
Abstract
Two novel compounds including a cyclohelminthol type polyketide (namely oxaleimide K, 1) and a maleimide derivative (namely peniroquefortine A, 2), and a new natural product (namely 2-(acetylamino)-N-[(1E)-2-phenylethenyl]-acetamide, 3), together with four known compounds (4–7), were isolated and identified from fungus Penicillium roqueforti, which was separated from the root soil of Hypericum beanii N. Robson collected from the Shennongjia Forestry District, Hubei Province. Their structures including absolute configurations were mainly established by the NMR spectroscopy analyses and single-crystal X-ray diffraction experiment. Compound 1 represents the second example of a cyclohelminthol type polyketide, which features a rare 6/6/5/5 tetracyclic system and a branched aliphatic chain containing a terminal olefin (oct-1-en-3-yl) moiety, and compound 2 possesses an unprecedented carbon skeleton that is uniquely defined by a maleimide moiety linked to the respective 4-methylene-2-(3-methylbut-2-en-1-yl)-phenol and para-substituted aromatic moieties via the carbon-carbon bonds. Remarkably, the absolute configuration of a cyclohelminthol type polyketide as exemplified by compound 1 is determined by the single-crystal diffraction analysis for the first time, highlighting an E-configuration for the linkage of a succinimide moiety and a tetrahydrofuran moiety for 1 rather than a Z-configuration as previously reported in the biosynthesis study, which gives a new insight into the structural elucidation of this category of polyketides. Additionally, compound 1 exhibited significant cytotoxic activity against multiple tumor cells, especially against the Farage and SU-DHL-2 cells (IC50 < 20 µM, 48 h). Further mechanism study revealed that compound 1 significantly induced cell cycle arrest in Farage and SU-DHL-2 cells by causing abnormal ROS level and triggering oxidative stress.
Graphical Abstract

Similar content being viewed by others
Avoid common mistakes on your manuscript.
1 Introduction
Cancer is a life-threatening disease which has highly invasive and heterogeneous with different histological subtypes and mutation characteristics [1,2,3]. In 2020, research institutions revealed that there were 19.3 million cancer cases and nearly 10 million deaths worldwide, with time going by, the number will rapidly increase influenced by some factors such as the fast pace of life, high-fat diet, and so on [4]. As is known to all, microorganisms have played an indispensable role in drug development, and more than two-thirds of clinically used antitumor drugs are derived from natural products, such as taxol, romidepsin, salinomycin, and rapamycin et al. [5,6,7,8]. In recent decades, the increase in drug resistance, which caused the use of some antitumors greatly limited [9, 10]. Therefore, it is an emergency challenge to develop new antitumor agents with high efficacy and minimal side effects from microorganisms.
According to the extensive literature investigations, fungus Penicilliu roqueforti has been considered to be an irreplaceable resource of structurally diverse and bioactive natural products, including meroterpenoids, sesquiterpenoids, sesterterpenoids, alkaloids, etc., with attracting biological activities, such as antimicrobial, neurotoxic, and cytotoxic activities [11,12,13]. Therefore, strain P. roqueforti isolated from the root soil of Hypericum beanii N. Robson collected from the Shennongjia Forestry District of Hubei Province caught our attention and was systematically investigated, which led to the discovery of two novel secondary metabolites including a cyclohelminthol type polyketide (1) and a maleimide derivative (2), and a new natural product (3), as well as four known compounds (4–7). Compounds 1–7 were evaluated for the cytotoxic activities against six human tumor cell lines, including SU-DHL-2, RKO, A549, Jurkat, Farage, and HEPG2 cells, and the results revealed that compound 1 exhibited significant cytotoxic activity, especially against the Farage and SU-DHL-2 cells (IC50 < 20 µM, 48 h). Herein, the isolation, structural elucidation, and bioactivities of these compounds are disclosed (Fig. 1).
