
Abstract
Ten new germacrane-type sesquiterpenoids, artemyrianosins A–J (1–10), were isolated from the aerial parts of Artemisia myriantha. Their structures were elucidated by spectral analyses including UV, IR, HRESIMS, 1D and 2D NMR, ECD and the absolute configurations of compounds 1 and 7–9 were characterized using X-ray crystallography. All isolates were tested their cytotoxicity against three human hepatoma cell lines (HepG2, Huh7, and SK-Hep-1), and compounds 1–3, 7, and 10 showed cytotoxicity with IC50 values ranging from 43.7 to 89.3 μM. Among them, the most active compound 3 exhibited activity against three human hepatoma cell lines with IC50 values of 43.7 μM (HepG2), 47.9 μM (Huh7), and 44.9 μM (SK-Hep-1).
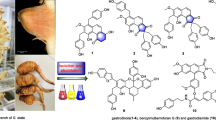
Graphical Abstract

Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
Hepatocellular carcinoma (HCC) as one of the most serious and common type of liver cancer is mainly caused by HBV or HCV infection, and heavy alcohol intake [1, 2]. HCC has resulted in nearly 0.83 million deaths worldwide in the year 2020 [3, 4], and suffered more than 1 million people will be affected by 2025 [5]. Clinically, four synthetic ones (sorafenib, regorafenib, lenvatinib and cabozantinib) and three monoclonal antibody ones (nivolumab, pembrolizumab and ramucirumab) are used to treat HCC [6, 7]. Although these drugs have obtained significant achievements, there are still some inevitable drawbacks, such as the low objective response rate, high incidence of adverse reactions, and drug resistance [8]. Therefore, new drugs with different targets for treating HCC are urgently needed (Fig. 1).

Chemical structures of compounds 1–10
Artemisia, one of the dominant genus within Asteraceae family, contains nearly 380 species globally with 186 species being dispersed in China [9]. Many Artemisia plants, such as A. annua, A. argyi, A. capillaris, and A. scoparia, have been recorded for the treatment of malaria, inflammation, hepatitis, cancer in the traditional Chinese medicine system [9,10,11,12]. Phytochemical investigation revealed that Artemisia genus are rich in sesquiterpenoids with antimalarial, antiinflammatory, antitumor, cytotoxic, antibacterial, and antihelminthic activities [13]. For example, artemisinin, a sesquiterpenoid lactone with an unusual peroxide bridge, which was obtained from A. annua by the Chinese scientist You-You Tu in 1972, showed antimalarial, anticancer and antiinflammatory activities [14]. Dihydroartemisinin, artemether, and artesunate which were chemically modified from artemisinin also exhibited antimalarial, antiviral, antifungal, anticancer, and antiinflammatory properties [15]. Arglabin, a guaiane-type sesquiterpenolide from A. glabella, inhibited of farnesyltransferase and its dimethylamino hydrochloride has been successfully developed into an anticancer drug in Kazakhstan for the treatment of colon, breast, ovarian, lung and liver cancers [16]. Arteminolides A–D from A. argyi were potential inhibitors of farnesyl protein transferase (FPTase) with IC50 values less than 1.0 μM in vitro, of which arteminolide C could prevent the development of lung tumor and human colon xenograft without causing weight loss in nude mice [17].
Our ongoing efforts to investigate bioactive sesquiterpenoids from the Artemisia plants, bioassay-guided isolation of A. atrovirens let to 26 guaiane dimers ([4 + 2] Diels–Alder cyclization) [18, 19], six rotundane-guaiane dimers ([4 + 2] Diels–Alder cyclization or containing a methylene-bridge) [19, 20], two guaiane-rotundane-guaiane trimers (containing a methylene-bridge) [20], two novel cagelike sesquiterpenoids (formed by intramolecular Diels–Alder cycloaddition) [21], and 16 undescribed guaiane sesquiterpenoids [9]. Among them, four guaiane-guaiane dimers (lavandiolide H and artematrolides A, J, and K) possessed significant cytotoxicity against HepG2, SMMC-7721, and Huh7 cell lines with IC50 values ranging from 3.8 to 9.6 μM, and lavandiolide H could induce G2/M cell cycle arrest and apoptosis in HepG2 cells via upregulating cleaved-PARP-1 and downregulating BCL-2 and PARP-1 [18]. Furthermore, artematrolide A was shown to activate the ROS/ERK/mTOR signaling pathway and promote metabolic shift in cervical cancer cells [22]. And the biomimetic synthesis via Diels–Alder reaction of the guaianolide dimers (artematrolide F and lavandiolides H, I, and K) and a battery of analogues were also achieved [23].
