016;17:159–77.
Reece AS. Hypothalamic opioid-Melanocortin appetitive balance and addictive craving. Med Hypotheses. 2011;76:132–7.
Yeomans MR, Gray RW. Effects of naltrexone on food intake and changes in subjective appetite during eating: Evidence for opioid involvement in the appetizer effect. Physiol Behav. 1997;62:15–21.
Yeomans MR, Gray RW. Selective effects of naltrexone on food pleasantness and intake. Physiol Behav. 1996;60:439–46. https://pubmed.ncbi.nlm.nih.gov/8840904/.
Wadden TA, Foreyt JP, Foster GD, Hill JO, Klein S, O’Neil PM, et al. Weight loss with naltrexone SR/bupropion SR combination therapy as an adjunct to behavior modification: The COR-BMOD trial. Obesity (Silver Spring). 2011;19:110–20.
Grilo CM, Lydecker JA, Fineberg SK, Moreno JO, Ivezaj V, Gueorguieva R. Naltrexone-Bupropion and Behavior Therapy, Alone and Combined, for Binge-Eating Disorder: Randomized Double-Blind Placebo-controlled Trial. Am J Psychiatry. 2022;179:927–37. https://doi.org/10.1176/appi.ajp.20220267.
Chao AM, Wadden TA, Walsh OA, Gruber KA, Alamuddin N, Berkowitz RI, et al. Effects of Liraglutide and Behavioral Weight Loss on Food Cravings, Eating Behaviors, and Eating Disorder Psychopathology. Obesity (Silver Spring). 2019;27:2005–10.
Farr OM, Sofopoulos M, Tsoukas MA, Dincer F, Thakkar B, Sahin-Efe A, et al. GLP-1 receptors exist in the parietal cortex, hypothalamus and medulla of human brains and the GLP-1 analogue liraglutide alters brain activity related to highly desirable food cues in individuals with diabetes: a crossover, randomised, placebo-controlled. Diabetologia. 2016;59:954–65.
Williams DL. Neural integration of satiation and food reward: role of GLP-1 and orexin pathways. Physiol Behav. 2014;136:194–9. https://doi.org/10.1016/j.physbeh.2014.03.013.
Sutton AK, Krashes MJ. Integrating Hunger with Rival Motivations. Trends Endocrinol. 2020;31(7):495–507. https://doi.org/10.1016/j.tem.2020.04.006.
Craig AD. An interoceptive neuroanatomical perspective on feelings, energy, and effort. Behav Brain Sci. 2013;36(6):685–6; discussion 707–26. https://doi.org/10.1017/S0140525X13001489.
Kremer Y, Flakowski J, Rohner C, Lüscher C. Context-dependent multiplexing by individual VTA dopamine neurons. J Neurosci. 2020;40:7489–509.
Hanssen R, Kretschmer AC, Rigoux L, Albus K, Edwin Thanarajah S, Sitnikow T, et al. GLP-1 and hunger modulate incentive motivation depending on insulin sensitivity in humans. Mol Metab. 2021;45:101163. https://doi.org/10.1016/j.molmet.2021.101163.
Wadden TA, Tronieri JS, Sugimoto D, Lund MT, Auerbach P, Jensen C, et al. Liraglutide 3.0 mg and Intensive Behavioral Therapy (IBT) for Obesity in Primary Care: The SCALE IBT Randomized Controlled Trial. Obesity (Silver Spring). 2020;28:529–36.
NCT03279731. Binge Eating Liraglutide Intervention - Tabular View - ClinicalTrials.gov. www.clinicaltrials.gov. 2017. https://clinicaltrials.gov/ct2/show/NCT03279731.
Tronieri JS, Wadden TA, Walsh O, Berkowitz RI, Alamuddin N, Gruber K, et al. Effects of liraglutide on appetite, food preoccupation, and food liking: results of a randomized controlled trial. Int J Obes. 2020;44:353–61.
Two-year Research Study Investigating How Well Semaglutide Works in People Suffering From Overweight or Obesity - Study Results - ClinicalTrials.gov. https://clinicaltrials.gov/ct2/show/NCT03693430.
Wharton S, Batterham RL, Bhatta M, Buscemi S, Christensen LN, Frias JP, et al. Two-year effect of semaglutide 2.4 mg on control of eating in adults with overweight/obesity: STEP 5. Obesity (Silver Spring); 2023; doi: https://doi.org/10.1002/oby.23673.
Gibbons C, Blundell J, Tetens Hoff S, Dahl K, Bauer R, Bækdal T. Effects of oral semaglutide on energy intake, food preference, appetite, control of eating and body weight in subjects with type 2 diabetes. Diabetes Obes Metab. 2021;23:581–8.
A Research Study Looking at How 50 mg Semaglutide Daily Affects Food Intake and Emptying of the Stomach in People With Obesity. ClinicalTrials.gov. 2022. https://www.clinicaltrials.gov/ct2/show/NCT05236517.