2 Results and discussion
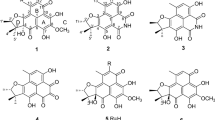
Compound 1 was purified as a colorless block crystal. It was established by the HRESIMS ion peak at m/z 538.2791 ([M + Na]+, calcd for 538.2775), which suggested a molecular formula of C29H41NO7 with 10 degrees of unsaturation. Conventional 13C NMR spectroscopy analyses (Table 1) of 1 revealed the presence of twenty-nine carbon resonances including three methyls (δC 21.1, 18.1, and 14.4), one methoxy (δC 52.1), seven methylenes (δC 116.7, 40.9, 35.3, 33.0, 31.5, 28.4, and 23.7), twelve methines (δC 140.9, 135.0, 127.3, 87.1, 75.8, 53.3, 49.8, 46.4, 45.6, 44.6, 43.3, and 38.5), and six non-protonated carbons (δC 179.7, 177.4, 173.8, 172.4, 99.7, and 79.8). Through extensive literature research and analysis of aforementioned data, compound 1 was deduced to be a structural analogue of cyclohelminthol CP-1 [14]. The three double bonds, one ester carbonyl, and two amide carbonyls accounted for six out of 10 degrees of unsaturation, implying that 1 possessed a tetracyclic system (Additional files 1, 2).
The 1D NMR data (Table 1) of 1 were closely similar to those of the known compound cyclohelminthol CP-1 [14], suggesting that both compounds shared the same core skeleton, and the differences were shown as follows: (1) a –COOH group at C-9 in cyclohelminthol CP-1 was replaced by a –COOCH3 group at the same position in 1; (2) a C-17/C-22 hexyl side chain linked at C-16 in cyclohelminthol CP-1 was replaced by a branched aliphatic chain containing a terminal olefin (oct-1-en-3-yl) moiety linked at the same position in 1, which were fully supported by the key 1H–1H COSY correlations of H3-24/H2-23/H2-22/H2-21/H2-20/H-19 (H-16)/H-18/H2-17 and HMBC correlations from H-19 to C-15/C-29 and from H-9/H3-26 to C-25 (Fig. 2). The relative configuration of 1 was partially verified by analyzing the ROESY data. The ROESY correlations (Fig. 3) from H-11 to H-7/H-9 determined that these protons were on the same side with β-orientations, while the ROESY cross-peak (Fig. 3) of H-12 to H-6 suggested that H-12 and H-6 were on the other side. However, the configurations of C-4, C-5, C-6, C-13, and C-19 could not be determined by analyzing the NMR data alone. Luckily, a crystal of 1 suitable for X-ray diffraction crystallographic analysis was furnished, which unequivocally confirmed the absolute configuration of 1 as 4R,5R,6S,7R,9R,11S,12S,13R,16R,19S with Cu Kα (Fig. 4) [Flack parameter = 0.02(5), CCDC 2,238,093]. Accordingly, the absolute structure of 1 was defined.

Chemical structures of compounds 1–7

Key 1H–1H COSY (blue lines) and HMBC (red arrows) correlations of compounds 1–3

Key ROESY correlations (black arrows) of compound 1

X-ray crystallographic structure of 1
Compound 2, obtained as a yellow oil, was established a molecular formula of C24H25NO5 on the basis of the positive HRESIMS ion peak at m/z 430.1631 [M + Na]+ (calcd for C24H25NO5Na+, 430.1625) in the HRESIMS spectrum, suggesting 13 degrees of unsaturation. The IR spectrum of 1 showed obvious absorptions for a hydroxy group (3442 cm−1) and an amide carbonyl group (1698 cm−1). The 1H NMR data (Table 1) indicated characteristic signals for an alkenyl proton at δH 5.21 (H-22), two methyl groups at δH 1.62 (H-24) and 1.68 (H-25), two sets of para-substituted aromatic protons at δH 7.43 (H-10/H-14) and 6.79 (H-11/H-13), and 1,2,4-trisubstituted aromatic protons at δH 6.81 (H-16), 6.61 (H-19), and 6.79 (H-20). The 13 C NMR data (Table 1) displayed the presence of 24 carbon resonances including two methyls (δC 26.0 and 17.8), four methylenes (δC 60.3, 41.4, 29.7, and 29.0), eight methines (δC 132.4, 132.4, 130.5, 127.6, 123.7, 116.6, 116.6, and 115.9), and ten non-protonated carbons (δC 173.8, 173.1, 161.0, 154.8, 138.9, 137.3, 133.2, 129.6, 129.4, and 121.2). Considering the degree of unsaturation, we could draw a conclusion that compound 2 was a maleimide derivative with a tricyclic system. By comparing the above spectral data and extensive literature research, the structure of 2 was somewhat similar to the reported compound 1-hydroxy-3-(4-hydroxyphenyl)-4-isobutyl-1 H-pyrrole-2,5-dione [15]. The differences were shown as follows: (1) the appearance of a 4-methylene-2-(3-methylbut-2-en-1-yl)-phenol moiety connected at C-4 in compound 2, which could be proved by the HMBC correlations (Fig. 2) from H2-6 to C-3/C-4/C-5 and from H-16/H-20 to C-6; (2) the existence of a -CH2CH2OH moiety linked at N-1 in compound 2, which was supported by the HMBC correlations (Fig. 2) from H2-7 to C-2/C-5. Accordingly, the structure of 2 was defined.