A. myriantha Wall. ex Bess. is commonly used for treating menorrhagia and inflammatory diseases in traditional Chinese medicine [24]. Phytochemical investigation on this species revealed the presence of sesquiterpenoids, flavonoids, and essential oils [25]. Among them, some sesquiterpenoids showed cytotoxicity against human colon cancer HCT-8, human gastric cancer BGC-823, and human liver cancer Bel-7402 cells [26, 27]. Our previous investigation reported 23 undescribed sesquiterpenolides with cytotoxicity against HepG2, SMMC-7721, and Huh7 from A. myriantha, classifying as germacranolide, guaianolide, and eudesmanolide, and revealed that artemyrianolide H displayed promising cytotoxicity against HepG2, SMMC-7721, and Huh7 with IC50 values of 4.9, 3.1, and 4.3 μM, respectively [25]. During our continuous search for antihepatic sesquiterpenoids from A. myriantha, 10 undescribed germacranolides (1‒10) were discovered (Fig. 1). Hence their isolation, structural identification, and cytotoxicity were discussed in this study.
2 Results and discussion
Artemyrianosin A (1) showed a molecular formula of C15H20O4 based on the analysis of the (+)-HRESIMS ion at m/z 287.1254 [M + Na]+ (calcd for C15H20O4Na, 287.1254) with six degrees of unsaturation. Its IR spectrum exhibited the presence of hydroxy (3414 cm‒1), carbonyl (1757 cm‒1), and double-bond (1643 and 1570 cm‒1) groups. The 1H and 13C NMR data (Tables 1 and 2) resembled those of artemyrianolide M [25], except for the only difference being that a ketone group at C-3 in artemyrianolide M was replaced by one oxygenated methine [δH 4.11 (1H, dd, J = 12.7, 5.1 Hz, H-3), δC 73.3 (C-3)]. This deduction was confirmed by the HMBC correlations from H-3 to C-15 (δC 117.2) and C-4 (δC 144.2) as well as the correlations of H-1/H2-2/H-3 in the 1H-1H COSY spectrum (Fig. 2). To determine its relative configuration, a ROESY experiment was carried out. The cross-peaks of H-3/H-7 and H-7/H-9 in the ROESY spectrum (Fig. 3) indicated that these protons were cofacial and β-oriented. However, the correlations of H-6/H-9 or H-6/H-3 were not observed in the ROESY spectrum, suggesting that H-6 was α-oriented. The absolute configuration of 1 was unambiguously verified to be (3R,6S,7R,9R) by Cu Kα X-ray crystallographic analysis (Fig. 4). Therefore, the structure of compound 1 was defined as (3R,6S,7R,9R)-3,9-dihydroxygermacra-4(15),10 (14),11(13)-trien-12,6-olide.

Key 1H-1H COSY and HMBC correlations of compounds 1–10

Key ROESY correlations of compounds 1–10

The X-ray ORTEP drawings of compounds 1 and 7–9
Artemyrianosin B (2) was deduced with the same molecular formula of C15H20O4 as 1 by the HRESIMS at m/z 265.1427 [M + H]+ (calcd for C15H21O4, 265.1434). The 1H and 13C NMR data (Tables 1 and 2) of compound 2 were closely related to those of 1, and further analyses of 2D NMR spectra implied that they possessed the same planar structure. Their only difference was the chemical shift changes of H-9 (δH 4.23 vs 4.28) and C-9 (δC 72.3 vs 77.6) and those surrounding the C-9 position (Tables 1 and 2), which might be caused by the different configurations at C-9. In the ROESY spectrum (Fig. 3), the correlations of H-3/H-7 and H-6/H-9 revealed β-orientations of H-3 and H-7, whereas α-orientations of H-6 and H-9. Its absolute configuration was assigned to be 3R,6S,7R,9S by comparison of its experimental ECD spectrum with the calculated one (Fig. 5). Therefore, the structure of compound 2 was established as (3R,6S,7R,9S)-3,9-dihydroxygermacra-4 (15), 10 (14), 11 (13)-trien-12,6-olide (Fig. 5).

