
Abstract
Mulberry Diels–Alder-type adducts (MDAAs) are unique phenolic natural products biosynthetically derived from the intermolecular [4 + 2]-cycloaddition of dienophiles (mainly chalcones) and dehydroprenylphenol dienes, which are exclusively distributed in moraceous plants. A total of 166 MDAAs with diverse skeletons have been isolated and identified since 1980. Structurally, the classic MDAAs characterized by the chalcone-skeleton dienophiles can be divided into eight groups (Types A − H), while others with non-chalcone dienophiles or some variations of classic MDAAs are non-classic MDAAs (Type I). These compounds have attracted significant attention of natural products and synthetic chemists due to their complex architectures, remarkable biological activities, and synthetic challenges. The present review provides a comprehensive summary of the structural properties, bioactivities, and syntheses of MDAAs. Cited references were collected between 1980 and 2021 from the SciFinder, Web of Science, and China National Knowledge Internet (CNKI).
Graphical Abstract

Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction to mulberry Diels–Alder-type adducts (MDAAs)
Mulberry Diels–Alder-type adducts (MDAAs), characteristic constituents of mulberry trees (Morus plants of the family Moraceae), are a group of structurally unique natural phenolic compounds biosynthetically derived from the intermolecular [4 + 2]-cycloaddition of dienophiles (mainly chalcones) and dehydroprenylphenol dienes (Scheme 1). Chalcomoracin.and kuwanons G and H (albanins F and G), the first representatives of MDAAs, were almost simultaneously reported from the well-known mulberry tree (Morus alba L.) by the two groups of Nomura and Takasugi in 1980 [1,2,3,4,5]. A total of 166 MDAAs have been obtained and characterized over the past four decades. MDAAs are not widely distributed in the plants of the family Moraceae, and until now, they have only found in seven genera (21 species) of this family, including Morus [13 species, M. alba (including the variant M. alba var. shalun), M. australis, M. bombycis, M. cathayana, M. insignis, M. lhou, M. macroura, M. mesozygia, M. mongolica (including M. yunanensis, a species revised as the present Latin name), M. multicaulis, M. nigra, M. notabilis, and M. wittiorum], Artocarpus (two species, A. heterophyllus and A. integer), Sorocea (two species, S. bonplandii and S. ilicifolia), Brosimopsis (one species, B. oblongifolia), Brosimum (one species, Brosimum rubescens), Chlorophora (one species, C. regia), and Dorstenia (one species, D. barteri). Among these 21 species, M. alba, M. mongolica, and M. macroura were the top three species rich in different MDAAs. Natural MDAAs were demonstrated to occur generally in root barks, stem barks, roots, stems or twigs, leaves, and callus cultures.

The biosynthesis pathway of classic MDAAs
A mini-review (in Chinese) on MDAAs and several comprehensive reviews on secondary metabolites from moraceous plants especially the Morus genus covered a limited number of MDAAs, but none has provided a complete and in-depth analysis of this group of natural products [6,7,8,9,10]. In this review, it provides a comprehensive summary of the structural classification, distribution, and biological functions of 166 naturally occurring MDAAs. The total synthetic investigations towards this family of compounds by various chemistry research groups are summarized for the first time.
2 Structural characteristics and classification of MDAAs
According to the structural characteristics, MDAAs can be divided into classic and non-classic types. Structurally, classic MDAAs share the same chalcone-skeleton dienophiles but differ in the dehydroprenylphenol dienes. In light of the structural types of dehydroprenylphenol dienes, classic MDAAs (Fig. 1) can be further classified into dehydroprenyl-2-arylbenzofuran type (Type A), dehydroprenylstilbene type (Type B), dehydroprenylchalcone type (Type C), dehydroprenylflavone type (Type D), dehydroprenyldihydroflavone type (Type E), dehydroprenylsanggenonflavone type (Type F), dehydroprenylcoumarin type (Type G), and simple or other dehydroprenylphenol type (Type H). Non-classic MDAAs (Type I) is considered as a kind of Diels–Alder adducts derived from cycloaddition of non-chalcone dienophiles and dehydroprenylphenol dienes or as variations of some classic MDAAs. All MDAAs are phenolic natural products, and the presence of adjacent phenolic hydroxyl groups has allowed different natural modifications of the ketone as well as of newly formed methylcyclohexene ring, resulting in compounds with complex structural features. In addition, the presence of intact or modified prenyl groups in the moiety of dienophiles or dienes also leads to a diversity of MDAAs.

Structures of eight important groups of classic MDAAs (Types A − H)
The occurrence and distribution details of 166 MDAAs together with their specific rotation values are summarized in Table 1.
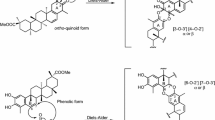
As shown in the biosynthetic pathway of classic MDAAs (Scheme 1), a new methylcyclohexene ring with three chiral carbons (C-3", C-4", and C-5") was produced in the Diels–Alder cycloaddition of the chalcone dienophile and the dehydroprenylphenol diene, in which the relative configuration of H-4" and H-5" would always maintain the original trans configuration while the stereochemistry of H-3" and H-4" would have cis and trans configurations. Therefore, this kind of Diels–Alder product has trans–trans and cis–trans types, both of which are found in natural products. From the structural characteristics of endo- and exo-adducts in the Diels–Alder reaction, the trans–trans type adducts have been found to be exo-addition products, while the cis–trans type adducts are attributed to endo-addition. According to the current data statistics, the ratio of the reported natural MDAAs with trans–trans and cis–trans types is about 1:1.5. It is noteworthy that natural MDAAs seem to be formed in the manner of in vitro [4 + 2] cycloaddition reactions.
The trans–trans and cis–trans types of classic MDAAs could be easily determined by analysis of the coupling constant of H-4" with H-3" and H-5" (generally, large J3"/4" and J4"/5" for trans–trans; small J3"/4" and large J4"/5" for cis–trans:) in the 1H NMR spectrum with resolvable signals. Sometimes this kind of compound usually exists as an equilibrium mixture of conformational isomers in solution, which requires NMR variable temperature experiments to obtain the resolvable signals of the methylcyclohexene ring [17, 22, 74, 85, 94, 110, 118, 124]. Based on X-ray crystallographic analysis and circular dichroism (CD) spectroscopic evidence, in 1988 the Nomura group proposed the empirical rules for determining the absolute configuration of the chiral centers on the methylcyclohexene ring in classic MDAAs [12]: a) the stereochemistry of C-3", C-4", and C-5" in the trans–trans type MDAAs was 3"R,4"R,5"S, while in the cis–trans type MDAAs was 3"S,4"R,5"S. b) The trans–trans type MDAAs exhibited negative optical rotations, and their Cotton effect at the maximum UV absorption tended to be negative, while the cis–trans type MDAAs showed the opposite optical rotations and Cotton effect. More reports of natural and synthetic MDAAs with clear absolute structures established by single crystals analysis, chemical methods, or ECD calculation methods have confirmed the practicability of Nomura’s empirical rules. In this review, all MDAAs with clear relative configurations have determined their absolute configurations according to the empirical rules.
2.1 Dehydroprenyl-2-arylbenzofuran type MDAAs (Type A)
Until now, 57 type A of MDAAs (1 − 57) have been discovered, which compose the biggest family in MDAAs. This type of MDAAs was found mostly in the plants of Morus; its diene moiety was the dehydroprenyl group of 2-arylbenzofuran, which is predominantly located on its B ring (C-4′), as illustrated by the structures of compounds 1–49 (Figs. 2 and 3). In addition, there were only eight type A of MDAAs (50–57) with the diene moiety on A ring (C-5) of 2-arylbenzofuran (Fig. 4).

Structures of dehydroprenyl-2-arylbenzofuran type MDAAs (Type A)

Structures of dehydroprenyl-2-arylbenzofuran type MDAAs (Type A, continued)

Structures of dehydroprenyl-2-arylbenzofuran type MDAAs (Type A, continued)
2.1.1 Type A with the diene moiety on ring B (Type A-I)
Mulberrofuran J (1) [11], mongolicin F (2) [25], albasin A (3) [14], and morushalunin (4) [26] were the only four type A-I MDAAs with trans–trans configuration found so far. Compound 1 was first isolated from the root barks of the cultivated mulberry tree M. lhou in 1985 by Hirakura et al. [11], and its absolute configuration was determined by the Nomura group in 1988 [12]. Albasin A (3) possessing a prenyl moiety at C-7″ was discovered from the root bark of M. alba [14]. MDAAs with a prenyl group or a modified prenyl group at C-7″ of the methylcyclohexene ring were rare, and only six examples have been reported. The others were macrourin E (60) [19] in type B, multicaulisin (99) [114] in type D, sanggenon G (124) [121], sanggenon T (125) [123], and sanggenol M (126) [122] in type E. Morushalunin (4) [26] isolated in M. alba was one of the only three methylated natural MDAAs, and the others were morbilisin D (36) [39] of the same type A from M. notabilis and kuwanon J 2,4,10"-trimethyl ether (82) [100] of type C from M. alba.
Compounds 5–23 represent a remarkable series of cis–trans type A-I MDAAs initially detected in plants of Morus [4, 14, 15, 22, 23, 39, 42, 47, 48, 51,52,53,54]. Chalcomoracin (5), which is the first example of MDAAs, was isolated from the diseased mulberry leaves (M. alba) by Takasugi et al. in 1980 [4]. Mulberrofuran C (6) and chalcomoracin (5) were the C-3" epimers of mulberrofuran J (1) and mongolicin F (2), respectively, all of which were detected in M. macroura [18, 19]. Both mulberrofuran U (11) [52] and mulberrofuran T (12) [53] have a prenyl unit at C-7, which were first obtained from M. insignis and M. alba, respectively. Morbilisins G and H (14 and 15), possessing isopropyldihydrobenzofuran rings, were a pair of the C-22" epimers firstly purified from the leaves of M. notabilis [39]. Due to their undefined absolute configurations of C-22", 14 and 15 were randomly assigned the R*- and S*-configurations, respectively, for the distinguishment of them. Morusalisin D (16) and its C-21" hydroxylated derive, morusalisin E (17), were recently obtained from cell cultures of M. alba, and the absolute configuraton of C-21" of 17 has not been confirmed [51]. The structures of compounds 18 − 22 [15, 22, 39, 48] were characteristic of a 6-membered oxygen ring, which might be formed by the intramolecular nucleophilic addition of the steric vicinal hydroxyl (3′- or 5′-OH) with the olefin (C-1") of the original methylcyclohexene ring. The successful transformation from chalcomoracin (5) to wittiorumin F (18) under 5% CF3COOH undoubtedly allowed the determination of the absolute configuration of 18 [22], and it was regarded as a model compound for ECD and specific rotations analysis to determine the absolute configuration of morbilisin E (19) [39], inethermulberrofuran C (20) [15], macrourin I (21) [48], and macrourin J (22) [48]. In addition, the ECD calculation of 20 using the TDDFT method further confirmed this way to determine the absolute configuration of these class compounds [15]. Yunanensin C (23) was another typical representative, which possessed a 6-membered oxygen ring with the same orientation of CH3-7" as H-5" formed by the intramolecular nucleophilic addition of 16″-OH with C-1" [23].
Compounds 24 − 30 [16, 19, 25, 46, 56,57,58], a series of type A-I MDAAs with a 6-membered oxygen ring similar to 23, which primarily differed in their methylcyclohexene ring and C-8″ carbon as well as the prenyl substituent. The appearance of the double bond (Δ2″) on the methylcyclohexene ring made the C-3" chirality of these compounds disappear, while the absolute configuration of other chiral carbons remained unchanged. Morusalbin A (27) [46] featured with an additional furan ring formed by cyclization between C-2" and C-5′ through an oxygen atom, which could structurally derive from mongolicin C (24) [25]. Mulberrofuran I (28) [56] and mulberrofurans S and Q (29 and 30) [57, 58] were first detected in M. bombycis and M. alba, respectively, which could also be regarded as the 4H-pyran derivatives of 24. Similarly, with 24 as the precursor, its 3′ or 5′-OH condensed with C-8" ketone to form a hemiketal intermediate, which was dehydrated to yield 28 and further oxidized to 6"-hydroxylated product 29 or the epoxy derivative 30. The orientations of 5-OH in 29 and the epoxy ring in 30 were not yet determined.
Compounds 31 − 48, a kind of ketalized type A-I MDAAs, were almost only discovered from the species of Morus [23, 25, 29, 39, 40, 46, 50, 51, 54, 64, 78,79,80]. All ketalized MDAAs were reported as cis–trans configuration based on the coupling constants of H-4" with H-3" and H-5" (small J3"/4" and large J4"/5"). The relative configuration of the new chiral carbon C-8" of this class of ketalized MDAAs could be determined by the NOE correlations of 8"-Ar–H with methylcyclohexene ring protons, and then its absolute configuration could be assigned bases on the empirical rules. Albanol A (mulberrofuran G, 31) was first isolated from M. alba bark by Rama Rao et al. in 1983, and its absolute configuration was confirmed by X-ray analysis of its pentamethyl ether [63]. In 1984, mulberrofuran F (32) and mulberrofuran G (31) were found from the root barks of M. lhou by Fukai et al. and mulberrofuran G was identified as albanol A by comparison of their NMR and physical data [64]. While the absolute configuration of 32 was determined by the Nomura group in 1988 [12]. In addition, compound 32 was also obtained in the above-mentioned conversion of 7 to 18 [22]. Mongolicin A (34), the only ketalized MDAAs possessing a prenyl unit in the moiety of diene, which was isolated from the stem and root bark of M. mongolica [25]. Morusalisin B (37) without analysis of the relative configuration of C-8″ was discovered from cell cultures of M. alba, and the stereochemistry of C-22″ was not defined [51]. Morusalbin B (42) being characteristic of a 4-membered oxygen ring were purified from the root bark of M. alba [46]. Yunanensin D (43), possessing the hydroxylated methyl and conjugated double bond on cyclohexene ring, was isolated from M. yunanensis [78]. If the cyclohexene ring with conjugated double bond continues to dehydrogenate, an aromatic ring will be obtained, as exemplified by compounds 44 − 49 [40, 46, 50, 79, 80]. Sorocenols C and D (47 and 48) were obtained from the root bark of Sorocea bonplandii, which were racemic mixtures due to their zero optical rations [40]. Compared with albanol B (46) [50], the specific optical rotation values of yunanensin A (44) [79] and mulberrofuran P (45) [80] are relatively small (Table 1), and they may also be racemic mixtures in which the ratio of (8"R)-44 or 45 was greater than its (8"S)-enantiomer. Morusalbin C (49), the first non-ketalized MDAAs with an aromatic ring instead of cyclohexene ring, was recently identified from M. alba [46].
2.1.2 Type A with the diene moiety on ring A (Type A-II)
Only eight MDAAs in type A-II have been discovered so far, and they also contained trans–trans (50 − 54) [17, 51, 78, 84, 85] and cis–trans (55 − 57) [16, 66, 85] configurations. Albafuran C (50), the first type A-II MDAAs to be reported, was isolated from M. alba by Mitsuo et al. in 1982 [84], and its C-3" epimer australisine C (55) was obtained from M. australis by Zhang et al. in 2007 [16]. Guangsangon J (51) [17] and guangsangon E (56) [85], another pair of the C-3" epimers in type A-II MDAAs, were isolated from M. macroura. The only one ketalized type A-II MDAAs isomulberrofuran G (57) [66] was discovered from M. alba, whose possible precursor was compound 55.
2.2 Dehydroprenylstilbene type MDAAs (Type B)
A total of 19 MDAAs (Fig. 5, 58 − 76) with dehydroprenylstilbene as the diene have been reported so far. Structurally, most of them (58 − 74) have the original dehydroprenyl group at the para-position of ring A, and only two compounds (75 and 76) at the meta-position of ring A.

Structures of dehydroprenylstilbene type MDAAs (Type B)
2.2.1 Type B with the diene moiety at the para-position of ring A (Type B-I)
Three compounds with a trans–trans configuration were found in the plant of M. macroura, and they were kuwanon X (58) [18], macrourin F (59) [19], and macrourin E (60) [19]. Compounds 61 − 68 were cis–trans type B-I MDAAs, which were mainly isolated from Morus and Sorocea [40, 46, 53, 55, 86, 88,89,90]. Kuwanon Y (61) [86] and macrourin E (62) [53] were the C-3" epimers of 58 and 59, respectively. Among the ketalized type B-I MDAAs (64 − 71), sorocenol E (69) [40], cathayanon C (70) [91], and cathayanon D (71) [91] were characterized by an aromatic ring rather than a cyclohexene ring. In addition, like sorocenols C and D (47 and 48), sorocenol E (69) with zero optical ration was also a racemic mixture [40]. In view of small specific optical rotation values of 70 and 71, they might be racemic mixtures with different ratio of (8"R) and (8"S)-enantiomers. Kuwanol B (72) [88] featured a 6-membered oxygen ring and a 4H-pyran ring, similar to compound 29 in type A-I. Kuwanon Z (73) could be considered as a highly oxidized derivative of 72, which was isolated from M. alba [86]. Sorocenol F (74) [40] obtained from S. bonplandii was a prenylated derivative of 73.
2.2.2 Type B with the diene moiety at the meta-position of ring A (Type B-II)
The only two MDAAs of type B-II, guangsangon B (75) [85] and kuwanon P (76) [92], were first obtained from M. macroura and M. lhou. They both possess the trans–trans configuration.
2.3 Dehydroprenylchalcone type MDAAs (Type C)
There are 15 MDAAs (Fig. 6, 77 − 91) formed by cycloaddition of chalcone dienophile with dehydroprenylchalcone diene, and the dienes and dienophiles of most type C MDAAs (77 − 90) are structurally derived from the same prenylatedchalcone. This type MDAAs is distributed in 11 species including A. heterophyllus, B. oblongifolia, D. barteri, M. alba (M. alba var. shalun), M. bombycis, M. macroura, M. mongolica, M. notabilis, M. nigra, S. bonplandii, and S. ilicifolia. According to the position of the dehydroprenyl group on chalcone skeleton, type C MDAAs could be divided into the following two subgroups.