Sbraccia P, D’Adamo M, Guglielmi V. Is type 2 diabetes an adiposity-based metabolic disease? From the origin of insulin resistance to the concept of dysfunctional adipose tissue. Eat Weight Disord. 2021;26(8):2429–41. https://doi.org/10.1007/s40519-021-01109-4.
Lingvay I, Sumithran P, Cohen RV, le Roux CW. Obesity management as a primary treatment goal for type 2 diabetes: time to reframe the conversation. Lancet. 2022;399(10322):394–405. https://doi.org/10.1016/S0140-6736(21)01919-X.
Hollander P, Gupta AK, Plodkowski R, Greenway F, Bays H, Burns C, et al. Effects of naltrexone sustained-release/bupropion sustained-release combination therapy on body weight and glycemic parameters in overweight and obese patients with type2 diabetes. Diabetes Care. 2013;36:4022–9.
Wharton S, Yin P, Burrows M, Gould E, Blavignac J, Christensen RAG, et al. Extended-release naltrexone/bupropion is safe and effective among subjects with type 2 diabetes already taking incretin agents: a post-hoc analysis of the LIGHT trial. Int J Obes. 2021;45:1687–95.
UK prospective diabetes study (UKPDS) - VIII. Study design, progress and performance. Diabetologia. 1991;34:877–90.
Vilsbøll T, Holst JJ. Incretins, insulin secretion and Type 2 diabetes mellitus. Diabetologia. 2004;47(3):357–66. https://doi.org/10.1007/s00125-004-1342-6.77.
Victoza | European Medicines Agency. Eur. Med. Agency. 2019. https://www.ema.europa.eu/en/medicines/human/EPAR/victoza.
Madsbad S. Liraglutide effect and action in diabetes (LEAD™) trial. Expert Rev Endocrinol Metab. 2009;4(2):119–29. https://doi.org/10.1586/17446651.4.2.119.
Tamborlane WV, Barrientos-Pérez M, Fainberg U, Frimer-Larsen H, Hafez M, Hale PM, et al. Liraglutide in Children and Adolescents with Type 2 Diabetes. N Engl J Med. 2019;381:637–46.
Dimitrios P, Michael D, Vasilios K, Konstantinos S, Konstantinos I, Ioanna Z, et al. Liraglutide as Adjunct to Insulin Treatment in Patients with Type 1 Diabetes: A Systematic Review and Meta-analysis. Curr Diabetes Rev. 2020;16:313–26.
Donath MY, Burcelin R. GLP-1 effects on islets: Hormonal, neuronal, or paracrine? Diabetes Care Diabetes Care. 2013;36(Suppl 2):S145–8. https://doi.org/10.2337/dcS13-2015.
Aroda VR, Saugstrup T, Buse JB, Donsmark M, Zacho J, Davies MJ. Incorporating and interpreting regulatory guidance on estimands in diabetes clinical trials: The PIONEER 1 randomized clinical trial as an example. Diabetes Obes Metab. 2019;21(10):2203–10. https://doi.org/10.1111/dom.13804.
Sorli C, Harashima S ichi, Tsoukas GM, Unger J, Karsbøl JD, Hansen T, et al. Efficacy and safety of once-weekly semaglutide monotherapy versus placebo in patients with type 2 diabetes (SUSTAIN 1): a double-blind, randomised, placebo-controlled, parallel-group, multinational, multicentre phase 3a trial. Lancet Diabetes Endocrinol. 2017;5:251–60.
Ahrén B, Masmiquel L, Kumar H, Sargin M, Karsbøl JD, Jacobsen SH, et al. Efficacy and safety of once-weekly semaglutide versus once-daily sitagliptin as an add-on to metformin, thiazolidinediones, or both, in patients with type 2 diabetes (SUSTAIN 2): a 56-week, double-blind, phase 3a, randomised trial. lancet Diabetes Endocrinol. 2017;5:341–54. https://pubmed.ncbi.nlm.nih.gov/28385659/.
Ahmann AJ, Capehorn M, Charpentier G, Dotta F, Henkel E, Lingvay I, et al. Efficacy and Safety of Once-Weekly Semaglutide Versus Exenatide ER in Subjects With Type 2 Diabetes (SUSTAIN 3): A 56-Week, Open-Label. Randomized Clinical Trial Diabetes Care. 2018;41:258–66.
Aroda VR, Bain SC, Cariou B, Piletič M, Rose L, Axelsen M, et al. Efficacy and safety of once-weekly semaglutide versus once-daily insulin glargine as add-on to metformin (with or without sulfonylureas) in insulin-naive patients with type 2 diabetes (SUSTAIN 4): a randomised, open-label, parallel-group, multicentre, multinational, phase 3a trial. Lancet Diabetes Endocrinol. 2017;5:355–66.
Rodbard HW, Lingvay I, Reed J, De La Rosa R, Rose L, Sugimoto D, et al. Semaglutide Added to Basal Insulin in Type 2 Diabetes (SUSTAIN 5): A Randomized. Controlled Trial J Clin Endocrinol Metab. 2018;103:2291–301.