The experimental and calculated ECD spectra of compounds 2–6, and 10
Artemyrianosin C (3) was assigned to have the same molecular formula C15H20O4 with compounds 1 and 2 from the HRESIMS at m/z 265.1431 [M + H]+ (calcd for C15H21O4, 265.1434). Compound 3 had the identical 2D structure as 1 and 2 based on their 1D and 2D NMR data (Tables 1 and 2, Fig. 2). The ROESY correlations of H-3/H-6, H-3/H-7, and H-7/H-9 revealed that these protons were β-oriented. Its absolute configuration was concluded to be 3R,6R,7R,9R by comparison of its experimental ECD spectrum with the calculated one (Fig. 5). Thus, the structure of compound 3 was established to be (3R,6R,7R,9R)-3,9-dihydroxygermacra-4 (15),10(14),11(13)-trien-12,6-olide.
Artemyrianosin D (4) had a molecular formula C15H22O4 according to the HRESIMS data at m/z 267.1579 [M + H]+ (calcd for C15H23O4, 267.1591), suggesting five degrees of unsaturation. The similarity of 1H and 13C NMR data (Tables 1 and 2) between compounds 4 and 1 indicated that they were structural analogs, and the main difference was that the exocyclic double bond between C-11 and C-13 in compound 1 was disappeared, and a doublet methyl [δH 1.19 (3H, d, J = 6.4 Hz, H-13), δC 13.5 (C-13)] and a methine [δH 2.36 (1H, m, H-11), δC 46.3 (C-11)] signals were appeared in compound 4. This deduction was confirmed by the 1H-1H COSY interactions of H-7/H-11/H3-13 and the HMBC correlations from H3-13 to C-7, C-11, and C-12. Its absolute stereochemistry was defined to be (3R,6S,7R,9R,11R) by the ROESY correlations of H-3/H-7, H-7/H-9, H-7/H-13, and H-6/H-11 and the similarity between the experimental and calculated ECD spectra. Consequently, the structure of compound 4 was assigned as (3R,6S,7R,9R,11R)-3,9-dihydroxygermacra-4 (15),10(14)-dien-12,6-olide.
Artemyrianosin E (5) was determined to possess a molecular formula of C18H26O6 with six indices of hydrogen deficiency by its HRESIMS ion at m/z 339.1792 [M + H]+ (calcd for C18H27O6, 339.1802). The IR spectrum of 5 showed the characteristic absorptions for hydroxy (3428 cm−1), carbonyl (1718 cm−1), and olefinic (1631 cm−1) functionalities. The 1H NMR spectrum (Table 1) of 5 displayed a methyl at δH 1.98 (3H, s), a methoxy δH 3.75 (3H, s), three oxygenated methine protons at δH 5.35 (1H, td, J = 8.0, 2.4 Hz, H-6), 4.36 (1H, dd, J = 6.3, 2.6 Hz, H-9), and 4.23 (1H, dd, J = 10.2, 4.0 Hz, H-3), and three pairs of olefinic methylene protons [δH 6.30 (1H, s, H-13a), 5.73 (1H, s, H-13b); 5.13 (1H, s, H-14a), 5.04 (1H, s, 14b); 5.22 (1H, s, H-15a), 5.19 (1H, s, 15b)]. The 13C NMR spectrum (Table 2) showed the existence of 18 carbons, including two methyl groups, seven methylenes (three terminal double bonds, and four aliphatic methylenes), four methines (three oxygenated and an aliphatic methine), and five quaternary carbons (two ester carbonyl and three olefinic carbons). The above characteristic signals indicated that compound 5 was a germacranolide-type sesquiterpenoid with acetoxy and methoxy groups. The above