Structures of dehydroprenylchalcone type MDAAs (Type C)
2.3.1 Type C with the diene moiety on ring B (Type C-I)
Fourteen type C-I MDAAs have been reported so far, whose dehydroprenyl groups are located on ring B of a chalcone skeleton. Four of them are provided with trans–trans configuration (77 − 80) and ten with cis–trans configuration (81 − 90).
Kuwanon I (78) [94] and kuwanon J (81) [28] together with dorstenone (79) [95] and kuwanon V (87) [47] are two pairs of 3"-epimers. Brosimone A (80) [96], the only one MDAAs being characteristic of two methylcyclohexene rings, could be considered to be derived from brosimone B (77) [93] via Deils-Alder cycloaddition between the prenyl moiety at C-13" and the chalcone. Brosimones A and B (80 and 77) in this type as well as brosimone D (98) in type D are the only three MDAAs isolated from the root of Brosimopsis oblongifolia (Table 2) [93, 96]. Kuwanon J 2,4,10"-trimethyl ether (82) [100] from M. alba is the trimethyl ether of 81. Comparion with kuwanons J and R (81 and 86) [28, 47], artonins C and X (84 and 85) [101, 102] from A. heterophyllus have a 3-methylbut-1-en-1-yl unit at C-11" instead of a prenyl group. Among the reported type C MDAAs, mongolicin G (89) is the only one with dehydroprenyldihydrochalcone as diene, which was identified from M. mongolica [104]. The only ketalized type C MDAAs, sorocein B (90), was obtained from the roots of S. bonplandii by Messana et al. in 1991 [90].
2.3.2 Type C with the diene moiety on ring A (Type C-II)
Only one member, namely guangsangon C (91), in this subgroup has been identified so far. As shown in Fig. 6, the position of its dehydroprenyl group is located at C-3 on ring A of the chalcone skeleton, and 91 has the trans–trans configuration. Guangsangon C (91) was isolated from the stem bark of M. macroura [85].
2.4 Dehydroprenylflavone type MDAAs (Type D)
Twenty-two MDAAs (Fig. 7, 92 − 113) with dehydroprenylflavone as the diene have been reported so far. On the basis of the position of dehydroprenyl unit, type D MDAAs are divided into two subgroups: type D-I (the diene moiety on ring A) and type D-II (the diene moiety on ring B), as illustrated by the structures of 92–104 and 105–113, respectively.

Structures of dehydroprenylflavone type MDAAs (Type D)
2.4.1 Type D with the diene moiety on ring A (Type D-I)
Among type D-I MDAAs 92–104, the diene moiety of six compounds (92–97) are located at C-8 of A ring and others (98–104) at C-6 of A ring. All six type D-I MDAAs with the diene moiety at C-8 of A ring, namely kuwanon G (92), kuwanon H (93), mongolicin D (94), moracenin D (95), moracenin E (96), kuwanon W (97), are provided with trans–trans configuration. The other seven type D-I MDAAs with the diene moiety at C-6 of A ring have trans–trans and cis–trans configurations, as exemplified by 98–99 and 100–104, respectively.
Kuwanon G (92) [1] and kuwanon H (93) [2], also known as albanin F and albanin G [3] as well as moracenin B [109] and moracenin A [112], respectively, are the first reported MDAAs by three research groups in 1980 from the same plant (M. alba). Their structures were further confirmed by partial synthesis in 1981 [5]. Moracenin D (95) was isolated from M. alba, and its structure was elucidated by oxidation of 92 [105]. Compounds 92–98 all possess a prenyl group or structure derived thereof at C-3. Multicaulisin (99) possessing an additional prenyl moiety at C-7″ was isolated from M. multicaulis by Ferrari et al. in 2000 [114]. Mesozygins A (104), B (100), and C (103) as well as artonin I (101) were all discovered in the leaves of M. mesozygia by Fozing et al. in 2012 [35]. Mesozygin C (103) is a prenylated derivative of mesozygin B (100), while mesozygin A (104) is a ketal form of compound 100.
2.4.2 Type D with the diene moiety on ring B (Type D-II)
There are only nine members (105–113) in this subgroup, and their diene moieties are all placed at C-3′ of B ring. Among type D-II MDAAs, compounds 105–110 are trans–trans configurations, while 111–113 are cis–trans configurations. Guangsangons G, I, and O (105, 106, and 111) from M. macroura [17, 119], wittiorumins A, B, and C (107, 108, and 112) from M. wittiorum [22], and australisine A (113) from M. australis [16] feature dehydroprenylflavonol dienes. Kuwanon K (109) and kuwanon N (110), possessing a prenyl unit at C-3 of dehydroprenylflavone diene, were discovered from M. lhou [110]. Australisine A (113) is the only ketal compound in type D-II MDAAs, which has been reported only once from moraceous plants [16].
2.5 Dehydroprenyldihydroflavone type MDAAs (Type E)
Fourteen type E MDAAs have been reported to date, as exemplified by 114 − 127 (Fig. 8), all sharing the dehydroprenyldihydroflavone diene. The location of the dehydroprenyl at dihydroflavone result in two subgroups: type E-I (the diene moiety on ring B) and type E-II (the diene moiety on ring A).

Structures of dehydroprenyldihydroflavone type MDAAs (Type E)
2.5.1 Type E with the diene moiety on ring B (Type E-I)
Among the reported type E-I MDAAs, all compounds having the diene moiety on ring B (all at C-3′) are trans–trans configuration. Guangsangons M and N (114 and 115) are a pair of the 2-epimers, which were isolated from the stem barks of M. macroura by Dai et al. in 2004 [74]. Wittiorumin D (116) and its prenylated derivative wittiorumin E (117) with the same 2S configuration were obtained from M. wittiorum by Tan et al. in 2009 [22]. Kuwanon L (118) [118] and its prenylated derivative kuwanon O (119) [110] wereisolated from M. alba in 1983 and M. lhou in 1984, respectively, in which the configurations of C-2 were not yet determined. Guangsangon D (120) [85], guangsangon H (121) [17], guangsangon K (122) [74], and guangsangon F (123) [74] all having a hydroyl group at C-2 are 2R,3R configurations. Their dienes also could be regarded as dehydroprenyldihydroflavonol.
2.5.2 Type E with the diene moiety on ring A (Type E-II)
There are only four compounds in type E-II MDAAs, two (124 and 125) of which feature the C-6 dehydroprenyl and two (126 and 127) feature the C-8 dehydroprenyl. Compounds 124 − 126 are trans–trans configurations, while 127 is cis–trans configuraion. Sanggenon G (124) [121] and sanggenon T (125) [123] have a prenyl group or structure derived thereof at C-7", in which the absolute configurations of C-2 were still not confirmed. Sanggenol M (126) is also characterized by the rare C-7" prenyl, which was isolated from the root barks of M. mongolica [122]. Wittiorumin G (127) obtained from M. wittiorum is the only type E MDAAs with cis–trans configuration [49].
2.6 Dehydroprenylsanggenonflavone type MDAAs (Type F)
Sanggenon-type flavanones are a kind of 3-hydroxy-2-prenylflavanones having an ether linkage between C-3 and C-2', which are characteristic constituents in Morus plants. Sanggenon-type flavanones are very rare in nature, so only several type F MDAAs (Fig. 9, 128 − 137) derived from sanggenon-type flavanone with the dehydroprenyl group have been reported so far. According to the position of the dehydroprenyl group on sanggenon-type flavanone, type F MDAAs could be divided into the following two subgroups.

Structures of dehydroprenylsanggenonflavone type MDAAs (Type F)
2.6.1 Type F with the diene moiety at C-6 of ring B (Type F-I)
Eight members (128 − 135) in type F-I MDAAs have so far been reported, which are distributed in five species of the family Moraceae including M. alba, M. cathayana, M. mongolica, M. nigra, and S. bonplandii [91, 99, 124,125,126, 129, 131]. Among this subgroup, two compounds (128 and 129) are provided with trans–trans configuration and six (130 − 135) with cis–trans configuration. Sanggenon D (128) [124], sanggenon E (129) [126], sanggenon C (130) [129], sanggenon O (131) [131], and sanggenon P (132) [126], isolated from M. alba by Nomura et al. in the 1980s, have been revised to be the present structures shown in Fig. 9 rather than the original structures derived from 2-hydroxy-3-prenylflavanones having an ether linkage between C-3 and C-2'. Except for compounds 130, 131, and 135, the absolute configurations of C-2 and C-3 of 128, 129, and 132 − 134 have not been determined. Sorocein C (134) belonging to the ketalize MDAAs, was isolated from the root bark of S. bonplandii [99]. Cathayanon E (135) has a 6-membered oxygen ring formed by the intramolecular nucleophilic addition of 5-OH with the olefinic C-1", which was obtained from the stem bark of M. cathayana by Zhang et al. in 2009 [91].
2.6.2 Type F with the diene moiety at C-8 of ring B (Type F-II)
Cathayanons A and B (136 and 137) are the only two type F MDAAs with their dehydroprenyl group at C-8 of ring B, which were isolated from M. cathayana by Shen et al. in 2001 [130]. The absolute structure of the cis–trans type adduct cathayanon A (136) was confirmed by X-ray crystallographic analysis. The stereochemistry of C-2 and C-3 in the trans–trans type adduct cathayanon B (137) was determined to be the same 2S,3R as compound 136.
2.7 Dehydroprenylcoumarin type MDAAs (Type G)
There are only seven members (Fig. 10, 138–144) in this type, in which the dehydroprenyl group is located on its phenyl ring (C-6). All these compounds were isolated from the bark of Brosimum rubescens by Shirota et al. [132], and they share the same cis–trans configuration. The common characteristic of palodesangrens A − E (138 − 142) is the presence of a 6-membered oxygen ring formed by an ether linkage between C-7 and C-8", and the main difference is the number and position of methoxy groups on the chalcone unit. Palodesagretins I and II (143 and 144) [133] have an additional 5-membered ring formed by the carbon bond of C-5 and C-8", and they are different in the position of methoxy groups. The absolute configurations of 138–144 were not determined.

Structures of dehydroprenylcoumarin type MDAAs (Type G) and simple or other dehydroprenylphenol type MDAAs (Type H)
2.8 Simple or other dehydroprenylphenol type MDAAs (Type H)
Compounds 145 − 149 (Fig. 10) are the simplest MDAAs found in moraceous plants, and their dienes are the simple phenolic compounds with the dehydroprenyl moiety. These simple phenolic compounds include 2,4-dihydroxybenzaldehyde, 3,5-dihydroxybenzaldehyde, methyl 2,4,6-trihydroxybenzoate, and resorcinol, as exemplified by 145, 146 and 147, 148, and 149, respectively. Guangsangon L (145) [74] from the stem bark of M. macroura is trans–trans configuration. The ketalized type H MDAAs, soroceal B (146) [13] from M. alba and soroceal (147) [90] from S. bonplandii, have cis–trans configuration. Morusalbanol A (148) [134] from M. alba, possessing the same 6-membered oxygen ring as 18 − 22, has cis–trans configuration. Sorocenol B (149), having the same methylcyclohexene ring and 6-membered oxygen ring as 24 − 26, was obtained from the root bark of S. bonplandii [87]. Sanggenon Q (150) [83] from M. mongolica is the only other dehydroprenylphenol type MDAAs, in which the diene is dehydroprenyl-2-oxo-3-prenylisoflavanone and the relative configuration of H-3″, H-4″, and H-5″ is cis–trans. The stereochemistry of its C-3 was not determined.
2.9 Non-classic MDAAs (Type I)
There are a few non-classic MDAAs (Fig. 11, 151 − 166) in moraceous plants, in which the dienophile moiety is the olefinic bond of other compounds rather than the α,β-unsaturated bond of a chalcone skeleton, or which might be derived from some classic MDAAs without the benzoyl at C-4" or the phenyl at C-8". The distribution of non-classic MDAAs in mulberry plants is limited, which were only found in the following three Morus plants, M. alba, M. lhou, and M. mongolica.