Pratley RE, Aroda VR, Lingvay I, Lüdemann J, Andreassen C, Navarria A, et al. Semaglutide versus dulaglutide once weekly in patients with type 2 diabetes (SUSTAIN 7): a randomised, open-label, phase 3b trial. Lancet Diabetes Endocrinol. 2018;6:275–86.
Thethi TK, Pratley R, Meier JJ. Efficacy, safety and cardiovascular outcomes of once-daily oral semaglutide in patients with type 2 diabetes: The PIONEER programme. Diabetes Obes Metab. 2020;22(8):1263–77. https://doi.org/10.1111/dom.14054.
Research Study to Look at How Well Cagrilintide Together With Semaglutide Works in People With Type 2 Diabetes - Full Text View - ClinicalTrials.gov. https://www.clinicaltrials.gov/ct2/show/NCT04982575.
Rosenstock J, Wysham C, Frías JP, Kaneko S, Lee CJ, Fernández Landó L, et al. Efficacy and safety of a novel dual GIP and GLP-1 receptor agonist tirzepatide in patients with type 2 diabetes (SURPASS-1): a double-blind, randomised, phase 3 trial. Lancet. 2021;398:143–55.
Frías JP, Davies MJ, Rosenstock J, Pérez Manghi FC, Fernández Landó L, Bergman BK, et al. Tirzepatide versus Semaglutide Once Weekly in Patients with Type 2 Diabetes. N Engl J Med. 2021;385:503–15.
Del Prato S, Kahn SE, Pavo I, Weerakkody GJ, Yang Z, Doupis J, et al. Tirzepatide versus insulin glargine in type 2 diabetes and increased cardiovascular risk (SURPASS-4): a randomised, open-label, parallel-group, multicentre, phase 3 trial. Lancet. 2021;398:1811–24.
Ludvik B, Giorgino F, Jódar E, Frias JP, Fernández Landó L, Brown K, et al. Once-weekly tirzepatide versus once-daily insulin degludec as add-on to metformin with or without SGLT2 inhibitors in patients with type 2 diabetes (SURPASS-3): a randomised, open-label, parallel-group, phase 3 trial. Lancet. 2021;398:583–98.
Dahl D, Onishi Y, Norwood P, Huh R, Bray R, Patel H, et al. Effect of Subcutaneous Tirzepatide vs Placebo Added to Titrated Insulin Glargine on Glycemic Control in Patients with Type 2 Diabetes: The SURPASS-5 Randomized Clinical Trial. JAMA. 2022;327:534–45.
Nissen SE, Wolski KE, Prcela L, Wadden T, Buse JB, Bakris G, et al. Effect of naltrexone-bupropion on major adverse cardiovascular events in overweight and obese patients with cardiovascular risk factors: A randomized clinical trial. JAMA. 2016;315:990–1004.
Cataldi M, Cignarelli A, Giallauria F, Muscogiuri G, Barrea L, Savastano S, et al. Cardiovascular effects of antiobesity drugs: are the new medicines all the same? Int J Obes Suppl. 2020;10:14–26.
Marso SP, Daniels GH, Brown-Frandsen K, Kristensen P, Mann JFE, Nauck MA, et al. Liraglutide and cardiovascular outcomes in type 2 diabetes. N Engl J Med. 2016;375(4):311–22. https://doi.org/10.1056/NEJMoa1603827.
Mann JFE, Ørsted DD, Brown-Frandsen K, Marso SP, Poulter NR, Rasmussen S, et al. Liraglutide and Renal Outcomes in Type 2 Diabetes. N Engl J Med. 2017;377:839–48.
Jorsal A, Kistorp C, Holmager P, Tougaard RS, Nielsen R, Hänselmann A, et al. Effect of liraglutide, a glucagon-like peptide-1 analogue, on left ventricular function in stable chronic heart failure patients with and without diabetes (LIVE)—a multicentre, double-blind, randomised, placebo-controlled trial. Eur J Heart Fail. 2017;19:69–77.
Hamad F, Elnour AA, Elamin A, Mohamed S, Yousif I, Don J, et al. Systematic Review of Glucagon-Like Peptide One Receptor Agonist Liraglutide of Subjects with Heart Failure with Reduced Left Ventricular Ejection Fraction. Curr Diabetes Rev. 2020;17:280–92.
Helmstädter J, Frenis K, Filippou K, Grill A, Dib M, Kalinovic S, et al. Endothelial GLP-1 (Glucagon-Like Peptide-1) Receptor Mediates Cardiovascular Protection by Liraglutide In Mice With Experimental Arterial Hypertension. Arterioscler Thromb Vasc Biol. 2020;40:145–58.