inference was supported by two proton spin systems of H2-1/H2-2/H-3 and H2-5/H-6/H-7/H2-8/H-9 in the 1H-1H COSY spectrum, as well as the HMBC correlations from H2-14 to C-1, C-9 and C-10 and from H2-15 to C-3, C-4 and C-5. In addition, the correlation from H-6 to C-1′ in the HMBC spectrum implied that the acetoxy group (δH 1.98; δC 172.2 and 21.0) was linked at C-6; the HMBC correlation of OMe to C-12 suggested that the methoxy group was pointed at C-12. In the ROESY spectrum (Fig. 3), the cross peaks of H-3/H-6, H-3/H-7, and H-7/H-9 determined its relative configuration. The absolute configuration of 5 was defined as (3R,6R,7R,9R) by the similarity between the experimental and calculated ECD curves. Therefore, the structure of compound 5 was established as (3R,6R,7R,9R)-6-acetoxy-3,9-dihydroxygermacra-4(15),10 (14),11(13)-trien-12-oic acid methyl ester.
Artemyrianosin F (6) was assigned to have the same molecular formula C18H26O6 with compound 5 from the HRESIMS data at m/z 339.1785 [M + H]+ (calcd for C18H27O6, 339.1802). The 1H and 13C NMR data (Tables 2 and 3) of compound 6 were very similar to those of 5, indicating that both were architecturally semblable. Further analyses of their 2D NMR spectra indicated that the planar structure of compound 6 was identical with that of compound 5. In the ROESY spectrum, the correlations of H-3/H-9 and H-7/H-9 indicated the homolateral orientation of H-3, H-7, and H-9. However, no correlation of H-6 with the former three protons was observed, but the correlation of H-6/H2-8 was obviously appeared in the ROESY spectrum, suggesting that H-6 was on the opposite side. Hence, the absolute stereochemistry of compound 6 was elucidated and named as (3R,6S,7R,9R)-6-acetoxy-3,9-dihydroxygermacra-4(15),10(14),11(13)-trien-12-oic acid methyl ester by comparing the calculated and experimental ECD curves.
Artemyrianosin G (7) was determined to have a molecular formula of C15H20O4 based on the HRESIMS data at m/z 265.1423 [M + H]+ (calcd for C15H21O4, 265.1434). The 1H and 13C NMR data (Tables 2 and 3) of compound 7 resembled to those of 1, the main difference was that the exocyclic double bond between C-4 and C-15 in 1 was absent and replaced by a trisubstituent double bond [δH 4.98 (1H, d, J = 10.6 Hz, H-5); δC 143.8 (C-4) and 123.8 (C-5)] and a singlet methyl [δH 1.75 (3H, d, J = 1.3 Hz, H-15); δC 16.7 (C-15)] in 7. This deduction was supported by the proton spin systems of H-5/H-6/H-7/H2-8/H-9 in the 1H-1H COSY spectrum and the HMBC correlations from H3-15 to C-3/C-4/C-5. Its relative configuration was proposed by the ROESY correlations of H-3/H-6 and H-7/H-9. In addition, the ROESY correlation of H-5 with H3-15 manifested that Δ4,5-double bond was Z-configuration. The absolute configuration of 7 was defined as 3R,6R,7R,9R by a single crystal X-ray crystallographic diffraction experiment with Cu Kα radiation (Fig. 4). Therefore, the structure of 7 was identified as (3R,6R,7R,9R,4Z)-3,9-dihydroxygermacra-4,10(14),11(13)-trien-12,6-olide.