Structures of non-classic MDAAs (Type I)
Kuwanon M (151) [135] from M. lhou and mongolicin E (152) [25] from M. mongolica are a pair of epimers, in which the dienophile and diene are delivered from the same prenylatedflavone, kuwanon C [33]. Specifically, after Diels–Alder reaction of the C-8 dehydroprenyl group in one molecule and the C-8 prenyl group in another molecule, a further intramolecular cyclodehydrogenation builds a 6-membered oxygen ring in the additions. Dimoracin (153) was isolated from M. alba [136], and its formation process is similar to that of 151 and 152, except that its dienophile and diene are derived from the same prenylated-2-arylbenzofuran, moracin C [136].
Morusalones A − D (154 − 159) from M. alba cell cultures [137], featuring unprecedented 6/7/6/6/6/6 hexacyclic core skeletons with a unique bridged cycloheptenone ring, could be regarded to be formed by Diels–Alder cycloaddition of quinostilbene dienophiles and dehydroprenyl-2-arylbenzofuran dienes and subsequent intramolecular nucleophilic addition. The absolute configurations of 154 − 159 were determined by X-ray analysis and ECD calculation [137]. Morusalones A and B are the first examples of Diels − Alder adduct enantiomers from the genus Morus.
Seven type I MDAAs might be variations of some classic MDAAs, in which the phenyl moieties at C-8" or the benzoyl moieties at C-4" is missing, as exemplified by compounds 160 − 161 [25, 51] or 162 − 166 [123, 138,139,140], respectively. Among these compounds, sanggenon S (164) [123] from M. alba is a typical intermediate in the formation of non-classic MDAAs, which seems to be a derivative induced from sanggenon D (128). Sanggenon B (165) [140] was also isolated from M. alba, which can be regarded as derived from 164 by hydrolyzing its 2,4-dihydroxybenzoyl group.
2.10 Overview on distribution of MDAAs in different plants
MDAAs were at least found in 21 species of the family Moraceae, most of which were isolated and identified from Morus plants (Tables 1 and 2). Dehydroprenyl-2-arylbenzofuran type (Type A) MDAAs with 57 compounds were widely distributed in 14 species of three genera (Morus, Sorocea, and Chlorophora), belonging to the richest class on MDAAs, followed by dehydroprenylflavone type (Type D) MDAAs with 22 structures distribution in 13 species of three genera (Morus, Artocarpus, and Brosimopsis) and dehydroprenylstilbene type (Type B) MDAAs with 19 compounds distribution in ten species of three genera (Morus, Sorocea, and Chlorophora) (Table 1). Dehydroprenyldihydroflavone type (Type E) MDAAs (14 compounds) and non-classic (Type I) MDAAs (16 compounds) were only identified in Morus plants, while dehydroprenylcoumarin type (Type G) MDAAs (seven compounds) was only found in one plant Brosimum rubescens. Although there were only 15 compounds in dehydroprenylchalcone type (Type C) MDAAs, they were distributed in five genera (Morus, Artocarpus, Sorocea, Brosimopsis, and Dorstenia), which was the type with the largest distribution of genera among all types. Most of the dehydroprenylsanggenonflavone type (Type F) MDAAs (ten compounds) were isolated from five Morus plants (M. alba, M. bombycis, M. cathayana, M. mongolica, and M. nigra), and simple or other dehydroprenylphenol type (Type H) MDAAs (five compounds) were distributed in five species including M. alba, M. macroura, M. mongolica (syn. M. yunanensis), M. nigra, and S. bonplandii (Table 1).
M. alba, M. mongolica, M. macroura, M. bombycis, M. australis, M. lhou, M. wittiorum, and S. bonplandii are rich in MDAAs (Table 2). Among them, the plant of M. alba has the largest number MDAAs, more than 70, covering eight types including Types A − F, H, and I. M. mongolica contains 45 MDAAs also involving in all eight types except for Type G. 38 and 16 MDAAs were isolated from M. macroura and M. bombycis, respectively. There are no more than 10 MDAAs in other species, such as D. barteri, M. insignis, and M. multicaulis (each contains one MDAAs), A. integer, Brosimopsis oblongifolia, and C. regia (each contains three MDAAs), M. mesozygia (contains five MDAAs), A. heterophyllus (contains six MDAAs), Brosimum rubescens (contains seven MDAAs), M. cathayana (contains eight MDAAs), and S. ilicifolia (contains nine MDAAs) (Table 2). In addition, according to the distribution of MDAAs, this kind of compound could be used as chemotaxonomy biomarker within moraceous plants.
3 Biological activities
As the characteristic components of Morus plants, MDAAs possess a variety of different biological activities, including antineoplastic, anti-inflammation, antimicrobial, antioxidant, antiviral, anti-neurodegenerative diseases, anti-cardiovascular diseases, as well as PTP1B, α-glucosidase, and tyrosinase inhibitory activities. In this section, we will focus predominantly on the biological and pharmacological activities of natural MDAAs.
3.1 Antineoplastic activity
The Yu and Chen groups have evaluated the cytotoxicity of 18 MDAAs, mulberrofuran J (1), chalcomoracin (5), mulberrofuran E (9), mulberrofuran O (13), mongolicin C (24), australisine B (25), mulberrofuran Q (30), mulberrofuran G (31), mulberrofuran F (32), mulberrofuran F1 (33), yunanensin A (44), albafuran C (50), australisine C (55), sorocein A (68), kuwanon J (81), kuwanon G (92), australisine A (113), and wittiorumin G (127), against five human cancer cell lines (A549, Bel-7402, BGC-823, HCT-8, and A2780). The results showed that nine compounds (5, 24, 25, 31 − 33, 44, 55, and 113) possessed potent cytotoxic properties in these tested cell lines (Table 3) [16, 29, 49, 79]. Mulberrofuran J (1), mulberrofuran C (6), mongolicin C (24), mulberrofuran G (31), artonin I (101), and soroceal B (146) were tested for their cytotoxicity against HL-60, Hela, HepG-2, A-549, and AGS cell lines. Compounds 24, 31, and 146 exhibited obvious cytotoxic activity against the tested five cell lines (Table 3) and 31 inhibited significantly selective cytotoxic activities towards HL-60 and AGS cells with IC50 of 3.4 and 3.5 μM [13]. Fitriani et al. found that morushalunin (4), chalcomoracin (5), mulberrofuran K (40), and guangsangon E (56), and kuwanon J (81) had significant cytotoxicity against murine leukemia P-388 cells with IC50 values 0.7, 1.7 (5.46 μM [34]), 0.6, and 2.5 (2.75 μg/mL [34]), and 5.9 μg/mL, respectively [26, 77]. Han et al. showed for the first time that chalcomoracin (5) treatment markedly promoted paraptosis along with extensive cytoplasmic vacuolation derived from the endoplasmic reticulum, rather than apoptosis, in PC-3 and MDA-MB-231cell lines [141]. Subsequently, Zhang et al. demonstrated that chalcomoracin (5) could inhibit cell proliferation and increase sensitivity to radiotherapy in human non-small cell lung cancer cells also via endoplasmic reticulum stress-mediated paraptosis mechanism [142]. Takashi et al. revealed that mulberrofuran G (31) could induce apoptotic cell death in HL-60 via both the cell death receptor pathway by stimulation of death receptor, and the mitochondrial pathway by Topo II inhibition through caspase-2 activation [59]. In addition, 31 had moderate cytotoxic activity against lung cancer cells NCI-H292 and A549 with IC50 value of 3.75 and 10.39 μM, respectively [68]. Mulberrofuran G (31), albafuran C (50), and kuwanon G (92) possessed antiproliferative activity against HePG2 and MCF-7 cell lines, and the IC50 values were equivalent to those of the positive control [76]. Phan et al. demonstrated for the first time that albanol B (46) exerted the anti-cancer effect by inducing apoptosis and cell cycle arrest at G2/M through mitochondrial ROS production in lung cancer cells [143]. Shu et al. reported that guangsangon E (56) could exert anti-lung and nasopharyngeal cancer cells through autophagy-mediated cell death [144]. The cytotoxicity of sanggenon G (124), sanggenon T (125), sanggenol M (126), sanggenon D (128), sanggenon C (130), and sanggenon B (165) from M. mongolica was evaluated. Of these MDAAs, 126 and 130 were the most potent agents against human oral tumor cell lines (HSC-2 and HSG) [122]. Park et al. have confirmed for the first time that sanggenon G (124) could suppress proliferation and induce apoptosis in lung cancer cells (A549) through caspase-3 activation and PL5-mediated inhibition of c-Myc, and its combination effect with adriamycin was more prominent [145]. Sanggenon D (128) was found to inhibit the growth of transplanted tumor and the proliferation of tumor cells in mice [146]. Chen et al. reported that sanggenon C (130) could induce apoptosis of colon cancer cells via inhibition of NO production, iNOS expression, and ROS activation of the mitochondrial pathway [147]. It was found that sanggenon O (131) induced apoptosis of A549 cells could be counterbalanced by protective autophagy, which indicated that 131 possesses great potential to be a promising candidate combined with autophagy inhibitor for the treatment of NSCLC [148].
Hypoxia-inducible factor-1 (HIF-1) inhibitor represents a promising anti-cancer agent. The eight MDAAs mulberrofuran G (31), kuwanon J (81), kuwanon Q (83), kuwanon R (86), kuwanon V (87), sanggenon C (130), sanggenon O (131), and mulberrofuran H (162) were reported for the first times to possess HIF-1 inhibitory effect in the Hep3B cell-based assay [71]. Additionally, these compounds were also active against hypoxia-induced vascular endothelial growth factor (VEGF) secretion in Hep3B cells (Table 3) [71].
3.2 Anti-inflammation activity
The Yu group have evaluated the inhibition effects of the obtained MDAAs (at a concentration of 10 μM) on the release of β-glucuronidase in rat polymorphonuclear (PMN) leukocytes induced by PAF and found that mongolicins C and E (24 and 152) showed potent anti-inflammatory activities with inhibitory ratios of 80.4% and 77.0% [25], respectively, while albafuran C (50), guangsangon J (51), guangsangon B (75), guangsangon I (106), and guangsangon H (121) displayed moderate effects [17, 85, 149]. Kimura et al. have found that mulberrofuran J (1), mulberrofuran Q (30), mulberrofuran G (31), kuwanon G (92), kuwanon H (93), sanggenon D (128), and sanggenon C (130) could affect arachidonate metabolism in rat platelets [150]. Kuwanon G (92) and kuwanon O (119) could significantly down-regulated the expressions of TNF-α, IL-1β, IL-6, COX-2, and NF-κB in dose-dependent manners in LPS-stimulated RAW264 cells [107]. Among albanol B (46), sanggenon D (128), and sanggenon B (165), 46 exhibited the best anti-inflammatory activity, which inhibited expression of inducible nitric oxide synthase (iNOS) and cyclooxygenase-2 (COX-2) and suppressed production of pro-inflammatory cytokines and mediators in LPS-induced RAW264.7 cells [81]. Mulberrofuran K (40) suppressed the production of NO, reactive oxygen species (ROS), and proinflammatory cytokines IL-1β, IL-6, and TNF-α in a dose-dependent manner in LPS-stimulated RAW264 cells [73]. Cathayanons A and B (136 and 137) displayed potent activities on the inhibition of HL-60 cell adhesion to BAEC (Bovine Arterial Endothalium cells) at 10 μM, with inhibitory rates of 44.72% and 39.02%, respectively [130]. Among the six MDAAs mulberrofuran J (1), mulberrofuran C (6), mongolicin C (24), mulberrofuran G (31), artonin I (101), and soroceal B (146), only 1 (IC50 = 21.4 μM) and 24 (IC50 = 8.8 μM) exhibited moderate inhibitory activity against NO production in LPS-activated RAW264.7 cells [13]. In TNF-α-stimulated HeLa cells, kuwanon J 2,4,10"-trimethyl ether (82) and kuwanon R (86) strongly inhibited NF-κB activity with the IC50 values of 4.65 and 7.38 μM, respectively [100]. Sanggenons C and O (130 and 131) could inhibit NO production and iNOS expression by suppressing NF-κB activity and IκBα activation in LPS-stimulated RAW264 cells [151]. In a high-throughput screening for ADAMTS1 (a disintegrin and metalloprotease with thrombospondin type I motifs-1) inhibitors by the fluorescence resonance energy transfer (FRET) method, four compounds mulberrofuran J (1), albafuran C (50), kuwanon X (58), and kuwanon P (76) were identified from a diverse library of 40,960 total compounds [152]. The results of kuwanon G (92) on ovalbumin (OVA)-induced allergic asthma in mice indicated that it could prevent the pathological progression of allergic asthma through the inhibition of lung destruction by inflammation and immune stimulation [153]. Kuwanon G (92) could inhibit chemokine production by blocking of STAT1 (signal transducer and activator of transcription 1) and NF-κB pathways in HaCaT keratinocytes and reduce the release of histamine and LTC4 (leukotriene C4) by suppressing the 5-LO (5-lipoxygenase) activation in MC/9 mast cells, which suggested it had anti-allergic and anti-inflammatory effects [154]. Among the five MDAAs sanggenons B − E and O, sanggenon E (129), sanggenon C (130), and sanggenon O (131) had inhibitory effects on two cyclooxygenase isoenzymes COX-1 and -2 with IC50 values of 10 − 14 μM and 40 − 50 μM, respectively [127].
3.3 Antibacterial activity
Fukai et al. found that chalcomoracin (5), mulberrofuran G (31), mulberrofuran F (32), and albanol B (46) showed considerable antibacterial activity against several vancomycin-resistant enterococci (VRE) and methicillin-resistant Staphylococcus aureus (MRSA) strains with MIC values ranging from 3.13 to 6.25 μg/mL. Among chalcomoracin (5), mulberrofuran G (31), mulberrofuran F (32), and albanol B (46), compound 5 (MIC = 0.78 μg/mL) showed antimicrobial activity comparable to that of positive drugs against MRSA (strains K3 and ST28) and methicillin-sensitive Staphylococcus aureus (MSSA) (strains FDA 209P and Smith), while 32 and 46 showed slightly weak antimicrobial activity against these MRSAs and MSSAs (MICs = 0.78–1.56 μg/mL) (Table 4) [155]. In addition to anti Staphylococcus aureus including MRSA, chalcomoracin (5) could strongly inhibited S. aureus enoyl-ACP reductase (saFabI) with a IC50 of 5.5 μM, a novel target for antibacterial drug development [30, 156]. Among the four MDAAs mulberrofuran G (31), albanol B (46), sanggenon D (128), and sanggenon B (165), compounds 31 and 46 showed strong antibacterial activity against Escherichia coli, Salmonella typhimurium, Staphylococcus epidermis, and Staphylococcus aureus with 5–30 μg/mL of MICs, while 128 and 165 were effective to Saccaromyces cerevisiae, Staphylococcus epidermis, and Staphylococcus aureus with 12.5–50 μg/mL of MICs [157]. Except for kuwanol A (64) and kuwanon L (118), all seven MDAAs mulberrofuran Q (30), mulberrofuran G (31), mulberrofuran K (40), albanol B (46), kuwanon G (92), kuwanon H (93), and kuwanon O (119) had better bioactivity against MRSA both in vitro and in vivo than antibiotics such as berberine, ampicillin, and kanamycin. Preliminary mechanism of action showed that these compounds could damage the bacterial cell membranes as well as inhibit the efflux of drugs such as methicillin and ethidium bromide [62]. Among the six MDAAs albanol B (46), albafuran C (50), kuwanon G (92), kuwanon H (93), kuwanon K (109), and kuwanon L (118), compounds 93 and 109 diminished the growth of a standard strain E. faecalis, three clinical isolates of VRE, and three clinical isolates of MRSA, with MIC values of 1–32 μg/mL [82]. Kuwanon G (92) significantly inhibited the growth of the cariogenic bacteria such as Streptococcus mutans (MIC = 8 μg/mL), Streptococcus sobrinus (MIC = 8 μg/mL), and Streptococcus sanguis (MIC = 8 μg/mL), and periodontal bacterium, Porpyromonas gingivalis (MIC = 8 μg/mL). Transmission electron microscopy (TEM) showed that 92 could remarkably cause morphological damage of the cell wall and condensation of the cytoplasm in S. mutans [106]. Wu et al. found that kuwanon G (92) and kuwanon H (93) could treat MRSA-associated infections by disrupting the proton motive force and membrane permeability [108]. Kuwanon G (92) was also identified as a good candidate for the development of novel antibacterial combination therapy [158, 159]. Kuwanon H (93), multicaulisin (99), and sanggenon G (124) were found to possess potent effect against ten clinical MRSA strains with MICs/MBCs of 2–8/16–128 mg/L, and also showed synergy with conventional antibacterial agents such as aminoglycosides [111]. Artonin I (101) could inhibit multidrug resistance in Staphylococcus aureus and potentiate the action of inactive antibiotics in vitro [116]. From the five MDAAs kuwanon L (118), sanggenon G (124), sanggenon D (128), sanggenon C (130), and sanggenon B (165), compound 124 was discovered to be a potent antibacterial agent against Streptococcus pneumoniae with a MIC of 5.44 μM [160]. Pang et al. found that sanggenon D (128) could inhibit the growth of Staphylococcus aureus by moderating the fatty acid biosynthesis system [161].
Protein tyrosine phosphatase B (PtpB) is a promising target for the development of novel anti-tuberculosis drugs. From an in house library of more than 800 natural substances, kuwanol E (62) was discovered by Mascarello et al. to be the most potent Mycobacterium tuberculosis PtpB inhibitor (IC50 = 1.9 μM and Ki = 1.6 μM) [162]. Subsequently, Mascarello et al. found that among the four MDAAs chalcomoracin (5), kuwanon G (92), kuwanon H (93), and kuwanon L (118), compounds 92 (IC50 = 0.83 μM) and 93 (IC50 = 0.36 μM) were the two most potent Mtb PtpB inbibitors with Ki values of 0.39 and 0.20 μM, respectively. The comprehensive research strategies including kinetics, mass spectrometry, and molecular docking demonstrated that both compounds were interacted with the active site of the enzyme [37]. Kuwanol E (64), kuwanon G (92), and kuwanon H (93) are the first non-peptidic PtpB inhibitors discovered from natural sources.
3.4 Antioxidant activity
MDAAs are phenolic natural products with multiple hydroxyl groups, which contribute their strong antioxidant properties. The Yu and Chen groups evaluated the antioxidant properties of their obtained MDAAs in Fe2+/cysteine-induced microsomal lipid peroxidation assay and found that most of the compounds at concentrations of 10 μM had good activities (Vitamine E as the positive control) [17, 20, 22, 25, 49, 50, 74, 78, 85, 91, 119, 149]. For example, guangsangon J (51), guangsangon I (106), and guangsangon H (121) were first reported to display potent antioxidant activities with the inhibitory rates of malondialdehyde being 91.1%, 93.9%, and 93.1%, respectively, compared to the positive control Vit E (33.4%) [17]. The other active MDAAs were listed in Table 5. In addition, kuwanol E (62) exhibited remarkable free radical scavenging properties with the IC50 value of 2.1 μg/mL (the standard trolox, IC50 = 1.1 μg/mL) [27]. Li et al. found that sanggenon C (130) and sanggenon D (128) may undergo an antioxidant approach to protect mesenchymal stem cells (MSCs) against oxidative stress, and the discrepancies in their antioxidant activities could be attributed to the steric effect [163].
3.5 Anti-neurodegenerative diseases
Ten MDAAs, mulberrofuran J (1), mulberrofuran C (6), inethermulberrofuran C (20), mulberrofuran G (31), mulberrofuran K (40), albafuran C (50), isomulberrofuran G (57), kuwanol A (64), kuwanon G (92), and kuwanon H (93), were systematically screened for their anti-Alzheimer's disease (anti-AD) properties on different targets such as tau aggregation, Aβ self-aggregation, and ChEs. Of these compounds, 6, 31, 40, and 57 were found to be potent multi-targeted agents for AD. The selected mulberrofuran K (40) with a good blood–brain barrier (BBB) permeability could play neuroprotective effects by up-regulating the level of glutathione (GSH) and inhibiting the production of reactive oxygen species (ROS) in glutamate-induced HT22 cell model [15]. Among the three MDAAs mulberrofuran C (6), mulberrofuran G (31), and sanggenon G (124), compounds 6 and 31 were found to have neuroprotective activity on glutamate-induced cell death in HT22 cells with EC50 values of 19.71 and 16.50 μM, respectively [44]. Mulberrofuran Q (30) showed inhibitory activity against oxygen glucose deprivation (OGD)-induced cell death of SH-SY5Y cells, and its protective effect was more potent than the positive control carnosine [60]. Hong et al. have investigated the neuroprotective effect of mulberrofuran G (31) in in vitro and in vivo models of cerebral ischemia. It was found that 31 could protect ischemic injury-induced cell death through the inhibition of NOX4-mediated ROS generation and ER stress [72]. Mulberrofuran G (31), albanol B (46), and kuwanon G (92) displayed potent inhibitory activities against ChEs and β-site amyloid precursor protein cleaving enzyme 1 (BACE1) [67]. Later, mulberrofuran G (31), albanol B (46), and kuwanon G (92) were identified as inhibitors of human monoamine oxidase (hMAO) and modulators of dopaminergic receptor [164]. Kuwanon V (87) could inhibit the proliferation of neural stem cells, promote cell survival, and increase neurogenesis [103]. Moracenin D (95) was found to possess protective effects in dopamine-induced SH-SY5Y cells via the up-regulation of nurr1 and down-regulation of α-synuclein expressions [165]. Zhao et al. demonstrated that sanggenon C (130) had neuroprotective effects on ischemic stroke by inhibiting inflammation and oxidative stress by regulating RhoA-ROCK signaling pathway [166]. The neuroprotective activity of morusalbanol A (148) in H2O2-induced PC12 cells was evaluated, and the result showed that pretreatment with 148 could significantly attenuate the H2O2-induced cell damage in a dose-dependent manner [134].
3.6 PTP1B inhibitory activity
Protein tyrosine phosphatase 1B (PTP1B) is a key negative regulator of insulin and leptin signaling pathways. It is currently considered as a valid therapeutic target for type 2 diabetes mellitus and obesity [167]. In 2006, Cui et al. first found four MDAAs mulberrofuran C (6), kuwanon L (118), sanggenon G (124), and sanggenon C (130) showed significant PTP1B inhibitory activities with IC50 values of 4.9, 16.9, 1.6, and 2.6 μM, respectively. Analysis of inhibition kinetics by Lineweaver–Burk plots suggested that the inhibition mode of the most active compounds (6, 124, and 130) on the activity of PTP1B was mixed-type [43]. Subsequently, kuwanon J (81), kuwanon R (86), and kuwanon V (87) were indentified as mixed-type inhibitors of PTP1B with IC50 values ranging from 2.7 to 13.8 μM [98]. Since 2015, a large number of MDAAs have been reported to have significant inhibitory activity against PTP1B (Table 6) [14, 39, 46, 51, 61, 137, 168]. Mechanism research suggested that the potent PTP1B inhibitors ( +)-morusalone A (154) and morusalone C (158) could significantly increase the insulin induced phosphorylation of IRβ in HepG2 cells, and then upregulate the phosphorylation level of downstream Akt [137].
3.7 α-Glucosidase inhibitory activity
α-Glucosidase is a key enzyme involved in carbohydrate digestion, and its inhibitors can be effective drugs in the management of postprandial blood glucose and insulin levels in type 2 diabetic patients [169]. Ha et al. found that all the PTP1B inhibitors albasin B (8), macrourin G (26), morusalbin A (27), mulberrofuran G (31), mulberrofuran K (40), morusalbin B (42), yunanensin A (44), albanol B (46), morusalbin C (49), and morusalbin D (67) exhibited strong inhibitory activities against α-glucosidase with IC50 ranging from 2.29 to 5.91 μM. The type of α-glucosidase inhibition of the active MDAAs 8, 26, 44, and 67 with Ki values of 0.42, 2.42, 1.19, and 0.64 μM, respectively, were determined to be competitive-type by enzyme kinetic studies [46]. In addition to these ten α-glucosidase inhibitors, mongolicin C (24) and mulberrofuran H (162) also had significant inhibitory effects on α-glucosidase [46]. In 2018, two additional MDAAs sanggenon G (124, IC50 = 11.96 μM) and sanggenon O (131, IC50 = 3.06 μM) with significant inhibitory activity against α-glucosidase were identified from crude extracts of Sang-Bai-Pi by a novel screening strategy based on the ligand fishing combined with HPLC-QTOF-MS and molecular docking [69]. The other MDAAs with potent inhibitory effects on α-glucosidase were listed in Table 7 [32, 48, 61, 68, 97, 168].
3.8 Tyrosinase inhibitory activity
Tyrosinase is a rate-limiting enzyme in the formation of melanin pigments in mammals and the key enzyme for enzymatic browning of many plant-derived food products [170]. Therefore, tyrosinase inhibitors are crucial in the pharmaceutical, skin whitening cosmetic, and food industries. In 2004, Lee et al. first discovered that the natural MDAA sanggenon D (128, IC50 = 7.3 μM) was a potent tyrosinase inhibitor (the positive control kojic acid, IC50 = 24.8 μM) [171]. Subsequently, more than 20 MDAAs were reported to have significant inhibitory activity against tyrosinase (Table 8) [19, 21, 32, 36, 70, 172, 173].
3.9 Antiviral activity
According to the anti-HBV assay on the HepG 2.2.15 cell line in vitro, mulberrofuran G (31) exhibited moderate inhibitory activity against hepatitis B virus (HBV) DNA replication (IC50 = 3.99 μM) [66]. At non-toxic concentrations, kuwanon X (58) possessed prominent activities against herpes simplex virus type 1 and 2 (HSV-1 and HSV-2). The IC50 values of 58 to the tested strains HSV-1 (15,577), HSV-1 (clinical), and HSV-2 (333) were 2.2, 1.5 and 2.5 μg/mL, respectively. Mechanism studies revealed that 58 could inhibit HSV-1 adsorption and penetration, HSV-1 IE and L genes expression, viral DNA biosynthesis, and the HSV-induced nuclear factor (NF)-κB activation [174]. In the antiviral investigation of Morus spp. plant extracts, the antiviral activity against human coronavirus (HCoV 229E) of their common component kuwanon G (92) was also evaluated [175]. Kuwanon L (118) was found to have the inhibition of HIV-1 integrase (IN) catalytic activity in the absence and in the presence of LEDGF/p75 protein, and could inhibit the IN dimerization, the IN/LEDGF binding, as well as HIV-1 replication [176]. Further study suggested that kuwanon L (118) might exhibit its antiviral activity via binding to multiple viral targets, which may be a promising natural HIV-1 IN inhibitor [177]. Sanggenon G (124) had a certain inhibitory effect on influenza A virus (IC50 = 30.9 μM) [160].
3.10 Anti-cardiovascular diseases
Nomura et al. found some MDAAs such as mulberrofuran C (6), mulberrofuran G (31), mulberrofuran F (32), kuwanon G (92), kuwanon H (93), sanggenon D (128), and sanggenon C (130) had clear hypotensive effects [1, 2, 5, 33, 124, 129]. In an in vitro evaluation carried out by Nikaido et al., these compounds and several other MDAAs showed strong inhibitory activities against beef heart cAMP phosphodiesterase with IC50 values ranging 1.0 − 64.0 μM. Nikaido et al. proposed that the possible correlation between the mode of inhibition activity of these compounds against cAMP phosphodiesterase and their hypotensive effects deserves further study [178]. Liu et al. found that kuwanon G (92) could attenuate atherosclerosis by upregulation of LXRα-ABCA1/ABCG1 and inhibition of NF-κB activity in macrophages [179]. Gu et al. demonstrated that sanggenon C (130) exerted direct cytoprotective effects against hypoxia injury in cardiac cells via signaling mechanisms involving the activation of AMPK and concomitant inhibition of target of rapamycin (mTOR) and forkhead box O3a (FOXO3a) [180]. In the same year, Xiao et al. found that sanggenon C (130) could protect against cardiac hypertrophy and fibrosis via suppression of the calcineurin/NFAT2 pathway [181].
3.11 Other activities
Some MDAAs have also been found as potential inhibitors of disease-related enzymes, such as phosphodiesterase 1 (PDE1) inhibitors [chalcomoracin (5), mesozygin B (100), artonin I (101), mesozygin C (103), and mesozygin A (104)] [35], human carboxylesterase 2 (hCE2) inhibitors [kuwanon G (92), sanggenon D (128), and sanggenon C (130)] [182], pancreatic lipase inhibitors [kuwanon G (92) and sanggenon D (128)] [183], β-glucuronidase inhibitors [kuwanon G (92) and sanggenon C (130)] [184, 185], and 5α-reductase inhibitors [palodesangrens C − E (140 − 142)] [132]. Mulberrofuran C (6) and mulberrofuran G (31) exhibited potent hepatoprotective activity on t-BHP-induced oxidative stress in HepG2 cells, with EC50 values of 0.41 and 15.31 μM, respectively [44]. Sanggenons C and D (130 and 128) were identified as positive γ-aminobutyric acid type A (GABAA) receptor modulators [186].
4 Chemical and chemoenzymatic total syntheses of MDAAs
MDAAs exhibit kinds of structurally unique frameworks and a variety of promising bioactivities, and so these natural products have attracted extensive attention from synthetic chemists. Since the first report of total syntheses of several dehydroprenyl-2-arylbenzofuran type (Type A) MDAAs in 2010 [187], an amount of total syntheses of these molecules (including Types A − D and F − G) were disclosed over the past decade. Among all the synthetic strategies for MDAAs, the biomimetic intermolecular [4 + 2]-cycloaddition between a diene and a dienophile is the key step, which might be driven by Lewis acids, organocatalysts, and even enzymes. To the best of our knowledge, there have been no reviews focusing on the total synthesis of these MDAAs until now. In this section, we will cover all chemical and chemoenzymatic total syntheses of the MDAAs and their methyl ether derivatives.
4.1 Chemical total syntheses of dehydroprenyl-2-arylbenzofuran type MDAAs (Type A)
4.1.1 Total syntheses of ( ±)-mulberrofuran J hexamethyl ether, ( ±)-mongolicin F hexamethyl ether, ( ±)-chalcomoracin heptamethyl ether, and ( ±)-mulberrofuran C heptamethyl ether
In 2010, the Rizzacasa group reported firstly the racemic total syntheses of four methyl ether derivatives of dehydroprenyl-2-arylbenzofuran type MDAAs including mulberrofuran J hexamethyl ether (1a), mongolicin F hexamethyl ether (2a), chalcomoracin heptamethyl ether (5a), and mulberrofuran C heptamethyl ether (6a), in which the unit of dehydroprenyl diene was proved to be a challenging intermediate due to its unstable. This diene unit was established by using a Suzuki–Miyaura coupling as the key step, and it was used immediately after rapid purification. They also proved that the presence of the H-bonded phenol in the chalcone dienophile was essential for the success of the [4 + 2]-cycloaddition [187].
As outlined in Scheme 2, the authors started the preparation of the chalcone dienophile S3 from ketone S1 and aldehyde S2 via Claisen-Schmidt condensation. As for the synthesis of dienophile S5, chalcone S3 was prenylated under standard conditions and the resultant prenyl ether S4 was subjected to a Florisil promoted [1, 3]-sigmatropic rearrangement to afford the prenylated chalcone S5 in 28% yield. Except for the [1, 3]-rearranged product, the corresponding [1, 5]-rearranged isomer (not shown) as well as S3 were also produced in a significant amount.