Neeland IJ, Marso SP, Ayers CR, Lewis B, Oslica R, Francis W, et al. Effects of liraglutide on visceral and ectopic fat in adults with overweight and obesity at high cardiovascular risk: a randomised, double-blind, placebo-controlled, clinical trial. Lancet Diabetes Endocrinol. 2021;9:595–605.
Marso SP, Bain SC, Consoli A, Eliaschewitz FG, Jódar E, Leiter LA, et al. Semaglutide and Cardiovascular Outcomes in Patients with Type 2 Diabetes. N Engl J Med. 2016;375:1834–44.
Husain M, Birkenfeld AL, Donsmark M, Dungan K, Eliaschewitz FG, Franco DR, et al. Oral Semaglutide and Cardiovascular Outcomes in Patients with Type 2 Diabetes. N Engl J Med. 2019;381:841–51.
Ryan DH, Lingvay I, Colhoun HM, Deanfield J, Emerson SS, Kahn SE, et al. Semaglutide Effects on Cardiovascular Outcomes in People With Overweight or Obesity (SELECT) rationale and design. Am Heart J. 2020;229:61–9.
Sattar N, McGuire DK, Pavo I, Weerakkody GJ, Nishiyama H, Wiese RJ, et al. Tirzepatide cardiovascular event risk assessment: a pre-specified meta-analysis. Nat Med. 2022;28:591–8.
Wilson JM, Lin Y, Luo MJ, Considine G, Cox AL, Bowsman LM, et al. The dual glucose-dependent insulinotropic polypeptide and glucagon-like peptide-1 receptor agonist tirzepatide improves cardiovascular risk biomarkers in patients with type 2 diabetes: A post hoc analysis. Diabetes, Obes Metab. 2022;24:148–53.
Gastaldelli A, Cusi K, Pettiti M, Hardies J, Miyazaki Y, Berria R, et al. Relationship Between Hepatic/Visceral Fat and Hepatic Insulin Resistance in Nondiabetic and Type 2 Diabetic Subjects. Gastroenterology. 2007;133:496–506.
Marchesini G, Roden M, Vettor R. “EASL-EASD-EASO Clinical Practice Guidelines for the management of non-alcoholic fatty liver disease”. J. Hepatol. European Association for the Study of the Liver. 2017. p. 466–7. https://linkinghub.elsevier.com/retrieve/pii/S0168827816306481.
Targher G, Byrne CD, Tilg H. NAFLD and increased risk of cardiovascular disease: clinical associations, pathophysiological mechanisms and pharmacological implications. Gut. 2020;69:1691–705.
Targher G, Tilg H, Byrne CD. Non-alcoholic fatty liver disease: a multisystem disease requiring a multidisciplinary and holistic approach. Lancet Gastroenterol Hepatol. 2021;6(7):578–88. https://doi.org/10.1016/S2468-1253(21)00020-0.
Bajaj HS, Burrows M, Blavignac J, Paron E, Camacho F, Gould E, et al. Extended-release naltrexone/bupropion and liver health: Pooled, post hoc analysis from four randomized controlled trials. Diabetes Obes Metab. 2021;23:861–5.
Mantovani A, Byrne CD, Targher G. Efficacy of peroxisome proliferator-activated receptor agonists, glucagon-like peptide-1 receptor agonists, or sodium-glucose cotransporter-2 inhibitors for treatment of non-alcoholic fatty liver disease: a systematic review. Lancet Gastroenterol Hepatol. 2022. p. 367–78.
Cusi K, Isaacs S, Barb D, Basu R, Caprio S, Garvey WT, et al. American Association of Clinical Endocrinology Clinical Practice Guideline for the Diagnosis and Management of Nonalcoholic Fatty Liver Disease in Primary Care and Endocrinology Clinical Settings: Co-Sponsored by the American Association for the Study of. Endocr Pract Endocr Pract. 2022;28:528–62.
Gupta NA, Mells J, Dunham RM, Grakoui A, Handy J, Saxena NK, et al. Glucagon-like peptide-1 receptor is present on human hepatocytes and has a direct role in decreasing hepatic steatosis in vitro by modulating elements of the insulin signaling pathway. Hepatol. 2010;51:1584–92.
Mahapatra MK, Karuppasamy M, Sahoo BM. Therapeutic Potential of Semaglutide, a Newer GLP-1 Receptor Agonist, in Abating Obesity, Non-Alcoholic Steatohepatitis and Neurodegenerative diseases: A Narrative Review. Pharm Res. 2022;39(6):1233–48. https://doi.org/10.1007/s11095-022-03302-1.
Vanderheiden A, Harrison LB, Warshauer JT, Adams-Huet B, Li X, Yuan Q, et al. Mechanisms of action of liraglutide in patients with type 2 diabetes treated with high-dose insulin. J Clin Endocrinol Metab Endocrine Society. 2016;101:1798–806.
Feng W, Gao C, Bi Y, Wu M, Li P, Shen S, et al. Randomized trial comparing the effects of gliclazide, liraglutide, and metformin on diabetes with non-alcoholic fatty liver disease. J Diabetes. 2017;9:800–9.