Artemyrianosin H (8) was deduced to have a molecular formula of C15H22O4 with five indices of hydrogen deficiency by its HRESIMS at m/z 267.1585 [M + H]+ (calcd for C15H23O4, 267.1591). The similarity of 1H and 13C NMR data (Tables 2 and 3) of 8 and 4 implied structurally closely related, but the major differences were that the oxygenated methine at C-3 and terminal double bond between C-4 and C-15 in compound 4 were absent, meanwhile, a trisubstituent double bond [δH 5.27 (1H, d, J = 9.7 Hz, H-3); δC 132.5 (C-3) and 133.5 (C-4)], a singlet methyl [δH 1.74 (3H, s, H-15); δC 20.8 (C-15)], and an oxygenated methine [δH 4.54 (1H, td, J = 10.3, 6.0 Hz, H-2); δC 70.8 (C-2)] were appeared in 8. The spin coupling of H2-1/H-2/H-3 in the 1H-1H COSY spectrum, together with the HMBC correlations from H3-15 to C-3/C-4/C-5 and from H2-5 to C-3/C-4/C-6/C-7/C-15 verified the above inference. In the ROESY spectrum (Fig. 3), the cross-peak of H-2/H-7 indicated that H-2 and H-7 were β-oriented, while the cross-peaks of H-6/H3-13 and H-6/H-9 supported their α-orientation. In addition, the correlation of H-2/H3-15 was clearly observed in the ROESY spectrum, indicating Δ3-double bond was E-configuration. Ultimately, the structure of compound 8 was elucidated as (2R,6S,7R,9S,11S,3E)-2,9-dihydroxygermacra-3,10(14)-dien-12,6-olide by a single-crystal X-ray diffraction experiment with Cu Kα radiation (Fig. 4).
Artemyrianosin I (9) shared the same molecular formula C15H22O4 with 8 according to the HRESIMS at m/z 267.1576 [M + H]+ (calcd for C15H23O4, 267.1591). Its 1H and 13C NMR data (Tables 2 and 3) were similar to those of 8, and detailed interpretation of the 1H-1H COSY and HMBC spectra revealed the same planar structures. The relative configuration was established through the ROESY cross-peaks of H-2/H-7, H-7/H-13, and H-6/H-9. Likely, the ROESY correlation of H-2/H3-15 implied Δ3-double bond was E-configuration. Subsequently, the structure of compound 9 was assigned as (2R,6S,7R,9S,11R,3E)-2,9-dihydroxygermacra-3,10 (14)-dien-12,6-olide by Cu Kα radiation X-ray crystallographic analysis (Fig. 4).
Artemyrianosin J (10) had a molecular formula of C15H20O4 as defined by the HRESIMS at m/z 265.1423 [M + H]+, which indicated two hydrogen atoms less than compound 9. The 1H and 13C NMR data of compound 10 were closely similar to those of 9, and the main differences were that a doublet methyl at C-13 and a methine at C-11 in compound 9 were replaced by an pair of exocyclic double bond between C-11 and C-13 [δC 141.8 (C-11), 124.7 (C-13); δH 6.24 (1H, d, J = 2.8 Hz, H-13a), 5.98 (1H, d, J = 2.8 Hz, H-13b)] in compound 10. This deduction was verified by the spin coupling of H-7/H-11/H2-13 in 1H-1H COSY spectrum and the correlations from H2-13 to C-7/C-11/C-12 in the HMBC spectrum. In the ROESY spectrum, the correlation of H-9 with H-2/H-6/H-7 allowed these β-orientation. The additional ROESY correlation of H-2/H3-15 implied Δ3-double bond was E-configuration. Therefore, the structure of compound 10 was assigned as (2R,6R,7R,9R,3E)-2,9-dihydroxygermacra-3,10(14),11(13)-trien-12,6-olide based on the similar experimental and calculated ECD curves (Fig. 5).