Syntheses of mulberrofuran J hexamethyl ether (1a), mongolicin F hexamethyl ether (2a), chalcomoracin heptamethyl ether (5a), and mulberrofuran C heptamethyl ether (6a) by the Rizzacasa group
With the use of Cs2CO3 rather than amine bases, Sonogashira coupling of S8 and phenylalkyne S7, prepared by Seyferth-Gilbert homologation of 4-iodo-3,5-dimethoxybenzaldehyde S6 with Bestman-Ohira reagent, successfully gave the bisphenylalkyne S9 in good yield. After methanolysis of the acetate alkyne S9, the resulting phenol S10 was subsequently subjected to cyclization into the benzofuran S11 in 82% yield using TBAF instead of gold or platinum catalysis. The challenging formation of the dehydroprenyl-2-arylbenzofuran diene S15 was achieved in excellent yield via the Suzuki–Miyaura coupling of iodide S11 and pinacolboronate S14 (preparation by simple hydroboration of enyne S13 with pinacolborane S12) after extensive experimentation.
The intermolecular Diels–Alder cycloaddition reaction between dienophile S3 and diene S15 succeeded to proceed at 180 °C in toluene in a sealed tube to give the exo and endo adducts, trans,trans-1a [the hexamethyl ether derivative of mulberrofuran J (1)] and cis,trans-S16, respectively, in a 1:1 ratio. Cycloaddition of S5 and S15 under the same conditions afforded a 1:2 ratio of the exo adduct mongolicin F hexamethyl ether (2a) and the endo adduct S17. Subsequently, methylation of S16 and S17 obtained the previously reported mulberrofuran C heptamethyl ether (6a) and the permethylation product of chalcomoracin (5), chalcomoracin heptamethyl ether (5a), respectively, which assisted to confirm the stereochemistry of the exo and endo adducts. Unfortunately, the authors did not obtain the natural products mulberrofuran J (1), mongolicin F (2), chalcomoracin (5), and mulberrofuran C (6) by deprotection of either their hexamethyl ether derivatives or heptamethyl ether ones.
Claisen-Schmidt condensation of aldehyde S2 and ketone S18 instead of S1 give the fully methylated dienophile S19, which failed to undergo clean cycloaddition with S15. This demonstrated that a H-bonded ortho OH substituent on the chalcone was critical for the success of the [4 + 2]-cycloaddition. Subsequent detailed studies including a computational investigation showed the acceleration of the cycloaddition reaction by the OH group arises both from the LUMO-lowering effect of the OH–carbonyl hydrogen bond and from better coplanarity between the diene and its aryl substituent in the transition structures [188]. Based on the Rizzacasa group's research, the subsequent synthesis of all chalcone dienophiles retained the presence of the free phenol in its C-2 position.
4.2 Chemical total syntheses of dehydroprenylstilbene type MDAAs (Type B)
4.2.1 Enantioselective total syntheses of kuwanon X, kuwanon Y, and kuwanol A
In 2014, the Lei group developed a new strategy to forge the desired cyclohexene core unit of dehydroprenylchalcone type MDAAs. This new strategy featured an asymmetric Diels−Alder cycloaddition, catalyzed by a chiral ligand/boron Lewis acid to construct the core structure (see Sect. 4.3.4). In 2016, the same group adopted this strategy to construct the cyclohexene moiety of dehydroprenylstilbene type MDAAs. The biosynthesis-inspired asymmetric Diels − Alder cycloaddition shows high exo selectivity, in which the ratio of exo/endo could up to 13:1. The implementation of this strategy allowed for the first asymmetric total syntheses of the natural products kuwanons X and Y (58 and 61) and kuwanol A (64) in 12 and 11 steps, respectively [189].
The synthesis of dienes S28 (acetyl-protection) and S31 (MOM-protection) started with the preparation of S25 from methyl 3,5-dihydroxy-benzoate S20 in high yield over five steps including aromatic C-H iodination, TBS protection, DIBAL reduction, alkane C–OH iodination, and Michaelis-Arbuzov reaction (Scheme 3). The acetyl protected diene S28 was obtained over four steps by the Horner-Wadsworth-Emmons reaction of S25 with S26 followed by deprotection of the silyl protecting groups and reprotection with acetyl groups, and then via the Suzuki–Miyaura reaction. The authors found that an additional step of acetyl reprotection of the crude mixture produced by the Suzuki reaction could improve the yield of diene S28. In a branch procedure, precursors S25 and S26 were subjected to the same Horner-Wadsworth-Emmons reaction and deprotection conditions, and then reprotected with MOM groups to produce iodide S30, which performed the Heck reaction with 2-methyl-but-3-en-2-ol followed by dehydration to provide the desired MOM protected diene S31.

Syntheses of dienes S28 and S31 by the Lei group
The critical factor for the symmetric Diels − Alder cycloaddition between diene S28 and the known dienophile S32 was to find a suitable chiral ligand. After screening of chiral boron ligands for catalytic [4 + 2] cycloaddition, two chiral ligands (S)-VAPOL and (R)-6,6′-dibromo-VANOL could be used directly for the enantioselective total synthesis of acetyl ether precursors of 58 and 61, respectively, due to their very high exo- or endo-selectivity and ee value. As shown in Scheme 4, (S)-VAPOL effectively catalyzed the cycloaddition with high exo selectivity (exo/endo = 13:1) and ee value (97%), while the chiral ligand (R)-6,6′-dibromo-VANOL gave a good endo-selectivity (exo/endo = 3.5:1) and satisfying ee value (96%). Deacetylation of the corresponding exo-S33 and endo-S34 in the presence of K2CO3 in the mixture of THF and MeOH furnished the natural products kuwanons X and Y (58 and 61), respectively. The biomimetic intramolecular ketalization of 58 and 61 was performed under the catalysis of sulfuric acid, but only 61 formed a ketalized product kuwanol A (64).

Syntheses of kuwanons X and Y (58 and 61) and kuwanol A (64) by the Lei group
The Lei group had further investigated whether the two acetyl groups near the dehydroprenyl group had obvious effect on the exo/endo stereoselectivity. As shown in Scheme 4, by using the MOM-protected diene S31 instead of the acetyl-protected diene S28 in the synthesis of kuwanol A (64) from the same dienophile S32 catalyzed by chiral (R)-VANOL-boron Lewis acid, the exo/endo stereoselectivity was changed from 5.3:1 to 1.2:1 without losing the enantioselectivity, but the total yield increased from 3.6% to 17.6% with one step shorter. Combined with other cases, the authors suggested that different substitutions in diene may contribute to different stereoselectivity in the asymmetric Diels − Alder cycloaddition.
4.2.2 Total syntheses of ( ±)-kuwanol E and ( ±)-kuwanon Y heptamethyl ether
Also in 2016, Iovine and coworkers completed their total syntheses of ( ±)-kuwanol E (62) and ( ±)-kuwanon Y heptamethyl ether (61a) via a convergent strategy in nine steps. The synthesis featured a Lewis acid-mediated biomimetic intermolecular Diels−Alder cycloaddition for creating a cyclohexene core unit from dienophiles S3 or S5 and dehydroprenylstilbene diene S46. Another key point in this synthesis was that its exo/endo diastereoselectivity was controlled by the reaction temperature [190].
The synthesis commenced with the preparation of dienophile S5 over several classical reported reactions involving Claisen-Schmidt condensation, prenylation, and sigmatropic rearrangement (Scheme 5). Different from the previous report, the use of montmorillonite K10 as the catalyst in [1, 3]-rearrangement could improve the yield of S5. As depicted in Scheme 5, the synthetic route towards the key intermediate S46 started with commercially available 4-bromo-3,5-dihydroxybenzoic acid S36, which proceeded via Fischer esterification and methyl protection to obtain bromide S38. For exploring the effects of different halogen atoms (Br or I) on the subsequent reactions, iodide S39 was obtained by aromatic Finkelstein iodination of bromide S38. Next, compounds S38 or S39 were reduced with LiAlH4, and then converted into benzyl bromides S42 or S43 using PBr3, which exhibited different yields over the two steps. The one-pot Arbuzov and Horner − Wadsworth − Emmons reactions of S42 or S43 with S2 were performed to install the stilbene halides S44 or S45, in which their yields differed by more than one time. After Suzuki − Miyaura coupling of S44 or S45 with boronate S14, diene S46 was obtained in equivalent yield from the two building blocks, respectively. Therefore, the use of bromine substituted substrates in these reactions would give a better combined yield of diene S46. In addition, compared with traditional ligands such as PPh3 or AsPh3, the use of S-Phos as a bulky and electron-rich ligand in this Suzuki−Miyaura coupling step was proved to be a key condition affecting the yield of the product.

Syntheses of ( ±)-kuwanol E (62) and ( ±)-kuwanon Y heptamethyl ether (61a) by Iovine and coworkers
The Diels − Alder cycloaddition of dienophile S5 and diene S46 in dry o-xylene with or without the catalyst BH3·THF at various temperatures was performed, which exhibited different reactivity and diastereoselectivity. For example, at low temperatures (25 and 50 °C), the cycloaddition reaction had no reactivity, but as the temperature increased to 100 °C, a mixture of the exo- and endo-adducts (S47 and S48) with a 1:4 ratio was obtained. However, when the temperature was further heated to 160 °C, it yielded S47 and S48 in the opposite ratio (4:1). Therefore, the production of the endo-isomers such as ( ±)-kuwanol E heptamethyl ether (S48) and ( ±)-kuwanon Y heptamethyl ether (61a) was carried out under the same conditions as shown in Scheme 5. Subsequently, ( ±)-kuwanol E (62) was obtained by demethylation of the corresponding endo-isomer S48 with BBr3 in DCM. In this study, the cleavage of the methoxy groups of the heptamethyl ether precursor of ( ±)-kuwanon Y was not attempted due to its unavailable as well as because of the completed total synthesis of kuwanon Y by Lei group.
4.3 Chemical total syntheses of dehydroprenylchalcone type MDAAs (Type C)
4.3.1 Total syntheses of ( ±)-dorsterone pentamethyl ether and ( ±)-kuwanon V pentamethyl ether
In 2011, the Rahman group reported the total synthesis of the pentamethyl ether derivatives 79a and 87a of dorsterone (79) and kuwanon V (87), respectively, via a Diels–Alder cycloaddition catalyzed by AgOTf/Bu4NBH4 [191]. The synthesis of diene S54 started with commercially available 1-(2,4-dihydroxyphenyl)ethanone (S49) that underwent iodination, methylation, and Claisen–Schmidt condensation with 4-methoxybenzaldehyde (S51) in three steps to yield S52 (Scheme 6). Next, Heck coupling of S52 with 2-methylbut-3-en-2-ol occurred under the treatment of Pd(OAc)2, (o-tolyl)3-P, and Et3N in DMF and the resultant S53 was dehydrated to afford diene S54 in the presence of AcCl/pyridine. The preparation of dienophile S58 was shown in Scheme 6. After the 2-methoxy group of the known chalcone S55 was selectively demethylated, at the position of this OH group, a prenyl was introduced under standard conditions. Next, the resulting prenyl ether S57 was subjected to a montmorillonite K10 promoted [1, 3]-rearrangement to afford the desired dienophile S58 in 45% yield. After screening of the reaction conditions, the key AgOTf/Bu4NBH4-catalyzed [4 + 2] cycloaddition of chalcones S54 and S58 occurred smoothly to deliver the exo- and endo-adducts 79a and 87a in 65% yield with a diastereomeric ratio of 2:3.

Syntheses of ( ±)-dorsterone pentamethyl ether (79a) and ( ±)-kuwanon V pentamethyl ether (87a) by the Rahman group
4.3.2 Total syntheses of ( ±)-kuwanon I heptamethyl ether and ( ±)-kuwanon J heptamethyl ether
In the studies of H-bond accelerated Diels–Alder cycloadditions of chalcones, in 2012 the Rizzacasa group had also completed the total syntheses of two heptamethyl ethers of the dehydroprenylchalcone type MDAAs to examine the cycloaddition reaction [188]. As shown in Scheme 7, the synthesis of the diene S61 started with the conversion of ketone S1 to iodide S59, which was then subjected to Claisen–Schmidt condensation with aldehyde S2 followed by methylation to afford a fully esterified chalcone S60. The subsequent Suzuki coupling with boronate S14 gave diene S61 in low yield, which underwent the [4 + 2]-cycloaddition reaction with the prepared dienophile S5 only to afford kuwanon I heptamethyl ether (78a) and the endo-isomer kuwanon J heptamethyl ether (81a) in a 1:1 ratio without the product resulting from the Diels–Alder reaction of diene S61 with itself as the dienophile. This result was also a proof to the importance of the H-bond in chalcone dienophile.