Armstrong MJ, Gaunt P, Aithal GP, Barton D, Hull D, Parker R, et al. Liraglutide safety and efficacy in patients with non-alcoholic steatohepatitis (LEAN): a multicentre, double-blind, randomised, placebo-controlled phase 2 study. Lancet. 2016;387:679–90.
Müller TD, Finan B, Bloom SR, D’Alessio D, Drucker DJ, Flatt PR, et al. Glucagon-like peptide 1 (GLP-1). Mol Metab. 2019;30:72–130. https://doi.org/10.1016/j.molmet.2019.09.010.
Pyke C, Heller RS, Kirk RK, Ørskov C, Reedtz-Runge S, Kaastrup P, et al. GLP-1 receptor localization in monkey and human tissue: Novel distribution revealed with extensively validated monoclonal antibody. Endocrinology. 2014;155:1280–90.
Newsome P, Francque S, Harrison S, Ratziu V, Van Gaal L, Calanna S, et al. Effect of semaglutide on liver enzymes and markers of inflammation in subjects with type 2 diabetes and/or obesity. Aliment Pharmacol Ther. 2019;50:193–203.
Flint A, Andersen G, Hockings P, Johansson L, Morsing A, Sundby Palle M, et al. Randomised clinical trial: semaglutide versus placebo reduced liver steatosis but not liver stiffness in subjects with non-alcoholic fatty liver disease assessed by magnetic resonance imaging. Aliment Pharmacol Ther. 2021;54:1150–61.
Newsome PN, Buchholtz K, Cusi K, Linder M, Okanoue T, Ratziu V, et al. A Placebo-Controlled Trial of Subcutaneous Semaglutide in Nonalcoholic Steatohepatitis. N Engl J Med. 2021;384:1113–24.
Patel Chavez C, Cusi K, Kadiyala S. The Emerging Role of Glucagon-like Peptide-1 Receptor Agonists for the Management of NAFLD. J Clin Endocrinol Metab. 2022;107:29–38.
Alkhouri N, Herring R, Kabler H, Kayali Z, Hassanein T, Kohli A, et al. Safety and efficacy of combination therapy with semaglutide, cilofexor and firsocostat in patients with non-alcoholic steatohepatitis: A randomised, open-label phase II trial. J Hepatol. 2022;77:607–18. https://clinicaltrials.gov/ct2/show/NCT03987074.
A Research Study on How Semaglutide Works in People With Fatty Liver Disease and Liver Damage - Full Text View - ClinicalTrials.gov. https://clinicaltrials.gov/ct2/show/NCT03987451.
Research Study on Whether Semaglutide Works in People With Non-alcoholic Steatohepatitis (NASH) - Full Text View - ClinicalTrials.gov. https://clinicaltrials.gov/ct2/show/NCT04822181.
Research Study on Whether a Combination of 2 Medicines (NNC0194 0499 and Semaglutide) Works in People With Non-alcoholic Steatohepatitis (NASH) - ClinicalTrials.gov. https://clinicaltrials.gov/ct2/show/NCT05016882.
Hartman ML, Sanyal AJ, Loomba R, Wilson JM, Nikooienejad A, Bray R, et al. Effects of Novel Dual GIP and GLP-1 Receptor Agonist Tirzepatide on Biomarkers of Nonalcoholic Steatohepatitis in Patients With Type 2 Diabetes Care. 2020;43:1352–5. https://pubmed.ncbi.nlm.nih.gov/32291277/.
Gastaldelli A, Cusi K, Fernández Landó L, Bray R, Brouwers B, Rodríguez Á. Effect of tirzepatide versus insulin degludec on liver fat content and abdominal adipose tissue in people with type 2 diabetes (SURPASS-3 MRI): a substudy of the randomised, open-label, parallel-group, phase 3 SURPASS-3 trial. Lancet Diabetes Endocrinol. 2022;10:393–406.
A Study of Tirzepatide (LY3298176) in Participants With Nonalcoholic Steatohepatitis (NASH) - Full Text View - ClinicalTrials.gov. https://clinicaltrials.gov/ct2/show/NCT04166773.
Young T, Palta M, Dempsey J, Skatrud J, Weber S, Badr S. The Occurrence of Sleep-Disordered Breathing among Middle-Aged Adults. N Engl J Med. 1993;328:1230–5.
Lévy P, Kohler M, McNicholas WT, Barbé F, McEvoy RD, Somers VK, et al. Obstructive sleep apnoea syndrome. Nat Rev Dis Primers. 2015;1:15015. https://doi.org/10.1038/nrdp.2015.15.
Vanuxem D, Badier M, Guillot C, Delpierre S, Jahjah F, Vanuxem P. Impairment of muscle energy metabolism in patients with sleep apnoea syndrome. Respir Med. 1997;91:551–7.