Compound 3 possessed a molecular formula of C12H14N2O2 as determined by the HRESIMS and NMR data, indicating seven degrees of unsaturation. The 1H NMR data (Table 1) exhibited signals for a mono-substituted phenyl ring at δH 7.32 (H-1/H-5), 7.27 (H-2/H-4), and 7.16 (H-3), one methyl at δH 2.03 (H-12), one methylene at δH 3.93 (H-10), and two alkenyl protons at δH 7.45 (H-8) and δH 6.26 (H-7). The 13C NMR data (Table 1) displayed twelve carbon resonances that were assigned to six phenyl C-atoms (δC 137.8, 129.7, 129.7, 127.7, 126.5, and 126.5), two olefinic C-atoms (δC 129.7 and 127.7), two amide C-atoms (δC 173.9 and 169.3), one methylene C-atom (δC 43.5), and one methyl C-atom (δC 22.4). Detailed 2D NMR data analyses (Fig. 2) designated the planar structure of 3, which was the same as a known synthetic product 2-(acetylamino)-N-[(1E)-2-phenylethenyl]-acetamide [16]. Accordingly, the structure of 3 was assigned to be a new natural product.
The four known compounds, compared with the literature, were identified as hydroxypropan-2',3'-diol orsellinate (4) [17], vomifoliol (5) [18], citreoisocoumarin (6) [19], and (–)-brevicolide A (7) [20].
We evaluated the cytotoxic activities of compounds 1–7 against six human tumor cell lines, including SU-DHL-2 (Diffuse large B-cell lymphoma), RKO (Colonic adenocareinoma), A549 (Lung cancer), Jurkat (T cell acute leukemia), Farage (Diffuse large B-cell lymphoma), and HEPG2 (Hepatocellular carcinoma). As a result, only compound 1 exhibited significant cytotoxic activity against multiple tumor cells, especially against the Farage and SU-DHL-2 (IC50 < 20 µM, 48 h) cells (Fig. 5A–B). Subsequently, we examined whether compound 1 could cause cell cycle arrest in Farage and SU-DHL-2 cells at a series of concentrations of 10, 20, and 30 µM, and the positive drug VP16 (Etoposide, 10 µM) concentration gradient for 24 h. The above results showed that compound 1 could significantly block G0/G1 phase (the stationary and pre-synthetic phase of the cell cycle) in both Farage and SU-DHL-2 cells, and the cell cycle arrest was dose-dependent (Fig. 5C–D). It is well known that tumor cells have a unique metabolic pattern and the highly activated oxidative metabolism in tumor cells makes them sensitive to ROS-induced oxidative stress, and abnormal ROS levels can lead to tumor cell cycle arrest or apoptosis [21, 22]. Therefore, we suspected that compound 1 exerted its antitumor activity by regulating ROS levels in Farage and SU-DHL-2 cells. Meanwhile, for the sake of discovering whether compound 1 had the ability to regulate ROS levels, we detected the level of oxidative stress in compound 1-treated tumor cells. As shown in Fig. 6A–B, in the more sensitive Farage cells, compound 1 significantly attenuated cellular ROS levels, which might represent an inhibition of cellular respiration or a state of frequent cell death. In contrast, in the SU-DHL-2 cells, low-dose treatment of compound 1 induced a significant increase of ROS levels, while high-dose treatment of compound 1 significantly attenuated cellular ROS levels. Consequently, we speculated that the disruption of redox homeostasis in SU-DHL-2 cells by compound 1 involved a dynamic process distinct from that in Farage cells, which ROS first rose and then fell, with elevated ROS inducing cellular oxidative stress and cell damage, followed by a decrease in ROS levels below normal levels and impaired proliferation. Finally, we examined the expression levels of some genes mRNA related to oxidative stress. The heat map showed that the expression levels of ATF4, SESN2, Gdf15, Trib3, and TXNIP were highly up-regulated after compound 1-treatment, and the cells were in a significant oxidative stress state after compound 1-treatment (Fig. 6C). Overall, we found that compound 1 significantly induced cell cycle arrest in Farage and SU-DHL-2 cells by causing abnormal ROS levels and triggering oxidative stress, which can be further investigated as a clinical candidate lead compound for treating tumor-related diseases.