Syntheses of ( ±)-kuwanon I heptamethyl ether (78a) and ( ±)-kuwanon J heptamethyl ether (81a) by the Rizzacasa group
4.3.3 Asymmetric total syntheses of ( +)-brosimones A and B
In 2013, the Porco group described their asymmetric total syntheses of brosimone A (80) and brosimone B (77) through biomimetic dehydrogenative Diels–Alder cycloadditions. The key steps towards these target molecules involved Pt/C-cyclopentene or 2,3-dichloro-5,6-dicyano-1,4-benzoquinone (DDQ) to effect dehydrogenation of prenylchalcones in combination with silver nanoparticles (AgNPs) to catalyze subsequent Diels − Alder cycloaddition [192].
Brosimones A and B were homodimers derived from prenyl chalcone. Therefore, the synthesis started with the easily prepared acetophone S62 that underwent a two-step procedure to yield the benzyl-protected prenyl chalcone S65 (Scheme 8). Based on the model reaction established by the authors, the cycloadducts exo-S67 and endo-S66 with a ratio of 1.2:1 were obtained in 64% yield by dehydrogenative Diels–Alder cycloaddition/dimerization of S65 using the optimized Pt/C-AgNP conditions with cyclopentene as H2 scavenger. Next, hydrogenolysis of exo-S67 produced ( +)-brosimone B (77), whose structure was confirmed by X-ray crystal analysis of its methyl-protected derivative S68 produced by methylation using Me2SO4. As shown in Scheme 8, the precursor exo-exo S69 of brosimone A also could be accessed by dehydrogenative cycloaddition of exo-S67 under different conditions including changing the temperature. For example, in the presence of DDQ and AgNPs as catalyst in chlorobenzene (PhCl) solvent, exo-S67 was converted into the cycloadduct exo-endo-S71 (17% yield) and the DDQ adduct S72 (34% yield) at 90 °C. Both exo-endo-S71 and DDQ adduct S72 could be converted exclusively into exo-exo S69 under AgNP-promoted conditions at 130 °C in excellent yield. When the reaction temperature was increased to 130 °C, the dehydrogenative Diels–Alder cycloaddition of exo-S67 predominantly afforded exo-exo S69 in 62% yield in the presence of AgNPs. Finally, the hydrogenolysis product of exo-exo S69 was determined to be ( +)-brosimone A (80) on the basis of the X-ray structure of its methyl derivative S70.

Asymmetric total syntheses of ( +)-brosimones A and B (80 and 77) by the Porco group
4.3.4 Enantioselective biomimetic total syntheses of kuwanons I and J and brosimones A and B
In 2014, the Lei group reported the first enantioselective total syntheses of dehydroprenylchalcone type MDAAs kuwanon I (78), kuwanon J (81), brosimone A (80), and brosimone B (77) by a common intermediate based on a concise synthetic strategy. The key feature of the synthesis included a biosynthesis-inspired asymmetric Diels–Alder cycloaddition mediated by a chiral ligand/boron Lewis acid. Another important progress involved regioselective Schenck ene reaction, reduction, and dehydration to realize a biomimetic dehydrogenation for generation of the required diene precursor. Furthermore, a remarkable process involved a tandem inter-/intramolecular asymmetric Diels–Alder cycloaddition of a diene with itself as the dienophile [193].
As shown in Scheme 9, base-mediated Claisen-Schmidt condensation of the MOM-protected aldehyde S73 and ketone S74 readily produced 2' hydroxychalcone S75. The subsequent prenylation and sigmatropic rearrangement resulted in the formation of two isomers, para- and ortho-prenylated chalcones (S77 and S78). Next, the acetyl-protected dienophile S79 was obtained from the para-prenylated chalcone S77 in 33% yield by replacing MOM groups with acyl groups. After a brief optimization, the key visible-light-mediated regioselective Schenck ene reaction of S79 by using TPP as photosensitizer and MeOH as solvent occurred smoothly to deliver the tertiary allylic alcohol S80 and secondary allylic alcohol (not shown in Scheme 9) in 3.2:1 ratio. Then dehydration of S80 with SOCl2/DBU produced the diene or dienophile S81 in 68% yield. In a parallel procedure, deprotection of the MOM groups followed by reprotection of the ortho-isomer S78 with acetyl groups delivered the required dienophile triacetate S82 in 35% yield. Next, the visible-light-mediated regioselective Schenck ene reaction of S82 using Ru(bpy)3Cl2·6H2O and MeOH gave an excellent ratio for the tertiary alcohol S84, a better substrate for dehydration, which smoothly provided the diene S85 under SOCl2/DBU in 75% yield.

Syntheses of dienophiles (S79, S81, and S82) and dienes (S81 and S85) by the Lei group
With dienophiles S79, S81, and S82 and dienes S81 and S85 in hand, a series of asymmetric Diels–Alder reactions promoted by different ligand/boron Lewis acid for the synthesis of the target dehydroprenylchalcone type MDAAs were investigated. As depicted in Scheme 10, the preferred (S)-VANOL ligand catalyzed the cycloaddition of S79 and S85 to afford the exo- and endo-adducts (S86 and S87 with a 1:1.2 ratio) in 71% combined yield with excellent ee values for both. Similarly, the using of (S)-8,8’-dimethyl-VANOL was proved to be the best chiral ligand to obtain both exo-S88 and endo-S89 with good ee values. Final deprotection of the acetyl groups of exo-S86, exo-S87, and endo-S89 with K2CO3 as a base efficiently furnished the desired natural products brosimone B (77), kuwanon I (78), and kuwanon J (81), respectively, each in 70% yield.

Enantioselective total syntheses of brosimone B (77), kuwanon I (78), and kuwanon J (81) by the Lei group
It is worth mentioning that a one-pot inter-/intramolecular Diels–Alder cycloaddition cascade was smoothly occurred to afford the three expected products including exo,exo-S90 in 13% yield, endo,endo-S91 in 28%, and exo,endo-S92 in 20% yield under the condition of a small amount of excess (S)-VANOL ligand (Scheme 11). Next, deprotection of exo,exo-S90 under mild basic conditions efficiently gave the target natural product brosimone A (80) in 70% yield. In addition, after removing the acetyl groups of S91 and S92 followed by methylation, S94 and S96 with definite structures were obtained over two steps, respectively.

Enantioselective total synthesis of brosimone A (80) by the Lei group
4.4 Chemical total syntheses of dehydroprenylflavone type MDAAs (Type D)
4.4.1 Total syntheses of kuwanons G and H
In 2021, the Tang group reported a convergent route towards the total synthesis of two MDAAs named kuwanons G and H (92 and 93) with unique dehydroprenylflavone dienes. The key features of this approach included the use of Baker-Venkataraman rearrangement, alkylation of β-diketone, intramolecular cyclization, and Suzuki–Miyaura coupling to achieve the unstable dehydroprenylflavone diene [194].
As outlined in Scheme 12, the synthesis of the key intermediate diene S107 started with a selective methyl protection of 2′,4′,6′-trihydroxyacetophenone (S97). The addition of two methyls onto the acetophenone S97 was performed using (CH3O)2SO2 in acetone to give S98. The following regioselective iodination of S98 resulted in the formation of iodobenzene S99 in 92% yield. After acylation of S100 with thionyl chloride, the crude product benzoyl chloride S101 was directly conducted with S99 to form an acyloxy ketone, which was then converted into β-diketone S102 through a base-catalyzed Baker–Venkataraman rearrangement. Next, alkylation of β-diketone S102 with prenyl bromide gave the desired S104 and by-product S103, and the later could be effectively hydrolyzed to the former. The treatment of S104 with concentrated sulfuric acid in anhydrous ethanol enabled intramolecular cyclization onto the target cyclic product S106 in 42% yield as well as an ethylated by-product S105, which also could be further converted to S106 under acidic condition. With the aim of installing the diene moiety at C-8, the iodoflavonoid S106 was subjected to the Suzuki–Miyaura coupling reaction with the easily prepared S14 to dehydroprenylflavone diene S107 in 41% yield.

Syntheses of kuwanons G and H (92 and 93) by the Tang group
The chalcone dienophiles S3 and S5 were easily furnished by the established route. As shown in Scheme 12, thermal-mediated intramolecular Diels–Alder cycloaddition of diene (S107) and dienophiles (S3 and S5) in a sealed tube with toluene smoothly occurred to the endo- and exo-adducts with a ratio of 1:1. Deprotection of the corresponding exo-isomers ( ±)-kuwanon G heptamethyl ether (S110) and ( ±)-kuwanon H heptamethyl ether (S111) finally yielded ( ±)-kuwanons G (92) and H (93), respectively, which were separated by a chiral HPLC to obtain the two natural products (–)-kuwanon G and (–)-kuwanon H.
4.5 Chemical total syntheses of dehydroprenylsanggenonflavone type MDAAs (Type F)
4.5.1 Asymmetric total syntheses of sanggenons C and O
In 2016, the Porco group completed the asymmetric total syntheses of sanggenons C and O (130 and 131). The syntheses relied on a Lewis acid-promoted double Claisen rearrangement of a bis-allyloxyflavone to install the hydrobenzofuro[3,2-b]chromenone core structure of sanggenonflavone diene precursors, and a stereodivergent reaction of a racemic mixture (stereodivergent RRM) involving the B(OPh)3/BINOL complexes catalytic enantioselective [4 + 2] cycloaddition to furnish the target molecules [195].
The synthesis of the diene precursor S120 started with tetra-MOM group protection of the commercially available morin S112 (Scheme 13). Subsequent 5-allylation of the MOM-protected flavonoid S113 to afford the intermediate S114, which was transformed to S115 by a chemoselective deprotection of the 3-MOM group using NaI and a catalytic amount of aqueous HCl. After allylation of the C-3 free hydroxyl group of S115, the obtained product S116 was subjected to global deprotection to afford the desired bis-allyloxyflavone S117. After evaluation of a number of rare earth metal triflates for double rearrangement, it was found that Yb(OTf)3, in the presence of CH2Cl2/HFIP (4:1), was used for producing the desired hydrobenzofuro[3,2-b]chromenone core structure S118 in 72% yield. With S118 in hand, the authors carried out silylation and crossmetathesis to afford the tri-silyl-protected ( ±)-sanggenol F [( ±)-S119] in excellent yield. The prenyl product ( ±)-S119 was then treated with 2,3-dichloro-5,6-dicyano-1,4-benzoquinone (DDQ) in tetrahydrofuran (THF) to transform to chromene ( ±)-S120 in 73% yield.

Asymmetric total syntheses of sanggenons C and O (130 and 131) by the Porco group
The Porco group had applied two different strategies to construct the cyclohexene core unit. First, AgNPs-mediated intermolecular Diels–Alder cycloaddition of the TBS-protected diene precursor ( ±)-S120 with acetylated 2′-hydroxychalcone S32 smoothly occurred to yield a mixture of two endo cycloadducts and minimal production of exo diastereomers. The mixture of endo cycloadducts was sequentially treated with aqueous NaHCO3 and NEt3·3HF to yield a mixture of ( ±)-sanggenon C (130) and ( ±)-sanggenon O (131) in 36% combined yield over three steps. Next, in order to synthesize enantioenriched sanggenons C and O, a verified catalytic system to a stereodivergent RRM strategy was applied. Based on the model reaction, the asymmetric [4 + 2] cycloaddition of diene precursor ( ±)-S120 with dienophile S32 using B(OPh)3/(R)-BINOL as catalyst was carried out under the conditions of PhCF3. After sequential deprotection of both acetate and silyl protecting groups, the promising enantioselectivities sanggenon C (130, with 98% ee) and sanggenon O (131, with 93% ee) were obtained in 2:1 ratio.
4.6 Chemical total syntheses of dehydroprenylcoumarin type MDAAs (Type G)
4.6.1 Total syntheses of palodesangren B trimethyl ether and palodesangren D dimethyl ether
In 2019, the Ploypradith group reported the diastereoconvergent total synthesis of the palodesangrens B and D methyl ethers (139a and 141a), which were completed by installing the final 2H-pyran-2-one ring onto the tricyclic 9-methyl-6,7-diphenyl-6a,7,8,10a-tetrahydro-6H-benzo[c]chromene core constructed from appropriate chalcones and dienes. At the early stage of the synthetic route, the Diels–Alder reaction was utilized to assemble the cyclohexene moiety of the tricyclic core. Next, a novel diastereoconvergent LiAlH4-mediated isomerization of the mixture of exo- and endo-adducts to install the desired stereochemistry, and subsequent acid-mediated stereoselective cyclization was employed to set up the pyran ring. Finally, the formation of its 2H-pyran-2-one ring was achieved by four consecutive steps including the regioselective MgCl2-mediated Casnati−Skattebøl ortho-formylation of phenol, Wittig methylenation, acryloylation, and Ru(II)-catalyzed ring-closing metathesis. Overall, palodesangrens D dimethyl ether and B trimethyl ether were successfully obtained for the first time via this strategy over nine steps starting from the Diels−Alder reactions [196].
The synthesis commenced with the preparation of aryldiene S123 from bis-MOM-protected benzaldehyde S121 by the Claisen − Schmidt condensation and subsequent Wittig methylenation (Scheme 14). With diene S123 and known chalcone dienophile S124 in hand, their intermolecular Diels–Alder cycloaddition gave the corresponding cyclohexene S126 as a mixture of exo- and endo-adducts with a ratio of 1:1.4 in 63% yield. Subsequent acetyl group deprotection with LiAlH4 followed by methylation with MeI provided S128 in 89% yield over two steps. The ensuing LiAlH4-mediated isomerization of the exo- and endo-adducts mixture S128 afforded a single endo isomer, which was directly subjected to acid-mediated chroman cyclization to furnish the desired tricyclic core S130 in 63% yield over two steps. After a brief optimization of the Casnati−Skattebøl reaction conditions, the key ortho-formylation of S130 proceeded smoothly to deliver the desired S132 in 46% yield and a small amount of the byproduct S134 (9%) together with 29% recovery of S130. Next, Wittig methylenation of S132 with MePPh3Br occurred smoothly in the presence of LiHMDS, and the resultant styrene S136 was subjected to acryloylation to provide the styrene acrylate S138. Finally, ring-closing metathesis of compound S138 using Grubbs II as the catalyst in toluene led to the formation of the desired palodesangren D dimethyl ether (141a).

Synthesis of palodesangrens B and D methyl ethers (139a and 141a) by the Ploypradith group
Similarly, palodesangren B trimethyl ether (139a) could be readily synthesized from aryldiene S123 and known chalcone dienophile S125 by using the same reaction sequences as 141a. The relative configurations of 139a and 141a were the same as those of natrual MDAAs palodesangrens B and D (139 and 141).
4.7 Chemical total syntheses of simple or other dehydroprenylphenol type MDAAs (Type H)
4.7.1 Total synthesis of ( ±)-sorocenol B
In 2012, the Porco group developed a concise route towards the total synthesis of ( ±)-sorocenol B (149). This synthesis featured a silver nanoparticle (AgNP)-catalyzed Diels−Alder cycloaddition to form the cyclohexene core unit and a late-stage Pd(II)-catalyzed oxidative cyclization to install the requisite bicyclo[3.3.1] framework of sorocenol B [197].
The synthesis started with the preparation of chalcone S141 from the Claisen−Schmidt condensation between chromene S140 and benzaldehyde S73 with NaH as a preferred base in THF (Scheme 15). A hydrolysis of the MOM-protected S141 delivered a polyphenol, which then gave the acetyl-protected dienophile S142 under the presence of acetic anhydride. Next, the requisite diene S147 was prepared in four steps from resorcinol S143. After protection of S143 with MOMCl, the resulting MOM-ether S144 was subjected to a regioselective formylation to afford benzaldehyde S145 in 83% yield over two steps. Sequential a base-catalyzed aldol condensation of S145 with acetone followed by the Wittig olefination yielded the desired diene S147. The key Diels−Alder cycloaddition between dienophile S142 and diene S147 was then implemented by utilizing silica-supported silver nanoparticles (AgNP’s), which efficiently allowed the formation of the desired cycloadducts exo-S148 and endo-S149 in 90% combined yield with a 1:2 ratio of exo/endo diastereomers. By unmasking the acetyl-protected phenols of S149, the obtained S150 was submitted to an oxidative Wacker cyclization catalyzed by Pd(OAc)2 to construct the bicyclo[1, 3, 3] product. As a result, the desired S151 and its C-4 epimer S152 were produced with a ratio of 2:1 in 50% combined yield (Scheme 15). The total synthesis of ( ±)-sorocenol B (149) was fulfilled after hydrolysis of S151 using aqueous HCl in MeOH.

Synthesis of ( ±)-sorocenol B (149) by the Porco group
4.7.2 Total synthesis of ( ±)-morusalbanol A pentamethyl ether
In 2016, the Chee group described their biomimetic synthesis of morusalbanol A pentamethyl ether (148a). The key steps towards the target molecule included a hydrogen-bond-assisted Diels–Alder cycloaddition to build the cyclohexene unit, and a selective cleavage of the ortho-methyl ether of the resulting cycloadduct to allow the C3–C21 bond rotation for intramolecular cyclization of the oxabicyclic [3.3.1] core [198]. The construction of this core was based on the previous research on its model by the same group [199].
The synthesis started with commercially available 2,4,6-trihydroxybenzoic acid S153 that underwent esterification and methyl ether protection to yield ester S154 in 75% yield (Scheme 16). Regioselective iodination of S154 followed by methylation of the remaining ortho-hydroxy group with iodomethane gave permethylated benzoic ester S156. Next, the Heck coupling of S156 with 2-methylbut-3-en-2-ol proceeded best with Pd(OAc)2 as a catalyst to form allylic alcohol S157 in 85% yield. Upon dehydration of the allylic alcohol with acetyl chloride/pyridine, the desired diene S158 was obtained in 90% yield. The synthesis of dienophile S3 in a higher yield was completed by the Claisen–Schmidt condensation of S1 with S2 using NaH as the base in THF instead of conventional KOH/MeOH conditions. The thermal Diels–Alder reaction of dienophile S3 with diene S158 in toluene at 135 °C afforded the endo-adduct S160 and exo-adduct S159 with a ratio of 3:2 in 55% combined yield. Finally, as shown in Scheme 16, the selective cleavage of the ortho-methyl ether of endo-S160 was achieved under the treatment of MgI2 and Et2O/THF, and subsequent intramolecular cyclization yielded ( ±)-morusalbanol A pentamethyl ether (148a) in a stereocontrolled manner in 50% yield.