Da Silva RP, Martinez D, Pedroso MM, Righi CG, Martins EF, Silva LMT, et al. Exercise, Occupational Activity, and Risk of Sleep Apnea: A cross-sectional study. J Clin Sleep Med. 2017;13:197–204.
Ursavas A, Ilcol YO, Nalci N, Karadag M, Ege E. Ghrelin, leptin, adiponectin, and resistin levels in sleep apnea syndrome: Role of obesity. Ann Thorac Med. 2010;5:161–5.
Ferber C, Duclaux R, Mouret J. Naltrexone improves blood gas patterns in obstructive sleep apnoea syndrome through its influence on sleep. J Sleep Res. 1993;2:149–55.
Reutrakul S, Mokhlesi B. Obstructive Sleep Apnea and Diabetes: A State of the Art Review. Chest. 2017;152(5):1070–86. https://doi.org/10.1016/j.chest.2017.05.009.
Matsumoto T, Harada N, Azuma M, Chihara Y, Murase K, Tachikawa R, et al. Plasma incretin levels and dipeptidyl peptidase-4 activity in patients with obstructive sleep apnea. Ann Am Thorac Soc. 2016;13:1378–87.
Blackman A, Foster GD, Zammit G, Rosenberg R, Aronne L, Wadden T, et al. Effect of liraglutide 3.0 mg in individuals with obesity and moderate or severe obstructive sleep apnea: The scale sleep apnea randomized clinical trial. Int J Obes. 2016;40:1310–9.
Obstructive Sleep Apnea Master Protocol GPIF: A Study of Tirzepatide (LY3298176) in Participants With Obstructive Sleep Apnea - Full Text View - ClinicalTrials.gov. https://clinicaltrials.gov/ct2/show/NCT05412004.
Naderali EK, Ratcliffe SH, Dale MC. Obesity and Alzheimer’s disease: a link between body weight and cognitive function in old age. Am J Alzheimers Dis Other Demen. 2009;24(6):445–9. https://doi.org/10.1177/1533317509348208.
Elias MF, Elias PK, Sullivan LM, Wolf PA, D’Agostino RB. Lower cognitive function in the presence of obesity and hypertension: The Framingham heart study. Int J Obes Relat Metab Disord. 2003;27:260–8.
Diehl T, Mullins R, Kapogiannis D. Insulin resistance in Alzheimer’s disease. Transl Res. 2017;183:26–40. https://doi.org/10.1016/j.trsl.2016.12.005.
Athauda D, Foltynie T. Insulin resistance and Parkinson’s disease: A new target for disease modification? Prog Neurobiol. 2016;145–146:98–120.
Gómez-Apo E, Mondragón-Maya A, Ferrari-Díaz M, Silva-Pereyra J. Structural Brain Changes Associated with Overweight and Obesity. J Obes. 2021. https://doi.org/10.1155/2021/6613385.
Deture MA, Dickson DW. The neuropathological diagnosis of Alzheimer’s disease. Mol Neurodegener. 2019. https://doi.org/10.1186/s13024-019-0333-5.
Gejl M, Gjedde A, Egefjord L, Møller A, Hansen SB, Vang K, et al. In Alzheimer’s disease, 6-month treatment with GLP-1 analog prevents decline of brain glucose metabolism: Randomized, placebo-controlled, double-blind clinical trial. Front Aging Neurosci. 2016;8:108. https://doi.org/10.3389/fnagi.2016.00108.
Salcedo I, Tweedie D, Li Y, Greig NH. Neuroprotective and neurotrophic actions of glucagon-like peptide-1: An emerging opportunity to treat neurodegenerative and cerebrovascular disorders. Br J Pharmacol. 2012;166(5):1586–99. https://doi.org/10.1111/j.1476-5381.2012.01971.x.
Hölscher C. Potential role of glucagon-like peptide-1 (GLP-1) in neuroprotection. CNS Drugs. 2012;26(10):871–82. https://doi.org/10.2165/11635890-000000000-00000.
Yan J, Yao B, Kuang H, Yang X, Huang Q, Hong T, et al. Liraglutide, Sitagliptin, and Insulin Glargine Added to Metformin: The Effect on Body Weight and Intrahepatic Lipid in Patients With Type 2 Diabetes Mellitus and Nonalcoholic Fatty Liver Disease. Hepatology. 2019;69:2414–26.
Zhang M, Yan W, Yu Y, Cheng J, Yi X, Guo T, et al. Liraglutide ameliorates diabetes-associated cognitive dysfunction via rescuing autophagic flux. J Pharmacol Sci. 2021;147:234–44.
Vargas-Soria M, Carranza-Naval MJ, del Marco A, Garcia-Alloza M. Role of liraglutide in Alzheimer’s disease pathology. Alzheimers Res Ther. 2021;13(1):112. https://doi.org/10.1186/s13195-021-00853-0.