Compound 1 blocked the cell cycle to exert antitumor effects. A IC50 calculated by cell viability assays against six human tumor cells exposed to the increasing doses of compound 1 over the course of 48 h. Mean ± s.d., n = 3. B Cell viability assays against the Farage and SU-DHL-2 cells exposed to the increasing doses of compound 1 over the course of 48 h. Mean ± s.d., n = 3. C–D Cell cycle assays against the Farage and SU-DHL-2 cell lines exposed to the increasing doses of compound 1 (10 ~ 30 µM) or VP16 over the course of 24 h. Mean ± s.d., n = 3 (*p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001, compared to the control group DMSO)

Compound 1 decreased the intracellular ROS level and triggered oxidative stress in tumor cells. A–B Intracellular ROS assays in Farage and SU-DHL-2 cells exposed to the increasing doses of compound 1 over the course of 24 h. Mean ± s.d., n = 3. C Oxidative stress-related gene mRNA analysis in Farage and SU-DHL-2 cells exposed to compound 1 (20 or 30 µM, 24 h, n = 3)
The chemical investigation on fungus P. roqueforti afforded two novel compounds including a cyclohelminthol type polyketide (1) and a maleimide derivative (2), and a new natural product (3), as well as four known compounds (4–7). Compound 1 represents the second example of a cyclohelminthol type polyketide, which features a rare 6/6/5/5 tetracyclic system and a branched aliphatic chain containing a terminal olefin (oct-1-en-3-yl) moiety, and compound 2 possesses an unprecedented carbon skeleton that is uniquely defined by a maleimide moiety linked with the respective 4-methylene-2-(3-methylbut-2-en-1-yl)-phenol and para-substituted aromatic moieties via the carbon-carbon bonds. Compound 1 showed significant cytotoxic activity against the Farage and SU-DHL-2 cells, which induced apoptosis in a dose-dependent manner and cell growth arrest at the G0/G1 phase. The mechanism study revealed that compound 1 could regulate the ROS level in tumor cells, thereby inducing oxidative stress and cell damage and destroying the redox homeostasis. This work provides new insights into the chemical diversity of fungus P. roqueforti, and highlights compound 1 as a potential clinical antitumor candidate.
References
Chan SH, Chui CH, Chan SW, Kok SHL, Chan D, Tsoi MYT, Leung PHM, Lam AKY, Chan ASC, Lam KH, Tang JCO. Synthesis of 8-hydroxyquinoline derivatives as novel antitumor agents. ACS Med Chem Lett. 2013;4:170–4.
Guo C, Dong E, Lai Q, Zhou S, Zhang G, Wu M, Yue X, Tao Y, Peng Y, Ali JM, Lu Y, Fu Y, Lai W, Zhang Z, Ma F, Yao Y, Gou L, Yang H, Yang J. Effective antitumor activity of 5T4-specific CAR-T cells against ovarian cancer cells in vitro and xenotransplanted tumors in vivo. MedComm. 2020;1:338–50.