Synthesis of ( ±)-morusalbanol A pentamethyl ether (148a) by the Chee group
4.8 Chemoenzymatic total syntheses of MDAAs (Types A − D)
4.8.1 MaDA-mediated chemoenzymatic total syntheses of chalcomoracin, 18″-O-methychalcomoracin, guangsangon E, kuwanol E, kuwanon J, and deoxyartonin I
In 2020, Lei and coworkers first reported the chemoenzymatic total syntheses of several MDAAs including chalcomoracin (5), 18″-O-methychalcomoracin (5b), guangsangon E (56), kuwanol E (62), kuwanon J (81), and deoxyartonin I (102). The synthesis featured an enzymatic intermolecular [4 + 2] cycloaddition of different substrates to produce the natural MDAAs and their derivates with a high efficiency and enantioselectivity. Morus alba Diels–Alderase (MaDA), a flavin adenine dinucleotide-dependent enzyme identified from Morus cell cultures, was the enzyme functionally responsible for the cycloaddition reaction in the biosynthesis of MDAAs [117].
As depicted in detail in Schemes 17 and 18, the key building blocks, two main dienophiles (S166 and 168) and different dienes (S176, S182, and S186 − S188), were all obtained by chemical synthesis. For the synthesis of prenyl chalcones morachalcone A (S166) and 4′-methylmorachalcone A (S168), the intermediate ketone S163 prepared from S161 over two steps was subjected to Claisen-Schmidt condensation with aldehydes S64 and S164, respectively, to obtain the benzyl-protected precursors S165 and S167. Benzyl group deprotection finally yielded the two dienophiles S166 and 168. As shown in Scheme 18, the phosphonium salts S173 and S178 were prepared from the same known compound 4-acetoxy-2-hydroxybenzyl acetate S172 over one and two steps, respectively. The NIS-mediated iodination of 3,5-dihydroxybenzoic acid S169 followed by acetyl protection provided iodide S170 in 72% yield over two steps, which was acylated with thionyl chloride to give S171. Next, the preparation of 2-arylbenzofuran iodides (S174 and S179) were achieved by coupling phosphonium salts (S173 and S178) with acyl chlorides (S171 and the resultant of acylation of S177 with oxalyl chloride) followed by intramolecular Wittig reaction. Suzuki–Miyaura coupling of 2-arylbenzofuran iodide S174 with diene borate S14 followed by an additional acetyl reprotection allowed the formation of acetyl-protected precursor S175 in excellent yield. While Stille cross-coupling of 2-arylbenzofuran iodide S179 with the known diene stannane S180 led to the construction of acetyl-protected precursor S181 in 93% yield. Finally, global acetyl deprotection of S175 and S181 delivered the desired dienes S176 and S182, respectively. After phenol-directed ortho-iodination of the known flavone S183 using BTMA•ICl2, the resultant iodide S184 was subjected to Suzuki–Miyaura coupling with diene borate S14 followed by a further acetyl reprotection to afford acetyl-protected precursor S185 in 15% combined yield over three steps. Diene S186 was prepared from its precursor S185 by deprotection (Scheme 18), so were dienes S187 and S188 from their precursors S28 and S85, respectively (Scheme 19).

Syntheses of dienophiles (S166 and 168) by the Lei group

Syntheses of dienes (S176, S182, and S186− S188) by the Lei group

MaDA-mediated chemoenzymatic total syntheses of chalcomoracin (5), 18″-O-methychalcomoracin (5b), guangsangon E (56), kuwanol E (62), kuwanon J (81), and deoxyartonin I (102) by the Lei group
As show in Scheme 19, during the final MaDA-mediated chemoenzymatic Diels–Alder cycloaddition, chalcone dienophiles was directly subjected to the corresponding mixture produced by deprotection of diene precursors to yield target MDAAs with high enantioselectivity. Therefore, natural MDAAs and their analogues chalcomoracin (5), 18″-O-methychalcomoracin (5b), guangsangon E (56), kuwanol E (62), kuwanon J (81), and deoxyartonin I (102) were obtained from the cycloaddition of S166 with S176, S168 with S176, S166 with S182, S166 with S187, S188 with S188, and S166 with S186, respectively. Their combined yields were 51%, 13%, 62%, 45%, 20%, and 47%, respectively.
4.8.2 MaDA-mediated chemoenzymatic total syntheses of artonin I and dideoxyartonin I
In 2020, the Lei group also completed the chemoenzymatic total syntheses of artonin I (101) and dideoxyartonin I (101a) with the MaDA catalyzed Diels–Alder reaction as the key step [200]. Based on their previous results, the authors embarked on the preparation of flavonoid dienes. With S196 as main target diene, the authors started the synthesis from the benzyl-protected S189 and S64. The formation of chalcone S190 through Claisen–Schmidt condensation reaction of ketone S189 with aldehyde S64 followed by iodinemediated oxidative cyclization resulted in flavonoid S191, which underwent debenzylation with BBr3 to afford flavonoid S192, Next, a selective acetylation was performed to yield S193, allowing for a phenol-directed ortho-iodination using BTMA•ICl2. The resulting iodide S194 (87% yield) was obtained in a regioselective manner. Stille cross-coupling reaction between iodide S194 and diene stannane S180 with Pd2(dba)3 as catalyst in the presence of AsPh3 in THF solvent delivered the desired diene precursor S195. After mild hydrolysis of S195, the product diene S196 directly underwent Diels–Alder reaction with S166 under the catalysis of MaDA to yield the natural product artonin I (101) with 99% ee in 90% yield over two steps (Scheme 20).