Alvarez A, Alarcón R, Opazo C, Campos EO, Muñoz FJ, Calderón FH, et al. Stable Complexes Involving Acetylcholinesterase and Amyloid-β Peptide Change the Biochemical Properties of the Enzyme and Increase the Neurotoxicity of Alzheimer’s Fibrils. J Neurosci. 1998;18:3213–23.
Hölscher C. Novel dual GLP-1/GIP receptor agonists show neuroprotective effects in Alzheimer’s and Parkinson’s disease models. Neuropharmacology. 2018;136(Pt B):251–9. https://doi.org/10.1016/j.neuropharm.2018.01.040.
Salameh TS, Rhea EM, Talbot K, Banks WA. Brain uptake pharmacokinetics of incretin receptor agonists showing promise as Alzheimer’s and Parkinson’s disease therapeutics. Biochem Pharmacol. 2020;180. https://pubmed.ncbi.nlm.nih.gov/32755557/.
Chang YF, Zhang D, Hu WM, Liu DX, Li L. Semaglutide-mediated protection against Aβ correlated with enhancement of autophagy and inhibition of apotosis. J Clin Neurosci. 2020;81:234–9. https://doi.org/10.1016/j.jocn.2020.09.054.
Novo Nordisk to enter phase 3 development in Alzheimer’s disease with oral semaglutide - Benzinga https://www.benzinga.com/pressreleases/20/12/tr18813500/novo-nordisk-to-enter-phase-3-development-in-alzheimers-disease-with-oral-semaglutide.
A Research Study Investigating Semaglutide in People With Early Alzheimer’s Disease (EVOKE) - ClinicalTrials.gov. https://clinicaltrials.gov/ct2/show/NCT04777396.
Tolosa E, Garrido A, Scholz SW, Poewe W. Challenges in the diagnosis of Parkinson’s disease. Lancet Neurol. 2021;20(5):385–97. https://doi.org/10.1016/S1474-4422(21)00030-2.
Kalia LV, Lang AE. Parkinson’s disease. Lancet. 2015;386(9996):896–912. https://doi.org/10.1016/S0140-6736(14)61393-3.
Li Y, Perry TA, Kindy MS, Harvey BK, Tweedie D, Holloway HW, et al. GLP-1 receptor stimulation preserves primary cortical and dopaminergic neurons in cellular and rodent models of stroke and Parkinsonism. Proc Natl Acad Sci U S A. 2009;106(4):1285–90. https://doi.org/10.1073/pnas.0806720106.
Kim DS, Choi H Il, Wang Y, Luo Y, Hoffer BJ, Greig NH. A New Treatment Strategy for Parkinson’s Disease through the Gut–Brain Axis: The Glucagon-Like Peptide-1 Receptor Pathway. Cell Transplant. 2017; 26(9):1560–1571. https://doi.org/10.1177/0963689717721234.
Badawi GA, Abd El Fattah MA, Zaki HF, El Sayed MI. Sitagliptin and liraglutide reversed nigrostriatal degeneration of rodent brain in rotenone-induced Parkinson’s disease. Inflammopharmacology. 2017;25(3):369–382. https://doi.org/10.1007/s10787-017-0331-6.
Ma D, Liu X, Liu J, Li M, Chen L, Gao M, et al. Long-term liraglutide ameliorates nigrostriatal impairment via regulating AMPK/PGC-1a signaling in diabetic mice. Brain Res. 2019;1714:126–32.
Safety and Efficacy of Liraglutide in Parkinson’s Disease - ClinicalTrials.gov. https://clinicaltrials.gov/ct2/show/NCT02953665.
Zhang L, Zhang L, Li L, Hölscher C. Neuroprotective effects of the novel GLP-1 long acting analogue semaglutide in the MPTP Parkinson’s disease mouse model. Neuropeptides. 2018;71:70–80.
Zhang L, Zhang L, Li L, Hölscher C. Semaglutide is neuroprotective and reduces α-synuclein levels in the chronic MPTP mouse model of Parkinson’s disease. J Parkinsons Dis. 2019;9:157–71.
GLP1R in Parkinson’s Disease - ClinicalTrials.gov. https://clinicaltrials.gov/ct2/show/NCT03659682.
Simon GE, Von Korff M, Saunders K, Miglioretti DL, Crane PK, Van Belle G, et al. Association between obesity and psychiatric disorders in the US adult population. Arch Gen Psychiatry. 2006;63(7):824–30. https://doi.org/10.1001/archpsyc.63.7.824.
Luppino FS, De Wit LM, Bouvy PF, Stijnen T, Cuijpers P, Penninx BWJH, et al. Overweight, obesity, and depression: A systematic review and meta-analysis of longitudinal studies. Arch Gen Psychiatry. 2010;67(3):220–9. https://doi.org/10.1001/archgenpsychiatry.2010.2.
Pi-Sunyer X, Apovian CM, McElroy SL, Dunayevich E, Acevedo LM, Greenway FL. Psychiatric adverse events and effects on mood with prolonged-release naltrexone/bupropion combination therapy: a pooled analysis. Int J Obes (Lond). 2019;43:2085–94. https://doi.org/10.1038/s41366-018-0302-z.