The cytotoxicity of all isolates against three human hepatoma cell lines (HepG2, Huh7, and SK-Hep-1) was evaluated at the concentration of 100 μM with sorafenib as the positive control. As shown in Fig. 6, compounds 1‒3, 7, and 10 containing an α-exo-methylene γ-butyrolactone group showed activity on HepG2, Huh7, and SK-Hep-1 with inhibitory ratios higher than 50%. The dose–response curves of the active compounds were further investigated to yield their respective IC50 values. As shown in Table 4, compounds 1‒3 exhibited cytotoxicity against HepG2 cells with IC50 values of 43.7‒46.5 μM, while compounds 7 and 10 showed weaker cytotoxicity (IC50: 55.1 and 66.1 μM). Meanwhile, compounds 1‒3 and 7 also displayed cytotoxicity against Huh7 cells with IC50 values ranging from 44.3 to 48.9 μM, but compound 10 showed weaker cytotoxicity with an IC50 value of 71.0 μM. For SK-Hep-1 cells, only compound 3 manifested cytotoxicity with an IC50 value of 44.9 μM, while other compounds (1, 2, 7 and 10) showed weaker cytotoxicity with IC50 values of 71.7‒89.3 μM. Significantly, compound 3 was the most active one with IC50 values of 43.7 (HepG2), 47.9 (Huh7), and 44.9 (SK-Hep-1) μM, respectively.

Inhibitory ratios of compounds 1–10 at 100 μM
3 Conclusion
In this study, 10 new germacrane-type sesquiterpenoids (1–10) were isolated and identified from A. myriantha. Their structures were elucidated by extensive analyses of spectral data, X-ray analyses, and ECD spectra. Compounds 1‒3 showed cytotoxicity against HepG2 cells with IC50 values of 43.7‒46.5 μM; compounds 1‒3 and 7 had cytotoxicity against Huh7 cell lines with IC50 values ranging from 44.3 to 48.9 μM; only compound 3 exhibited cytotoxicity against SK-Hep-1 cells with IC50 value of 44.9 μM. Interestingly, compound 3 displayed cytotoxicity against three human hepatoma cell lines with IC50 values of 43.7 (HepG2), 47.9 (Huh7), and 44.9 (SK-Hep-1) μM, respectively. This investigation provided valuable information for the understanding of antihepatoma parts of A. myriantha and germacrane-type sesquiterpenoids as the active constituents.
4 Materials and methods
General experimental procedures, the ECD calculation, and cytotoxicity assays were provided in Additional file 1.
4.1 Plant materials
Artemisia myriantha was collected from Lijiang, Yunnan province, China in September 2018, and identified by Dr. Zhuo Zhou (Kunming Institute of Botany, Chinese Academy of Sciences). A voucher specimen (No. 201809AM) was deposited in the laboratory of Antivirus and Natural Medicinal Chemistry, Kunming Institute of Botany, Chinese Academy of Sciences, Kunming, China.
4.2 Extraction and isolation