Shi R, Tang Y, Miao H. Metabolism in tumor microenvironment: implications for cancer immunotherapy. MedComm. 2020;1:47–68.
Pich O, Bailey C, Watkins TBK, Zaccaria S, Jamal-Hanjani M, Swanton C. The translational challenges of precision oncology. Cancer Cell. 2022;40:458–78.
Chen S, Zhen Y. Research progress in new anti-tumor drugs on different targets derived from microorganisms. Acta Pharm Sin. 2018;53:833–8.
Duan Y, Meng L. Research progress in multi-targeted anti-tumor natural products. Acta Pharm Sin. 2021;56:403–13.
Newman DJ, Cragg GM. Natural products as sources of new drugs over the nearly four decades from 01/1981 to 09/2019. J Nat Prod. 2020;83:770–803.
Xu J, Meng L, Qing C. The clinical application and development of traditional antitumor drug. Acta Pharm Sin. 2021;56:1551–61.
Wu Q, Qian W, Sun X, Jiang S. Small-molecule inhibitors, immune checkpoint inhibitors, and more: FDA-approved novel therapeutic drugs for solid tumors from 1991 to 2021. J Hematol Oncol. 2022;15:143–3.
Zhao S, Wang D, Zhao H, Gong J, Zhang J, Fang W, Ma F, Xu B, Li J, Hu X, Ba Y, Chen X, Yang Z, Shen L, Jiang J, Zhang L. Time to raise the bar: transition rate of phase 1 programs on anticancer drugs. Cancer Cell. 2022;40:233–5.
Moreau S, Cacan M, Lablache-Combier A, Eremofortin C. A new metabolite obtained from Penicillium roqueforti cultures and from biotransformation of PR toxin. J Org Chem. 1977;42:2632–4.
Omura S, Inokoshi J, Uchida R, Shiomi K, Masuma R, Kawakubo T, Tanaka H, Iwai Y, Kosemura S, Yamamura S. Andrastins A–C, new protein farnesyltransferase inhibitors produced by Penicillium sp. FO-3929. I. producing strain, fermentation, isolation, and biological activities. J Antibiot. 1996;49:414–7.
Wang J, Yu J, Shu Y, Shi Y, Luo P, Cai L, Ding Z. Peniroquesines A–C: sesterterpenoids possessing a 5-6-5-6-5-fused pentacyclic ring system from Penicillium roqueforti YJ-14. Org Lett. 2018;20:5853–6.
Inose K, Tanaka S, Tanaka K, Hashimoto M. Cyclohelminthol CPs: scope and limitations of density functional theory-based structural elucidation of natural products. J Org Chem. 2021;86:1501–15.
Stewart SG, Ho LA, Polomska ME, Percival AT, Yeoh GCT. Rapid evaluation of Antrodia camphorata natural products and derivatives in tumourigenic liver progenitor cells with a novel cell proliferation assay. ChemMedChem. 2009;4:1657–67.
Gooßen LJ, Blanchot M, Salih KSM, Gooßen K. Ruthenium-catalyzed addition of primary amides to alkynes: a stereoselective synthesis of secondary enamides. Synthesis. 2009;13:2283–8.
Talontsi FM, Facey P, Tatong MDK, Islam MT, Frauendorf H, Draeger S, Tiedemann VA, Laatsch H. Zoosporicidal metabolites from an endophytic fungus Cryptosporiopsis sp. of Zanthoxylum leprieurii. Phytochemistry. 2012;83:87–94.
Yoshida Y, Haraguchi D, Ukuda-Hosokawa R, Andou T, Matsuyama T, Kohama T, Eguchi T, Ohno S, Ono H, Nishida R. Synthesis and activity of 3-oxo-α-ionone analogs as male attractants for the solanaceous fruit fly, Bactrocera latifrons (Diptera: Tephritidae). Biosci Biotechnol Biochem. 2021;85:2360–7.
Zhao Y, Liu D, Proksch P, Yua S, Lin W. Isocoumarin derivatives from the sponge-associated fungus Peyronellaea glomerata with antioxidant activities. Chem Biodivers. 2016;13:1186–93.