McElroy SL, Guerdjikova AI, Kim DD, Burns C, Harris-Collazo R, Landbloom R, et al. Naltrexone/bupropion combination therapy in overweight or obese patients with major depressive disorder: Results of a pilot study. Prim Care Companion CNS Disord. 2013;15(3):PCC.12m01494. https://doi.org/10.4088/PCC.12m01494.
McIntyre RS, Loft H, Christensen MC. Efficacy of vortioxetine on anhedonia: Results from a pooled analysis of short-term studies in patients with major depressive disorder. Neuropsychiatr Dis Treat. 2021;17:575–85.
Detka J, Głombik K. Insights into a possible role of glucagon-like peptide-1 receptor agonists in the treatment of depression. Pharmacol Rep. 2021;73(4):1020–32. https://doi.org/10.1007/s43440-021-00274-8.
O’Neil PM, Aroda VR, Astrup A, Kushner R, Lau DCW, Wadden TA, et al. Neuropsychiatric safety with liraglutide 3.0 mg for weight management: Results from randomized controlled phase 2 and 3a trials. Diabetes Obes Metab. 2017;19:1529–36.
Weina H, Yuhu N, Christian H, Birong L, Feiyu S, Le W. Liraglutide attenuates the depressive- and anxiety-like behaviour in the corticosterone induced depression model via improving hippocampal neural plasticity. Brain Res Brain Res. 2018;1694:55–62.
Seo MK, Jeong S, Seog DH, Lee JA, Lee JH, Lee Y, et al. Effects of liraglutide on depressive behavior in a mouse depression model and cognition in the probe trial of Morris water maze test. J Affect Disord. 2023;324:8–15.
Mansur RB, Zugman A, Ahmed J, Cha DS, Subramaniapillai M, Lee Y, et al. Treatment with a GLP−1R agonist over four weeks promotes weight loss-moderated changes in frontal-striatal brain structures in individuals with mood disorders. Eur Neuropsychopharmacol. 2017;27:1153–62.
Nedunchezhiyan U, Varughese I, Sun ARJ, Wu X, Crawford R, Prasadam I. Obesity, Inflammation, and Immune System in Osteoarthritis. Front Immunol. 2022 Jul 4;13:907750. https://doi.org/10.3389/fimmu.2022.907750.
Nauck M. Incretin therapies: Highlighting common features and differences in the modes of action of glucagon-like peptide-1 receptor agonists and dipeptidyl peptidase-4 inhibitors. Diabetes Obes Metab. 2016;18(3):203–16. https://doi.org/10.1111/dom.12591.
Gudbergsen H, Overgaard A, Henriksen M, Wæhrens EE, Bliddal H, Christensen R, et al. Liraglutide after diet-induced weight loss for pain and weight control in knee osteoarthritis: A randomized controlled trial. Am J Clin Nutr. 2021;113:314–23.
Iepsen EW, Lundgren JR, Hartmann B, Pedersen O, Hansen T, Jørgensen NR, et al. GLP-1 receptor agonist treatment increases bone formation and prevents bone loss in weight-reduced obese women. J Clin Endocrinol Metab. 2015;100:2909–17.
Mei J, Sun J, Wu J, Zheng X. Liraglutide suppresses TNF-α-induced degradation of extracellular matrix in human chondrocytes: A therapeutic implication in osteoarthritis. Am J Transl Res. 2019;11(8):4800–8.
Meurot C, Martin C, Sudre L, Breton J, Bougault C, Rattenbach R, et al. Liraglutide, a glucagon-like peptide 1 receptor agonist, exerts analgesic, anti-inflammatory and anti-degradative actions in osteoarthritis. Sci Rep. 2022;12(1):1567. https://doi.org/10.1038/s41598-022-05323-7.
Research Study Looking at How Well Semaglutide Works in People Suffering From Obesity and Knee Osteoarthritis - ClinicalTrials.gov https://clinicaltrials.gov/ct2/show/NCT05064735.
Funding
Open access funding provided by Università degli Studi di Napoli Federico II within the CRUI-CARE Agreement. The authors did not receive support from any organization for the submitted work.
Author information
Authors and Affiliations
Contributions
GM had the idea for the article, GM, MP, MEG, SB, VG performed the literature search, GM, MEG, SB, VG drafted and critically revised the work, LB, AC, WY, PS critically revised the work.
Corresponding author
Ethics declarations
Competing Interests
No competiting interests to declare.
Employment
No employment to declare.
Financial Interests
No financial Interests to declare.
Non-financial Interests
No non-financial Interests to declare.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Guglielmi, V., Bettini, S., Sbraccia, P. et al. Beyond Weight Loss: Added Benefits Could Guide the Choice of Anti-Obesity Medications. Curr Obes Rep 12, 127–146 (2023). https://doi.org/10.1007/s13679-023-00502-7
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13679-023-00502-7