In connection with our previous paper [25], Fr. E (165 g) was chromatographed on a silica gel column (1.6 kg, 10 × 90 cm, MeOH–CHCl3, 2:98–10:90, v/v) to obtain fractions E1–E4 (25, 30, 46 and 55 g). Fraction E2 was subjected to MCI gel CHP 20P column (490 g, 5.0 × 50 cm) and eluted with a H2O–MeOH gradient (70:30, 50:50, 30:70, 0:100) to yield four subfractions E2.1–E2.4. Fraction E2.2 (18 g) was applied to Si CC (200 g, 5.0 cm × 25 cm) and eluted with an EtOAc-CHCl3 gradient (10:90, 20:80 and 30:70) to produce four subfractions E2.2.1–E2.2.4. The obtained fraction E.2.2.2 (2.2 g) was separated by preparative HPLC (H2O–MeCN, 76:24, 10.0 mL/min) to afford three fractions (E2.2.2.1–E2.2.2.3). Fraction E2.2.2.1 (218 mg) was purified by semipreparative HPLC (H2O–MeOH, 72:28, 3.0 mL/min) to yield compounds 1 (22 mg, tR = 28.3 min), 2 (13 mg, tR = 26.5 min), and 3 (25 mg, tR = 30.6 min). Fraction E2.2.3 (1.9 g) was isolated by repeated silica gel CC (50 g, 2.5 × 20 cm, EtOAC–CHCl3, 10:90–30:70) and semipreparative HPLC (H2O–MeCN, 75:25) to get compounds 4 (14 mg, tR = 27.8 min) and 8 (8 mg, tR = 29.3 min). Fraction E3 (46 g) was fractioned by MPLC on an MCI gel CHP 20P column (490 g, 5 cm × 50 cm) with a gradient solvent system of H2O–MeOH (80:20, 60:40, 40:60, 0:100) to provide four subfractions E3.1–E3.4 (16, 6.5, 8.9, and 18 g). Fraction E3.2 (6.5 g) was fractionated with Si CC (80 g, 3.5 × 35 cm) using EtOAc–CHCl3 (10:90–30:70) to afford three subfractions E3.2.1 − E3.2.3 (2.5, 1.6 and 1.8 g). The obtained fraction E3.2.2 (1.6 g) was further isolated by Sephadex LH-20 CC (120 g, 2.5 × 150 cm, MeOH) and semipreparative HPLC (H2O − MeCN, 82:18, 3.0 mL/min) to provide compounds 7 (18 mg, tR = 24.3 min), 9 (6 mg, tR = 21.2 min), and 10 (5 mg, tR = 22.8 min). Fraction E3.3 (8.9 g) was chromatographed over a silica gel column (110 g, 4.5 × 20 cm) eluted with an EtOAc − CHCl3 gradient (10:90, 20:80 and 30:70) to give fractions E3.3.1–E3.3.4. Fraction E3.3.2 (2.4 g) was conducted on preparative HPLC (H2O–MeCN, 82:18, 10 mL/min) and semipreparative HPLC (H2O − MeOH, 72:28, 3.0 mL/min) to yield compounds 5 (28 mg, tR = 35.3 min) and 6 (15 mg, tR = 32.8 min).
4.3 Spectroscopic data of compounds 1–10
4.3.1 Artemyrianosin A (1)
Colorless monoclinic crystals (MeOH-CHCl3, 95:5); mp 153.8–155.2 ℃; [α]25 D + 12.5 (c 0.11, MeOH); ECD (MeOH) λmax (Δε) 199 (− 4.0), 218 (+ 3.4), 250 (+ 1.3) nm; IR vmax 3414, 1757, 1643, 1570, 1457, 1414, 1384, 1276, 1155, 1016, 994 cm‒1; 1H and 13C NMR data see Tables 1 and 2; (+)-HRESIMS m/z 287.1254 [M + Na]+ (calcd for C15H20O4Na, 287.1254).
4.3.2 Artemyrianosin B (2)
White amorphous powder; [α] D26 + 37.9 (c 0.11, MeOH); ECD (MeOH) λmax (Δε) 196 (− 0.8), 220 (+ 4.6) nm; IR vmax 3430, 1742, 1644, 1449, 1384, 1276, 1160, 1001, 908 cm‒1; 1H and 13C NMR data see Tables 1 and 2; (+)-HRESIMS m/z 265.1427 [M + H]+ (calcd for C15H21O4, 265.1434).
4.3.3 Artemyrianosin C (3)
White amorphous powder; [α] D25 + 31.2 (c 0.11, MeOH); ECD (MeOH) λmax (Δε) 197 (+ 1.4), 212 (+ 3.4) nm; IR vmax 3424, 3388, 1767, 1648, 1632, 1426, 1384, 1323, 1286, 1116, 989 cm‒1; 1H and 13C NMR data see Tables 1 and 2; (+)-HRESIMS m/z 265.1431 [M + H]+ (calcd for C15H21O4, 265.1434).
4.3.4 Artemyrianosin D (4)
White amorphous powder; [α] D24 + 48.4 (c 0.11, MeOH); ECD (MeOH) λmax (Δε) 201 (− 3.6), 244 (+ 0.2) nm;IR vmax 3427, 1759, 1634, 1459, 1384, 1346, 1186, 1039, 991, 908 cm‒1; 1H and 13C NMR data see Tables 1 and 2; (+)-HRESIMS m/z 267.1579 [M + H]+ (calcd for C15H23O4, 267.1591).