MaDA-mediated chemoenzymatic total syntheses of artonin I (101) and dideoxyartonin I (101a) by the Lei group
Starting from the known flavonoid S197, an iodine group was smoothly introduced through phenol-directed ortho-iodination. The resulting product S198 underwent Stille cross-coupling reaction followed by a protection to yield diene precursor S199. Diene S200 was prepared from its precursor S199 by hydrolysis and then used in the enzymatic Diels–Alder reaction. As expected, MaDA catalyzed the intermolecular [4 + 2] cycloaddition of S166 with S200 to afford dideoxyartonin I (101a) exclusively (Scheme 20).
5 Conclusions
This review has summarized structural classification, distribution, bioactivities, together with chemical and chemoenzymatic total syntheses of naturally occuring MDAAs. MDAAs are a group of structurally unique natural phenolic compounds, which are the most characteristic components of Morus plants of the family Moraceae. Over the past four decades, a total of 166 MDAAs including 150 classic (90%) and 16 non-classic (10%) ones have been described, of which dehydroprenyl-2-arylbenzofuran-, dehydroprenylstilbene-, dehydroprenylchalcone-, dehydroprenylflavone-, dehydroprenyldihydroflavone-, dehydroprenylsanggenonflavone-, dehydroprenylcoumarin-, and simple or other dehydroprenylphenol-types, are the eight subtypes (Types A − H) of the classic MDAAs with the same chalcone-skeleton dienophiles. Most MDAAs have many interesting biological properties, especially antineoplastic, anti-inflammation, antimicrobial, antioxidant, and antiviral activities, which may provide some candidates for finding the corresponding innovative drugs. In particular, several compounds such as chalcomoracin, mulberrofurans F, G, J, K, and Q, kuwanons G, H, and L, sanggenons C, D, and G, and albanol B were found to have diverse biological and pharmacological activities, indicating that they have the potential to be developed into multifunctional drugs. Except for the structures of dehydroprenyldihydroflavone type (Type E) and non-classic (Type I) MDAAs, all other types of MDAAs have several examples of chemical or MaDA-mediated chemoenzymatic total syntheses. From all reported synthesis examples, their synthetic strategies included three steps: syntheses of dienophiles, syntheses of dienes, and their biomimetic intermolecular Diels–Alder cycloaddition. Chalcone dienophiles could be obtained directly from known compounds or by a simple synthetic route. The challenge for the synthesis group was to build different diene blocks or to complete the enantioselective biomimetic Diels–Alder cycloaddition. Among them, the enzymatic intermolecular [4 + 2] cycloaddition to produce natural MDAAs was the most efficient and applied strategy with an excellent enantioselectivity and yield. We hope that our newly proposed classification of natural MDAAs and summary of recent total synthetic investigations on them will provide a useful reference for researchers in natural products chemistry and organic synthesis.
References
Nomura T, Fukai T, Kuwanon G. a new flavone derivative from the root barks of the cultivated mulberry tree (Morus alba L). Chem Pharm Bull. 1980;28:2548–52.
Nomura T, Fukai T, Narita T. Hypotensive constituent, kuwanon H, a new flavone derivative from the root bark of the cultivated mulberry tree (Morus alba L). Heterocycles. 1980;14:1943–51.
Takasugi M, Ishikawa S-I, Nagao S, Masamune T, Shirata A, Takahashi K. Albanins F and G, natural Diels-Alder adducts from mulberry. Chem Lett. 1980;9:1577–80.
Takasugi M, Nagao S, Masamune T, Shirata A, Takahashi K. Chalcomoracin, a natural Diels-Alder adduct from diseased mulberry. Chem Lett. 1980;9:1573–6.
Nomura T, Fukai T, Narita T, Terada S, Uzawa J, Iitaka Y, Takasugi M, Ishikawa S-I, Nagao S, Masamune T. Confirmation of the structures of kuwanons G and H (albanins F and G) by partial synthesis. Tetrahedron Lett. 1981;22:2195–8.
Nomura T, Hano Y. Isoprenoid-substituted phenolic compounds of moraceous plants. Nat Prod Rep. 1994;11:205–18.
Dai SJ, Lu ZM, Chen RY, Yu DQ. Structure and spectral characteristics of Diels-Alder type adducts from Morus. Acta Pharm Sin. 2005;40:876–81.
Yang Y, Tan Y-X, Chen R-Y, Kang J. The latest review on the polyphenols and their bioactivities of Chinese Morus plants. J Asian Nat Prod Res. 2014;16:690–702.
Wei H, Zhu JJ, Liu XQ, Feng WH, Wang ZM, Yan LH. Review of bioactive compounds from root barks of Morus plants (Sang-Bai-Pi) and their pharmacological effects. Cogent Chem. 2016;2:1212320.
Yan J, Ruan J, Huang P, Sun F, Zheng D, Zhang Y, Wang T. The structure-activity relationship review of the main bioactive constituents of Morus genus plants. J Nat Med. 2020;74:331–40.
Hirakura K, Hano Y, Fukai T, Nomura T, Uzawa JUN, Fukushima K. Structures of three new natural Diels-Alder type adducts, kuwanons P and X, and mulberrofuran J, from the cultivated mulberry tree (Morus lhou Koidz.). Chem Pharm Bull. 1985;33:1088–96.
Hano Y, Suzuki S, Nomura T, Iitaka Y. Absolute configuration of natural Diels-Alder type adducts from the Morus Root Bark. Heterocycles. 1988;27:2315–25.
Qin J, Fan M, He J, Wu XD, Peng LY, Su J, Cheng X, Li Y, Kong LM, Li RT, Zhao QS. New cytotoxic and anti-inflammatory compounds isolated from Morus alba L. Nat Prod Res. 2015;29:1711–8.
Huang QH, Lei C, Wang PP, Li JY, Li J, Hou AJ. Isoprenylated phenolic compounds with PTP1B inhibition from Morus alba. Fitoterapia. 2017;122:138–43.
Xia CL, Tang GH, Guo YQ, Xu YK, Huang ZS, Yin S. Mulberry Diels–Alder-type adducts from Morus alba as multi-targeted agents for Alzheimer’s disease. Phytochemistry. 2019;157:82–91.
Zhang QJ, Tang YB, Chen RY, Yu DQ. Three new cytotoxic Diels–Alder-type adducts from Morus australis. Chem Biodivers. 2007;4:1533–40.
Dai SJ, Ma ZB, Wu Y, Chen RY, Yu DQ. Guangsangons F-J, anti-oxidant and anti-inflammatory Diels-Alder type adducts, from Morus macroura Miq. Phytochemistry. 2004;65:3135–41.
Dai S, Chen R, Yu D. Diels-Alder type adducts of Morus macroura Miq. J China Pharm Univ. 2006;37:119–22.
Wang Y, Xu L, Gao W, Niu L, Huang C, Yang P, Hu X. Isoprenylated phenolic compounds from Morus macroura as potent tyrosinase inhibitors. Planta Med. 2018;84:336–43.
Kang J, Chen RY, Yu DQ. Diels-Alder type adducts in stem bark of Morus mongolica. Chin Tradit Herb Drugs. 2006;37:976–9.
Zheng ZP, Cheng KW, Zhu Q, Wang XC, Lin ZX, Wang M. Tyrosinase inhibitory constituents from the roots of Morus nigra: a structure-activity relationship study. J Agric Food Chem. 2010;58:5368–73.
Tan YX, Yan RY, Wang HQ, Chen RY, Yu DQ. Wittiorumins A – F, antioxidant Diels–Alder-type adducts from Morus wittiorum. Planta Med. 2009;75:249–55.
Cui XQ, Doctor Dissertation, Chinese Academy of Medical Sciences & Peking Union Medical College, 2008.
Cui XQ, Chen H, Chen RY. Study on Diels-Alder type adducts from stem barks of Morus yunanensis. China J Chin Mater Med. 2009;34:286–90.
Kang J, Chen RY, Yu DQ. Five new Diels-Alder type adducts from the stem and root bark of Morus mongolica. Planta Med. 2006;72:52–9.
Fitriani R, Happyana N, Hakim EH. Potential cytotoxic Diels-Alder type adducts from liquid medium of Morus alba var. shalun root cultures. Nat Prod Res. 2021;35:2274–8.
Kyekyeku JO, Kusari S, Adosraku RK, Zuhlke S, Spiteller M. Prenylated 2-arylbenzofuran derivatives with potent antioxidant properties from Chlorophora regia (Moraceae). Fitoterapia. 2016;108:41–7.
Ueda S, Nomura T, Fukai T, Matsumoto J, Kuwanon J. a new Diels-Alder adduct and chalcomoracin from callus culture of Morus alba L. Chem Pharm Bull. 1982;30:3042–5.
Yang Y, Wang HQ, Chen RY. Flavonoids from the leaves of Morus alba L. Acta Pharm Sin. 2010;45:77–81.
Kim YJ, Sohn MJ, Kim WG. Chalcomoracin and moracin C, new inhibitors of Staphylococcus aureus enoyl-acyl carrier protein reductase from Morus alba. Biol Pharm Bull. 2012;35:791–5.
Lee YG, Seo KH, Hong EK, Kim DM, Kim YE, Baek NI. Diels-Alder type adducts from the fruits of Morus alba L. J Appl Biol Chem. 2016;59:91–4.
Jeon YH, Choi SW. Isolation, identification, and quantification of tyrosinase and α-glucosidase inhibitors from UVC-irradiated mulberry (Morus alba L.) leaves. Prev Nutr Food Sci. 2019;24:84–94.
Nomura T, Fukai T, Matsumoto J, Ohmori T. Constituents of the cultivated mulberry tree VIII. Components of root barks of Morus bombycis. Planta Med. 1982;46:28–32.
Happyana N, Hakim EH, Syah VM, Kayser O, Juliawaty LD, Mujahidin D, Ermayanti TM, Achmad SA. Diels-Alder type adducts from hairy root cultures of Morus macroura. Nat Prod Sci. 2019;25:233–7.
Fozing CD, Ali Z, Ngadjui BT, Choudhary MI, Kapche GD, Abegaz BM, Khan IA. Phosphodiesterase I-inhibiting Diels-Alder adducts from the leaves of Morus mesozygia. Planta Med. 2012;78:154–9.
Hu X, Yu MH, Yan GR, Wang HY, Hou AJ, Lei C. Isoprenylated phenolic compounds with tyrosinase inhibition from Morus nigra. J Asian Nat Prod Res. 2018;20:488–93.
Mascarello A, Orbem Menegatti AC, Calcaterra A, Martins PGA, Chiaradia-Delatorre LD, D’Acquarica I, Ferrari F, Pau V, Sanna A, De Logu A, Botta M, Botta B, Terenzi H, Mori M. Naturally occurring Diels–Alder-type adducts from Morus nigra as potent inhibitors of Mycobacterium tuberculosis protein tyrosine phosphatase B. Eur J Med Chem. 2018;144:277–88.
Hu X, Ji J, Wang M, Wu JW, Zhao QS, Wang HY, Hou AJ. New isoprenylated flavonoids and adipogenesis-promoting constituents from Morus notabilis. Bioorg Med Chem Lett. 2011;21:4441–6.
Wang M, Gao LX, Wang J, Li JY, Yu MH, Li J, Hou AJ. Diels-Alder adducts with PTP1B inhibition from Morus notabilis. Phytochemistry. 2015;109:140–6.
Hano Y, Yamanaka J, Momose Y, Nomura T. Sorocenols C-F, four new isoprenylated phenols from the root bark of Sorocea bonplandii Baillon. Heterocycles. 1995;41:2811–21.
Ferrari F, Messana I. Prenylated flavanones from Sorocea ilicifolia. Phytochemistry. 1995;38:251–4.
Nomura T, Fukai T, Matsumoto J, Fukushima K, Momose Y. Structure of mulberrofuran C, a natural hypotensive Diels-Alder adduct from root barks of the cultivated mulberry tree (Morus bombycis Koidzumi). Heterocycles. 1981;16:759–65.
Cui L, Na M, Oh H, Bae EY, Jeong DG, Ryu SE, Kim S, Kim BY, Oh WK, Ahn JS. Protein tyrosine phosphatase 1B inhibitors from Morus root bark. Bioorg Med Chem Lett. 2006;16:1426–9.
Jung JW, Ko WM, Park JH, Seo KH, Oh EJ, Lee DY, Lee DS, Kim YC, Lim DW, Han D, Baek NI. Isoprenylated flavonoids from the root bark of Morus alba and their hepatoprotective and neuroprotective activities. Arch Pharm Res. 2015;38:2066–75.
Qi SZ, Li N, Tuo ZD, Li JL, Xing SS, Li BB, Zhang L, Lee HS, Chen JG, Cui L. Effects of Morus root bark extract and active constituents on blood lipids in hyperlipidemia rats. J Ethnopharmacol. 2016;180:54–9.
Ha MT, Seong SH, Nguyen TD, Cho WK, Ah KJ, Ma JY, Woo MH, Choi JS, Min BS. Chalcone derivatives from the root bark of Morus alba L. act as inhibitors of PTP1B and α-glucosidase. Phytochemistry. 2018;155:114–25.
Ueda S, Matsumoto J, Nomura T. Four natural Diels-Alder adducts mulberrofuran E, kuwanon Q, R, and V from callus culture of Morus alba L. Chem Pharm Bull. 1984;32:350–3.
Wang YF, Yu MH, Xu LJ, Niu LX, Huang CY, Xu H, Yang PM, Hu X. Diels-Alder type adducts with potent alpha-glucosidase inhibitory activity from Morus macroura. Phytochem Lett. 2018;26:149–53.
Tan YX, Liu C, Zhang T, Chen RY, Yu DQ. Bioactive constituents of Morus wittiorum. Phytochem Lett. 2010;3:57–61.
Cui XQ, Wang HQ, Liu C, Chen RY. Study of anti-oxidant phenolic compounds from stem barks of Morus yunanensis. China J Chin Mater Med. 2008;33:1569–72.
Su C, Duan Y, Tian J, Liu J, Xie K, Chen D, Ye F, Chen R, Dai J. Morusalisins A-F, six new Diels-Alder type adducts, as potential PTP1B inhibitors from cell cultures of Morus alba. Fitoterapia. 2020;146: 104682.
Basnet P, Kadota S, Terashima S, Shimizu M, Namba T. Two new 2-arylbenzofuran derivatives from hypoglycemic activity-bearing fractions of Morus insignis. Chem Pharm Bull. 1993;41:1238–43.
Hano Y, Nomura T, Ueda S. Two new Diels-Alder type adducts, mulberrofuran T and kuwanol E, from callus tissues of Morus alba L. Heterocycles. 1989;29:2035–41.
Hano Y, Kohno H, Itoh M, Nomura T. Structures of three new 2-arylbenzofuran derivatives from the Chinese crude drug “Sang-Bai-Pi” (Morus Root Bark). Chem Pharm Bull. 1985;33:5294–300.
Ferrari F, Filho VC, Cabras T, Messana I. Sorocein L and sorocein M: Two Diels-Alder type adducts from Sorocea ilicifolia. J Nat Prod. 2003;66:581–2.
Hano Y, Fukai T, Nomura T, Uzawa J, Fukushima K. Structure of mulberrofuran I, a novel 2-arylbenzofuran derivative from the cultivated mulberry tree (Morus bombycis Koidz). Chem Pharm Bull. 1984;32:1260–3.
Hano Y, Miyagawa Y, Yano M, Nomura T. Correlation between albanol B and mulberrofuran I, and structure of mulberrofuran S, novel 2-arylbenzofuran derivative. Heterocycles. 1989;28:745–50.
Hano Y, Tsubura H, Nomura T. Structure of mulberrofuran Q, a novel 2-arylbenzofuran derivative from the cultivated mulberry tree (Morus alba L.). Heterocycles. 1986;24:1807–13.
Kikuchi T, Nihei M, Nagai H, Fukushi H, Tabata K, Suzuki T, Akihisa T. Albanol A from the root bark of Morus alba L. induces apoptotic cell death in HL60 human leukemia cell line. Chem Pharm Bull. 2010;58:568–71.
Lee HJ, da Lyu H, Koo U, Lee SJ, Hong SS, Kim K, Kim KH, Lee D, Mar W. Inhibitory effect of 2-arylbenzofurans from the Mori Cortex Radicis (Moraceae) on oxygen glucose deprivation (OGD)-induced cell death of SH-SY5Y cells. Arch Pharm Res. 2011;34:1373–80.
Zhao Y, Kongstad KT, Jager AK, Nielsen J, Staerk D. Quadruple high-resolution α-glucosidase/α-amylase/PTP1B/radical scavenging profiling combined with high-performance liquid chromatography-high-resolution mass spectrometry-solid-phase extraction-nuclear magnetic resonance spectroscopy for identification of antidiabetic constituents in crude root bark of Morus alba L. J Chromatogr A. 2018;1556:55–63.
Zhu M, Wang ZJ, He YJ, Qin Y, Zhou Y, Qi ZH, Zhou ZS, Zhu YY, Jin DN, Chen SS, Luo XD. Bioguided isolation, identification and bioactivity evaluation of anti-MRSA constituents from Morus alba Linn. J Ethnopharmacol. 2021;281: 114542.
Rama Rao AV, Deshpande VH, Shastri RK, Tavale SS, Dhaneshwar NN. Structures of albanols A and B, two novel phenols from Morus alba bark. Tetrahedron Lett. 1983;24:3013–6.
Fukai T, Hano Y, Hirakura K, Nomura T, Uzawa J, Fukushima K. Structures of mulberrofurans F and G, two natural hypotensive Diels-Alder type adducts from the cultivated mulberry tree (Morus lhou (Ser.) Koidz). Heterocycles. 1984;22:473–7.
Fukai T, Hano Y, Hirakura K, Nomura T, Uzawa J, Fukushima K. Structures of two natural hypotensive Diels-Alder type adducts, mulberrofurans F and G, from the cultivated mulberry tree (Morus lhou Koidz). Chem Pharm Bull. 1985;33:3195–204.
Geng CA, Ma YB, Zhang XM, Yao SY, Xue DQ, Zhang RP, Chen JJ. Mulberrofuran G and isomulberrofuran G from Morus alba L.: anti-hepatitis B virus activity and mass spectrometric fragmentation. J Agric Food Chem. 2012;60:8197–202.
Kuk EB, Jo AR, Oh SI, Sohn HS, Seong SH, Roy A, Choi JS, Jung HA. Anti-Alzheimer’s disease activity of compounds from the root bark of Morus alba L. Arch Pharm Res. 2017;40:338–49.
Li M, Wu X, Wang X, Shen T, Ren D. Two novel compounds from the root bark of Morus alba L. Nat Prod Res. 2018;32:36–42.
Wang Z, Li X, Chen M, Liu F, Han C, Kong L, Luo J. A strategy for screening of α-glucosidase inhibitors from Morus alba root bark based on the ligand fishing combined with high-performance liquid chromatography mass spectrometer and molecular docking. Talanta. 2018;180:337–45.
Zheng ZP, Tan HY, Wang M. Tyrosinase inhibition constituents from the roots of Morus australis. Fitoterapia. 2012;83:1008–13.
Dat NT, Jin X, Lee K, Hong YS, Kim YH, Lee JJ. Hypoxia-inducible factor-1 inhibitory benzofurans and chalcone-derived Diels-Alder adducts from Morus species. J Nat Prod. 2009;72:39–43.
Hong S, Kwon J, Kim DW, Lee HJ, Lee D, Mar W. Mulberrofuran G protects ischemic injury-induced cell death via inhibition of NOX4-mediated ROS generation and ER stress. Phytother Res. 2017;31:321–9.
Shim SY, Sung SH, Lee M. Anti-inflammatory activity of mulberrofuran K isolated from the bark of Morus bombycis. Int Immunopharmacol. 2018;58:117–24.
Dai SJ, Wu Y, Wang YH, He WY, Chen RY, Yu DQ. New Diels-Alder type adducts from Morus macroura and their anti-oxidant activities. Chem Pharm Bull. 2004;52:1190–3.
Heger V, Benesova B, Viskupicova J, Majekova M, Zoofishan Z, Hunyadi A, Horakova L. Phenolic compounds from Morus nigra regulate viability and apoptosis of pancreatic β-cells possibly via SERCA activity. ACS Med Chem Lett. 2020;11:1006–13.
Abdel Bar FM, Abbas GM, Gohar AA, Lahloub MFI. Antiproliferative activity of stilbene derivatives and other constituents from the stem bark of Morus nigra L. Nat Prod Res. 2019;34:3506–13.
Fitriani R, Happyana N, Hakim EH. Three new Diels-Alder type adducts from root cultures media of Morus alba var. shalun. Res J Chem Environ. 2019;23:96–102.
Kang J, Cui XQ, Wang HQ, Yu DQ, Chen RY. Two new Diels–Alder-type adducts from the stem barks of Morus yunanensis. J Asian Nat Prod Res. 2014;16:617–22.
Cui XQ, Wang L, Yan RY, Tan YX, Chen RY, Yu DQ. A new Diels-Alder type adduct and two new flavones from the stem bark of Morus yunanensis Koidz. J Asian Nat Prod Res. 2008;10:315–8.
Hano Y, Nomura T. Structure of mulberrofuran P, a novel 2-arylbenzofuran derivative from the cultivated mulberry tree (Morus alba L.). Heterocycles. 1986;24:1381–6.
Wu YX, Kim YJ, Kwon TH, Tan CP, Son KH, Kim T. Anti-inflammatory effects of mulberry (Morus alba L.) root bark and its active compounds. Nat Prod Res. 2018;34:1786–90.
Culenova M, Sychrova A, Hassan STS, Berchova-Bimova K, Svobodova P, Helclova A, Michnova H, Hosek J, Vasilev H, Suchy P, Kuzminova G, Svajdlenka E, Gajdziok J, Cizek A, Suchy V, Smejkal K. Multiple In vitro biological effects of phenolic compounds from Morus alba root bark. J Ethnopharmacol. 2020;248: 112296.
Sun JY, Hano Y, Nomura T. Constituents of the cultivated mulberry tree. Part XL. The structure of sanggenon Q, a new Diels-Alder type adduct from Morus mongolica Schneider. Heterocycles. 1989;29:195–202.
Takasugi M, Ishikawa SI, Nagao S, Masamune T. Albafuran C, a natural Diels-Alder adduct of a dehydroprenyl-2-phenylbenzofuran with a chalcone from mulberry. Chem Lett. 1982;11:1223–4.
Dai SJ, Mi ZM, Ma ZB, Li S, Chen RY, Yu DQ. Bioactive Diels-Alder type adducts from the stem bark of Morus macroura. Planta Med. 2004;70:758–63.
Hano Y, Tsubura H, Nomura T. Structures of kuwanons Y and Z, two new stilbene derivatives from the cultivated mulberry tree (Morus alba L.). Heterocycles. 1986;24:2603–10.
Hano Y, Yamanaka J, Nomura T, Momose Y. Constituents of the Moraceae plants. 23. Sorocenols A and B, two new isoprenylated phenols from the root bark of Sorocea bonplandii Baillon. Heterocycles. 1995;41:1035–43.
Hano Y, Itoh M, Nomura T. Structures of kuwanols A and B, two novel stilbene derivatives from the cultivated mulberry tree (Morus bombycis Koidz). Heterocycles. 1985;23:819–24.
Ferrari F, Delle MF. Sorocein I, a new Diels-Alder type adduct from Sorocea ilicifolia. Fitoterapia. 2001;72:301–3.
Messana I, Ferrari F, Delle Monache F, Yunes RA, Calixto JB, Bisognin T. Three new Diels-Alder type adducts from the roots of Sorocea bonplandii Baillon. Heterocycles. 1991;32:1287–96.
Zhang QJ, Ni G, Wang YH, Chen RY, Yu DQ. Three new Diels-Alder type adducts from the stem bark of Morus cathayana. J Asian Nat Prod Res. 2009;11:267–73.
Hano Y, Takizawa S, Mizuno E, Nomura T. Structure of kuwanon P, a new Diels-Alder type adduct from the root bark of the cultivated mulberry tree (Morus lhou (Ser.) Koidz). Chem Pharm Bull. 1983;31:2936–9.
Messana I, Ferrari F, Francisco de Mello J, Mesquita de Araujo MDC. Constituents of Brosimopsis oblongifolia. 4. Structures of two new Diels–Alder type adducts, brosimone B and brosimone D. Heterocycles. 1989;29:683–90.
Nomura T, Fukai T, Matsumoto J, Imashimizu A, Terada S, Hama M. Constituents of the cultivated mulberry tree X. Structure of kuwanon I, a new natural Diels-Alder adduct from the root bark of Morus alba. Planta Med. 1982;46:167–74.
Tsopmo A, Tene M, Kamnaing P, Ayafor JF, Sterner O. A new Diels−Alder-type adduct flavonoid from Dorstenia barteri. J Nat Prod. 1999;62:1432–4.
Messana I, Ferrari F, De Araujo MDCM. Constituents of Brosimopsis oblongifolia. 3 Structure of a new Diels-Alder type adduct, brosimone A. Tetrahedron. 1988;44:6693–8.
Duong TH, Nguyen HT, Nguyen CH, Tran NMA, Danova A, Tran TMD, Vu-Huynh KL, Musa V, Jutakanoke R, Nguyen NH, Sichaem J. Identification of highly potent α-glucosidase inhibitors from Artocarpus integer and molecular docking studies. Chem Biodivers. 2021;18: e202100499.
Hoang DM, Ngoc TM, Dat NT, Ha DT, Kim YH, Luong HV, Ahn JS, Bae KH. Protein tyrosine phosphatase 1B inhibitors isolated from Morus bombycis. Bioorg Med Chem Lett. 2009;19:6759–61.
Messana I, Ferrari F, Delle Monache F, Yunes RA, Gacs-Baitz E. Three new flavanone derivatives from the root bark of Sorocea bonplandii Baillon. Heterocycles. 1994;38:1287–97.