Chen L, Zhu T, Zhu G, Liu Y, Wang C, Pawinee P, Arthit C, Zhu W. Bioactive natural products from the marine-derived Penicillium brevicompactum OUCMDZ-4920. Chin J Org Chem. 2017;37:2752–62.
Vurusaner B, Poli G, Basaga H. Tumor suppressor genes and ROS: complex networks of interactions. Free Radical Bio Med. 2012;52:7–18.
Zhu C, Shi H, Wu M, Wei X, Lin W. A dual MET/AXL small-molecule inhibitor exerts efficacy against gastric carcinoma through killing cancer cells as well as modulating tumor microenvironment. MedComm. 2020;1:103–18.
Acknowledgements
We thank the Analytical and Testing Center at Huazhong University of Science and Technology for measuring ECD, IR, and single-crystal X-ray diffraction data. This project was financially supported by the National Program for Support of Top-notch Young Professionals (No. 0106514050), the National Natural Science Foundation of China (Nos. 82273811 and 31870326), the National Key R&D Program of China (No. 2021YFA0910500), the National Natural Science Foundation for Distinguished Young Scholars (No. 81725021), the Innovative Research Groups of the National Natural Science Foundation of China (No. 81721005), the Research and Development Program of Hubei Province (No. 2020BCA058), and the Chinese Medicine Research Foundation of Health Commission of Hubei Province (No. ZY2021Z019).
Author information
Authors and Affiliations
Contributions
All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1:
Figure S1. 1H NMR spectrum of compound 1 (Recorded in methanol-d4). Figure S2. 13C NMR and DEPT spectra of compound 1 (Recorded in methanol-d4). Figure S3. HSQC spectrum of compound 1 (Recorded in methanol-d4). Figure S4. HMBC spectrum of compound 1 (Recorded in methanol-d4). Figure S5. 1H–1H COSY spectrum of compound 1 (Recorded in methanol-d4). Figure S6. ROESY spectrum of compound 1 (Recorded in methanol-d4). Figure S7. HRESIMS spectrum of compound 1. Figure S8. UV spectrum of compound 1. Figure S9. IR spectrum of compound 1. Figure S10. ECD spectrum of compound 1. Figure S11. 1H NMR spectrum of compound 2 (Recorded in methanol-d4). Figure S12. 13C NMR and DEPT spectra of compound 2 (Recorded in methanol-d4). Figure S13. HSQC spectrum of compound 2 (Recorded in methanol-d4). Figure S14. HMBC spectrum of compound 2 (Recorded in methanol-d4). Figure S15. 1H–1HCOSY spectrum of compound 2 (Recorded in methanol-d4). Figure S16. NOESY spectrum of compound 2 (Recorded in methanol-d4). Figure S17. HRESIMS spectrum of compound 2. Figure S18. UV spectrum of compound 2. Figure S19. IR spectrum of compound 2. Figure S20. ECD spectrum of compound 2. Figure S21. 1H NMR spectrum of compound 3 (Recorded in methanol-d4). Figure S22. 13C NMR and DEPT spectra of compound 3 (Recorded in methanol-d4). Figure S23. HSQC spectrum of compound 3 (Recorded in methanol-d4). Figure S24. HMBC spectrum of compound 3 (Recorded in methanol-d4). Figure S25. 1H–1H COSY spectrum of compound 3 (Recorded in methanol-d4). Figure S26. ROESY spectrum of compound 3 (Recorded in methanol-d4). Figure S27. HRESIMS spectrum of compound 3. Figure S28. UV spectrum of compound 3. Figure S29. IR spectrum of compound 3. Figure S30. ECD spectrum of compound 3.
Additional file 2
: Additional data associated with this article including 1D and 2D NMR, HRESIMS, UV, IR of 1–3 and X-ray data of compound 1.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Mo, S., Zhao, Z., Ye, Z. et al. New secondary metabolites with cytotoxicity from fungus Penicillium roqueforti. Nat. Prod. Bioprospect. 13, 17 (2023). https://doi.org/10.1007/s13659-023-00381-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s13659-023-00381-4