Phung TXB, Tran THH, Dan TTH, Chau VM, Hoang TH, Nguyen TD. Chalcone-derived Diels-Alder adducts as NF-κB inhibitors from Morus alba. J Asian Nat Prod Res. 2012;14:596–600.
Hano Y, Aida M, Nomura T. Two new natural Diels–Alder-type adducts from the root bark of Artocarpus heterophyllus. J Nat Prod. 1990;53:391–5.
Shinomiya K, Aida M, Hano Y, Nomura T. A Diels–Alder-type adduct from Artocarpus heterophyllus. Phytochemistry. 1995;40:1317–9.
Kong SY, Park MH, Lee M, Kim JO, Lee HR, Han BW, Svendsen CN, Sung SH, Kim HJ. Kuwanon V inhibits proliferation, promotes cell survival and increases neurogenesis of neural stem cells. PLoS ONE. 2015;10: e0118188.
Kang J, Chen R-Y, Yu D-Q. A new Diels-Alder type adduct and a new flavone from the stem and root bark of Morus mongolica. Chin Chem Lett. 2005;16:1474–6.
Nomura T, Fukai T, Sato E, Fukushima K. The formation of moracenin D from kuwanon G. Heterocycles. 1981;16:983–6.
Park KM, You JS, Lee HY, Baek NI, Hwang JK. Kuwanon G: an antibacterial agent from the root bark of Morus alba against oral pathogens. J Ethnopharmacol. 2003;84:181–5.
Yang L, Zhao F, Zhang T, Guo F, Lu H. Isolation of active compounds from Cortex Mori and its mechanism on anti-inflammatory. Chin Arch Tradit Chin Med. 2016;34:3008–12.
Wu SC, Han F, Song MR, Chen S, Li Q, Zhang Q, Zhu K, Shen JZ. Natural flavones from Morus alba against methicillin-resistant Staphylococcus aureus via targeting the proton motive force and membrane permeability. J Agric Food Chem. 2019;67:10222–34.
Oshima Y, Konno C, Hikino H, Matsushita K. Structure of moracenin B, a hypotensive principle of Morus root barks. Tetrahedron Lett. 1980;21:3381–4.
Hano Y, Hirakura K, Nomura T, Terada S, Fukushima K. Components of root bark of Morus lhou 1. Structures of two new natural Diels-Alder adducts, kuwanons N and O. Planta Med. 1984;50:127–30.
Zuo GY, Yang CX, Ruan ZJ, Han J, Wang GC. Potent anti-MRSA activity and synergism with aminoglycosides by flavonoid derivatives from the root barks of Morus alba, a traditional Chinese medicine. Med Chem Res. 2019;28:1547–56.
Oshima Y, Konno C, Hikino H, Matsushita K. Structure of moracenin A, a hypotensive principle of Morus root barks. Heterocycles. 1980;14:1287–90.
Hirakura K, Fukai T, Hano Y, Nomura T, Kuwanon W. a natural Diels-Alder type adduct from the root bark of Morus lhou. Phytochemistry. 1985;24:159–61.
Ferrari F, Delle Monache F, Suarez AI, Compagnone RS. Multicaulisin, a new Diels-Alder type adduct from Morus multicaulis. Fitoterapia. 2000;71:213–5.
Hano Y, Aida M, Nomura T, Ueda S. A novel way of determining the structure of artonin I, an optically active Diels-Alder type adduct, with the aid of an enzyme system of Morus alba cell cultures. J Chem Soc Chem Commun. 1992;17:1177–8.
Farooq S, Wahab AT, Fozing CDA, Rahman AU, Choudhary MI. Artonin I inhibits multidrug resistance in Staphylococcus aureus and potentiates the action of inactive antibiotics in vitro. J Appl Microbiol. 2014;117:996–1011.
Gao L, Su C, Du X, Wang R, Chen S, Zhou Y, Liu C, Liu X, Tian R, Zhang L, Xie K, Chen S, Guo Q, Guo L, Hano Y, Shimazaki M, Minami A, Oikawa H, Huang N, Houk KN, Huang L, Dai J, Lei X. FAD-dependent enzyme-catalysed intermolecular [4+2] cycloaddition in natural product biosynthesis. Nat Chem. 2020;12:620–8.
Nomura T, Fukai T, Hano Y, Nemoto K, Terada S, Kuramochi T. Constituents of cultivated mulberry tree XII. Isolation of two new natural Diels-Alder adducts from root bark of Morus alba. Planta Med. 1983;47:151–6.
Dai SJ, Yu DQ. A new anti-oxidant Diels-Alder type adduct from Morus macroura. Nat Prod Res. 2006;20:676–9.
Rollinger JM, Spitaler R, Menz M, Marschall K, Zelger R, Ellmerer EP, Schneider P, Stuppner H. Venturia inaequalis-inhibiting Diels-Alder adducts from Morus root bark. J Agric Food Chem. 2006;54:8432–6.
Fukai T, Hano Y, Fujimoto T, Nomura T. Structure of sanggenon G, a new Diels-Alder adduct from the Chinese crude drug “Sang Bai Pi” (Morus Root Barks). Heterocycles. 1983;20:611–5.
Shi YQ, Fukai T, Sakagami H, Chang WJ, Yang PQ, Wang FP, Nomura T. Cytotoxic flavonoids with isoprenoid groups from Morus mongolica. J Nat Prod. 2001;64:181–8.
Hano Y, Ichikawa K, Okuyama M, Yamanaka J, Miyoshi T, Nomura T, Sanggenons R. S, and T, three new isoprenylated phenols from the Chinese crude drug “Sang-Bai-Pi” (Morus Root Bark). Heterocycles. 1995;40:953–65.
Nomura T, Fukai T, Hano Y, Uzawa J. Structure of sanggenon D, a natural hypotensive Diels-Alder adduct from Chinese crude drug “Sang-Bai-Pi” (Morus Root Barks). Heterocycles. 1982;17:381–9.
Fukai T, Pei YH, Nomura T, Xu CQ, Wu LJ, Chen YJ. Isoprenylated flavanones from Morus cathayana. Phytochemistry. 1998;47:273–80.
Hano Y, Kohno H, Suzuki S, Nomura T. Structures of sanggenons E and P, two new Diels–Alder-type adducts from the Chinese crude drug Sang-Bai-Pi (Morus Root Bark). Heterocycles. 1986;24:2285–91.
Rollinger JM, Bodensieck A, Seger C, Ellmerer EP, Bauer R, Langer T, Stuppner H. Discovering COX-inhibiting constituents of Morus root bark: Activity-guided versus computer-aided methods. Planta Med. 2005;71:399–405.
Zelová H, Hanáková Z, Čermáková Z, Šmejkal K, Dalĺ Acqua S, Babula P, Cvačka J, Hošek J. Evaluation of anti-inflammatory activity of prenylated substances isolated from Morus alba and Morus nigra. J Nat Prod. 2014;77:1297–303.
Nomura T, Fukai T, Hano Y, Uzawa J. Structure of sanggenon C, a natural hypotensive Diels-Alder adduct from Chinese crude drug “Sang Bai-Pi” (Morus Root Barks). Heterocycles. 1981;16:2141–8.
Shen RC, Lin M. Diels-Alder type adducts from Morus cathayana. Phytochemistry. 2001;57:1231–5.
Hano Y, Nomura T. Structure of sanggenon O, a natural Diels-Alder type adduct from the Chinese crude drug “Sang-Bai-Pi” (Morus Root Bark). Heterocycles. 1985;23:2499–503.
Shirota O, Takizawa K, Sekita S, Satake M, Hirayama Y, Hakamata Y, Hayashi T, Yanagawa T. Antiandrogenic natural Diels-Alder-type adducts from Brosimum rubescens. J Nat Prod. 1997;60:997–1002.
Shirota O, Sekita S, Hirayama Y, Hakamata Y, Hayashi T, Yanagawa T, Satake M. Two chalcone-prenylcoumarin Diels-Alder adducts from Brosimum rubescens. Phytochemistry. 1998;47:1381–5.
Chen HD, Ding YQ, Yang SP, Li XC, Wang XJ, Zhang HY, Ferreira D, Yue JM. Morusalbanol A, a neuro-protective Diels-Alder adduct with an unprecedented architecture from Morus alba. Tetrahedron. 2012;68:6054–8.
Nomura T, Fukai T, Hano Y, Ikuta H, Kuwanon M. a new Diels-Alder adduct from the root barks of the cultivated mulberry tree (Morus lhou (Ser.) Koidz.). Heterocycles. 1983;20:585–91.
Takasugi M, Nagao S, Masamune T. Structure of dimoracin, a new natural Diels-Alder adduct from diseased mulberry. Chem Lett. 1982;11:1217–20.
Su C, Tao X, Yin Z, Zhang X, Tian J, Chen R, Liu J, Li L, Ye F, Zhang PC, Zhang D, Dai J. Morusalones A-D, Diels-Alder adducts with 6/7/6/6/6/6 hexacyclic ring systems as potential PTP1B inhibitors from cell cultures of Morus alba. Org Lett. 2019;21:9463–7.
Fukai T, Hano Y, Hirakura K, Nomura T, Uzawa JUN. Structures of a novel 2-arylbenzofuran derivative and two flavone derivatives from the cultivated mulberry tree (Morus lhou Koidz). Chem Pharm Bull. 1985;33:4288–95.
Kohno H, Takaba K, Fukai T, Nomura T. Constituents of the cultivated mulberry tree. Part XXXIX. Structure of mulberrofuran R, a novel 2-arylbenzofuran derivative from the cultivated mulberry tree (Morus lhou Koidz). Heterocycles. 1987;26:759–62.
Nomura T, Fukai T, Hano Y, Urano S. Constituents of the Chinese crude drug “Sang Bai Pi” (Morus Root Bark). II. Structure of a new flavanone derivative, sanggenon B. Planta Med. 1983;47:95–9.
Han H, Chou C-C, Li R, Liu J, Zhang L, Zhu W, Hu J, Yang B, Tian J. Chalcomoracin is a potent anticancer agent acting through triggering oxidative stress via a mitophagy- and paraptosis-dependent mechanism. Sci Rep. 2018;8:9566.
Zhang SR, Zhang XC, Liang JF, Fang HM, Huang HX, Zhao YY, Chen XQ, Ma SL. Chalcomoracin inhibits cell proliferation and increases sensitivity to radiotherapy in human non-small cell lung cancer cells via inducing endoplasmic reticulum stress-mediated paraptosis. Acta Pharmacol Sin. 2020;41:825–34.
Phan TN, Kim O, Ha MT, Hwangbo C, Min BS, Lee JH. Albanol B from mulberries exerts anti-cancer effect through mitochondria ROS production in lung cancer cells and suppresses in vivo tumor growth. Int J Mol Sci. 2020;21:9502.
Shu YH, Yuan HH, Xu MT, Gao CC, Wu ZP, Han HT, Li SX, Tian JK, Shu YH, Zhang JB, Hong YT, Sun X, Yang SF, Gao RL, Li SX, Tian JK. A novel Diels-Alder adduct of mulberry leaves exerts anticancer effect through autophagy-mediated cell death. Acta Pharmacol Sin. 2021;42:780–90.
Park JE, Jung JH, Lee HJ, Sim DY, Im E, Park WY, Shim BS, Ko SG, Kim SH. Ribosomal protein L5 mediated inhibition of c-Myc is critically involved in sanggenon G induced apoptosis in non-small lung cancer cells. Phytother Res. 2021;35:1080–8.
Wang Y, Wei H, Yang G, Zhang M. Effect of sanggenon D on the growth of tumor cells and transplantation tumors. Nat Prod Res Dev. 2020;32:160–7.
Chen LD, Liu ZH, Zhang LF, Yao JN, Wang CF. Sanggenon C induces apoptosis of colon cancer cells via inhibition of NO production, iNOS expression and ROS activation of the mitochondrial pathway. Oncol Rep. 2017;38:2123–31.
Li ZR, Ma T, Guo YJ, Hu B, Niu SH, Suo FZ, Du LN, You YH, Kang WT, Liu S, Mamun M, Song QM, Pang JR, Zheng YC, Liu HM. Sanggenon O induced apoptosis of A549 cells is counterbalanced by protective autophagy. Bioorg Chem. 2019;87:688–98.
Wang L, Yang Y, Liu C, Chen RY. Three new compounds from Morus nigra L. J Asian Nat Prod Res. 2010;12:431–7.
Kimura Y, Okuda H. Effects of phenolic constituents from the mulberry tree on arachidonate metabolism in rat platelets. J Nat Prod. 1986;49:639–44.
Dat NT, Binh PT, le Quynh TP, Huong HT, Minh CV. Sanggenon C and O inhibit NO production, iNOS expression and NF-κB activation in LPS-induced RAW264.7 cells. Immunopharm Immunot. 2012;34:84–8.
Peng J, Gong L, Si K, Bai X, Du G. Fluorescence resonance energy transfer assay for high-throughput screening of ADAMTS1 inhibitors. Molecules. 2011;16:10709–21.
Jung HW, Kang SY, Kang JS, Kim AR, Woo ER, Park YK. Effect of kuwanon G isolated from the root bark of Morus alba on ovalbumin-induced allergic response in a mouse model of asthma. Phytother Res. 2014;28:1713–9.
Jin SE, Ha H, Shin HK, Seo CS. Anti-allergic and anti-inflammatory effects of kuwanon G and morusin on MC/9 mast cells and HaCaT keratinocytes. Molecules. 2019;24:265.
Fukai T, Kaitou K, Terada S. Antimicrobial activity of 2-arylbenzofurans from Morus species against methicillin-resistant Staphylococcus aureus. Fitoterapia. 2005;76:708–11.
Fukai T, Oku Y, Hano Y, Terada S. Antimicrobial activities of hydrophobic 2-arylbenzofurans and an isoflavone against vancomycin-resistant enterococci and methicillin-resistant Staphylococcus aureus. Planta Med. 2004;70:685–7.
Sohn HY, Son KH, Kwon CS, Kwon GS, Kang SS. Antimicrobial and cytotoxic activity of 18 prenylated flavonoids isolated from medicinal plants: Morus alba L., Morus mongolica Schneider, Broussnetia papyrifera (L.) Vent, Sophora flavescens Ait and Echinosophora koreensis Nakai. Phytomedicine. 2004;11:666–72.
Aelenei P, Rimbu CM, Horhogea CE, Lobiuc A, Neagu AN, Dunca SI, Motrescu I, Dimitriu G, Aprotosoaie AC, Miron A. Prenylated phenolics as promising candidates for combination antibacterial therapy: morusin and kuwanon G. Saudi Pharm J. 2020;28:1172–81.
Barker WT, Jania LA, Melander RJ, Koller BH, Melander C. Eukaryotic phosphatase inhibitors enhance colistin efficacy in gram-negative bacteria. Chem Biol Drug Des. 2020;96:1180–6.
Grienke U, Richter M, Walther E, Hoffmann A, Kirchmair J, Makarov V, Nietzsche S, Schmidtke M, Rollinger JM. Discovery of prenylated flavonoids with dual activity against influenza virus and Streptococcus pneumoniae. Sci Rep. 2016;6:27156.
Pang D, Wang W, Li E, Shen W, Mu L, Liao S, Liu F. Sanggenon D from root bark of mulberry inhibits the growth of Staphylococcus aureus by moderating the fatty acid biosynthesis system. Ind Crop Prod. 2019. https://doi.org/10.1016/j.indcrop.2019.111719.
Mascarello A, Mori M, Chiaradia-Delatorre LD, Menegatti AC, Delle Monache F, Ferrari F, Yunes RA, Nunes RJ, Terenzi H, Botta B, Botta M. Discovery of Mycobacterium tuberculosis protein tyrosine phosphatase B (PtpB) inhibitors from natural products. PLoS ONE. 2013;8: e77081.
Li X, Ren Z, Wu Z, Fu Z, Xie H, Deng L, Jiang X, Chen D. Steric effect of antioxidant Diels–Alder-type adducts: a comparison of sanggenon C with sanggenon D. Molecules. 2018;23:2610.
Paudel P, Park SE, Seong SH, Jung HA, Choi JS. Novel Diels-Alder type adducts from Morus alba root bark targeting human monoamine oxidase and dopaminergic receptors for the management of neurodegenerative diseases. Int J Mol Sci. 2019;20:6232.
Ham A, Lee HJ, Hong SS, Lee D, Mar W. Moracenin D from Mori Cortex radicis protects SH-SY5Y cells against dopamine-induced cell death by regulating nurr1 and α-synuclein expression. Phytother Res. 2012;26:620–4.
Zhao Y, Xu J. Sanggenon C ameliorates cerebral ischemia-reperfusion injury by inhibiting inflammation and oxidative stress through regulating RhoA-ROCK signaling. Inflammation. 2020;43:1476–87.
Johnson TO, Ermolieff J, Jirousek MR. Protein tyrosine phosphatase 1B inhibitors for diabetes. Nat Rev Drug Discov. 2002;1:696–709.
Paudel P, Yu T, Seong SH, Kuk EB, Jung HA, Choi JS. Protein tyrosine phosphatase 1B inhibition and glucose uptake potentials of mulberrofuran G, albanol B, and kuwanon G from root bark of Morus alba L. in insulin-resistant HepG2 cells: an in vitro and in silico study. Int J Mol Sci. 2018;19:1542.
van de Laar FA, Lucassen PL, Akkermans RP, van de Lisdonk EH, Rutten GE, van Weel C. α-Glucosidase inhibitors for patients with type 2 diabetes: Results from a cochrane systematic review and meta-analysis. Diabetes Care. 2005;28:154–63.
Zolghadri S, Bahrami A, Hassan Khan MT, Munoz-Munoz J, Garcia-Molina F, Garcia-Canovas F, Saboury AA. A comprehensive review on tyrosinase inhibitors. J Enzyme Inhib Med Chem. 2019;34:279–309.
Lee NK, Son KH, Chang HW, Kang SS, Park H, Heo MY, Kim HP. Prenylated flavonoids as tyrosinase inhibitors. Arch Pharm Res. 2004;27:1132–5.
Hu S, Zheng Z, Chen F, Wang M. The depigmenting effect of natural resorcinol type polyphenols kuwanon O and sanggenon T from the roots of Morus australis. J Ethnopharmacol. 2017;195:196–203.
Koirala P, Seong SH, Zhou Y, Shrestha S, Jung HA, Choi JS. Structure-activity relationship of the tyrosinase inhibitors kuwanon G, mulberrofuran G, and albanol B from Morus species: a kinetics and molecular docking study. Molecules. 2018;23:1413.
Ma F, Shen W, Zhang X, Li M, Wang Y, Zou Y, Li Y, Wang H. Anti-HSV activity of kuwanon X from mulberry leaves with genes expression inhibitory and HSV-1 induced NF-κB deactivated properties. Biol Pharma Bull. 2016;39:1667–74.
Thabti I, Albert Q, Philippot S, Dupire F, Westerhuis B, Fontanay S, Risler A, Kassab T, Elfalleh W, Aferchichi A, Varbanov M. Advances on antiviral activity of Morus spp. plant extracts: Human coronavirus and virus-related respiratory tract infections in the spotlight. Molecules. 2020;25:1876.
Esposito F, Tintori C, Martini R, Christ F, Debyser Z, Ferrarese R, Cabiddu G, Corona A, Ceresola ER, Calcaterra A, Iovine V, Botta B, Clementi M, Canducci F, Botta M, Tramontano E. Kuwanon-L as a new allosteric HIV-1 integrase inhibitor: molecular modeling and biological evaluation. ChemBioChem. 2015;16:2507–12.
Martini R, Esposito F, Corona A, Ferrarese R, Ceresola ER, Visconti L, Tintori C, Barbieri A, Calcaterra A, Iovine V, Canducci F, Tramontano E, Botta M. Natural product kuwanon-L inhibits HIV-1 replication through multiple target binding. ChemBioChem. 2017;18:374–7.
Nikaido T, Ohmoto T, Nomura T, Fukai T, Sankawa U. Inhibition of adenosine 3’,5’-cyclic monophosphate phosphodiesterase by phenolic constituents of mulberry tree. Chem Pharm Bull. 1984;32:4929–34.
Liu XX, Zhang XW, Wang K, Wang XY, Ma WL, Cao W, Mo D, Sun Y, Li XQ. Kuwanon G attenuates atherosclerosis by upregulation of LXRα-ABCA1/ABCG1 and inhibition of NFκB activity in macrophages. Toxicol Appl Pharmacol. 2018;341:56–63.
Gu Y, Gao L, Chen Y, Xu Z, Yu K, Zhang D, Zhang G, Zhang X. Sanggenon C protects against cardiomyocyte hypoxia injury by increasing autophagy. Mol Med Rep. 2017;16:8130–6.
Xiao L, Gu Y, Gao L, Shangguan J, Chen Y, Zhang Y, Li L. Sanggenon C protects against pressure overloadinduced cardiac hypertrophy via the calcineurin/NFAT2 pathway. Mol Med Reps. 2017;16:5338–46.
Liu YJ, Li SY, Hou J, Liu YF, Wang DD, Jiang YS, Ge GB, Liang XM, Yang L. Identification and characterization of naturally occurring inhibitors against human carboxylesterase 2 in White Mulberry root-bark. Fitoterapia. 2016;115:57–63.
Hou XD, Ge GB, Weng ZM, Dai ZR, Leng YH, Ding LL, Jin LL, Yu Y, Cao YF, Hou J. Natural constituents from Cortex Mori Radicis as new pancreatic lipase inhibitors. Bioorg Chem. 2018;80:577–84.
Wei B, Yang W, Yan ZX, Zhang QW, Yan R. Prenylflavonoids sanggenon C and kuwanon G from mulberry (Morus alba L.) as potent broad-spectrum bacterial β-glucuronidase inhibitors: biological evaluation and molecular docking studies. J Funct Foods. 2018;48:210–9.
Bai Y, Chen L, Cao YF, Hou XD, Jia SN, Zhou Q, He YQ, Hou J. Beta-Glucuronidase inhibition by constituents of mulberry bark. Planta Med. 2021;87:631–41.
Kim H, Baburin I, Zaugg J, Ebrahimi S, Hering S, Hamburger M. HPLC-based activity profiling–discovery of sanggenons as GABAA receptor modulators in the traditional Chinese drug Sang bai pi (Morus alba root bark). Planta Med. 2012;78:440–7.
Gunawan C, Rizzacasa MA. Mulberry Diels−Alder adducts: synthesis of chalcomoracin and mulberrofuran C methyl ethers. Org Lett. 2010;12:1388–91.
Boonsri S, Gunawan C, Krenske EH, Rizzacasa MA. Synthetic studies towards the mulberry Diels-Alder adducts: H-bond accelerated cycloadditions of chalcones. Org Biomol Chem. 2012;10:6010–21.
Gao L, Han J, Lei X. Enantioselective total syntheses of kuwanon X, kuwanon Y, and kuwanol A. Org Lett. 2016;18:360–3.
Iovine V, Benni I, Sabia R, D’Acquarica I, Fabrizi G, Botta B, Calcaterra A. Total synthesis of (±)-kuwanol E. J Nat Prod. 2016;79:2495–503.
Chee CF, Lee YK, Buckle MJC, Rahman NA. Synthesis of (±)-kuwanon V and (±)-dorsterone methyl ethers via Diels-Alder reaction. Tetrahedron Lett. 2011;52:1797–9.
Qi C, Cong H, Cahill KJ, Muller P, Johnson RP, Porco JA Jr. Biomimetic dehydrogenative Diels-Alder cycloadditions: total syntheses of brosimones A and B. Angew Chem Int Ed Engl. 2013;52:8345–8.
Han J, Li X, Guan Y, Zhao W, Wulff WD, Lei X. Enantioselective biomimetic total syntheses of kuwanons I and J and brosimones A and B. Angew Chem Int Ed Engl. 2014;53:9257–61.
Luo SY, Tang ZY, Li Q, Weng J, Yin S, Tang GH. Total synthesis of mulberry Diels–Alder-type adducts kuwanons G and H. J Org Chem. 2021;86:4786–93.
Qi C, Xiong Y, Eschenbrenner-Lux V, Cong H, Porco JA. Asymmetric syntheses of the flavonoid Diels-Alder natural products sanggenons C and O. J Am Chem Soc. 2016;138:798–801.
Tangdenpaisal K, Songthammawat P, Akkarasereenon K, Chuayboonsong K, Ruchirawat S, Ploypradith P. Total synthesis of palodesangren B trimethyl ether and D dimethyl ether via a late-stage formation of 2H-pyran-2-one of the tetrahydrobenzo[c]pyranochromenone core. J Org Chem. 2019;84:13410–29.
Cong H, Porco JA. Total synthesis of (±)-sorocenol B employing nanoparticle catalysis. Org Lett. 2012;14:2516–9.
Tee JT, Keane T, Meijer AJHM, Khaledi H, Abd Rahman N, Chee CF. A strategy toward the biomimetic synthesis of (+/−)-morusalbanol A pentamethyl ether. Synthesis. 2016;48:2263–70.
Tee JT, Chee CF, Buckle MJC, Lee VS, Chong WL, Khaledi H, Rahman NA. Model studies on construction of the oxabicyclic [3.3.1] core of the mulberry Diels-Alder adducts morusalbanol A and 441772–64–1. Tetrahedron Lett. 2015;56:5082–5.
Liu X, Yang J, Gao L, Zhang L, Lei X. Chemoenzymatic total syntheses of artonin I with an intermolecular Diels-Alderase. Biotechnol J. 2020;15:2000119.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (Nos. 81973203 and 81973195), the Guangdong Basic and Applied Basic Research Foundation, China (No. 2020A1515010841), the Open Program of Shenzhen Bay Laboratory (No. SZBL2021080601007), the Southern Marine Science and Engineering Guangdong Laboratory (Zhuhai) (No. SML2021SP301), and the Key-Area Research and Development Program of Guangdong Province, China (No. 2020B1111110003).
Author information
Authors and Affiliations
Contributions
S-YL, J-YZ, and M-FZ collected the related references and prepared chemical compounds structures; S Y provided writing suggestions; S-YL and G-HT wrote and edited the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare that there are no conflicts of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Luo, SY., Zhu, JY., Zou, MF. et al. Mulberry Diels–Alder-type adducts: isolation, structure, bioactivity, and synthesis. Nat. Prod. Bioprospect. 12, 31 (2022). https://doi.org/10.1007/s13659-022-00355-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s13659-022-00355-y