4.3.5 Artemyrianosin E (5)
Colorless oil; [α] D25 − 3.9 (c 0.10, MeOH); ECD (MeOH) λmax (Δε) 197 (− 5.7), 224 (+ 1.0) nm; IR vmax 3428, 1718, 1631, 1439, 1384, 1245, 1150, 1020, 909 cm‒1; 1H and 13C NMR data see Tables 1 and 2; (+)-HRESIMS m/z 339.1792 [M + H]+ (calcd for C18H27O6, 339.1802).
4.3.6 Artemyrianosin F (6)
Colorless oil; [α] D25 − 17.6 (c 0.12, MeOH); ECD (MeOH) λmax (Δε) 202 (− 10.3), 224 (+ 1.7) nm; IR vmax 3423, 1719, 1629, 1439, 1382, 1246, 1152, 1027, 908 cm‒1; 1H and 13C NMR data see Tables 2 and 3; (+)-HRESIMS m/z 339.1785 [M + H]+ (calcd for C18H27O6, 339.1802).
4.3.7 Artemyrianosin G (7)
Colorless monoclinic crystals (MeOH-CHCl3, 95:5); mp 154.2–156.1 ℃; [α] D25 + 66.8 (c 0.14, MeOH); ECD (MeOH) λmax (Δε) 203 (+ 8.5), 220 (+ 3.2) nm; IR vmax 3467, 3435, 1749, 1664, 1643, 1632, 1445, 1410, 1381, 1314, 1263, 1129, 1027 cm‒1; 1H and 13C NMR data see Tables 2 and 3; (+)-HRESIMS m/z 265.1423 [M + H]+ (calcd for C15H21O4, 265.1434).
4.3.8 Artemyrianosin H (8)
Colorless monoclinic crystals (MeOH-CHCl3, 95:5); mp 151.2–153.1 ℃; [α] D24 –83.8 (c 0.13, MeOH); ECD (MeOH) λmax (Δε) 202 (–27.1), 227 (+ 0.5) nm; IR vmax 3500, 3367, 1751, 1667, 1650, 1452, 1384, 1195, 1184, 1021, 1010 cm‒1; 1H and 13C NMR data see Tables 2 and 3; (+)-HRESIMS m/z 267.1585 [M + H]+ (calcd for C15H23O4, 267.1591).
4.3.9 Artemyrianosin I (9)
Colorless tetragonal crystals (MeOH-CHCl3, 95:5); mp 150.8–152.3 ℃; [α] D26 –2.6 (c 0.12, MeOH); ECD (MeOH) λmax (Δε) 202 (–3.9), 229 (+ 0.2) nm; IR vmax 3391, 3308, 1751, 1632, 1564, 1384, 1064 cm‒1; 1H and 13C NMR data see Tables 2 and 3; (+)-HRESIMS m/z 267.1576 [M + H]+ (calcd for C15H23O4, 267.1591).
4.3.10 Artemyrianosin J (10)
White amorphous powder; [α] D26 –11.4 (c 0.13, MeOH); ECD (MeOH) λmax (Δε) 201 (+ 4.0), 222 (+ 0.8) nm; IR vmax 3391, 1754, 1631, 1594, 1567, 1384, 1073 cm‒1; 1H and 13C NMR data see Tables 2 and 3; (+)-HRESIMS m/z 265.1423 [M + H]+ (calcd for C15H21O4, 265.1434).
4.4 X-ray crystallographic analysis of compounds 1 and 7‒9
Compounds 1 and 7‒9 were afforded by recrystallization in a mixture of MeOH–CHCl3 (95:5). X-ray diffraction analyses were performed on a Bruker D8 QUEST instrument using Cu Kα radiation and the intensity data were collected at 100 (2) K. The crystal structures were solved by using SHELXS-97 and difference Fourier techniques, and refinements were performed through the program and refined by full-matrix least-squares calculations on F2. All non-hydrogen atoms were anisotropically refined, and the positions of hydrogens bonded to carbons were initially determined through geometry and refined using a riding model. The crystallographic data for compounds 1 and 7‒9 in standard CIF format were deposited in the Cambridge Crystallographic Data Centre. The data can be accessed free of charge at http://www.ccdc.cam.ac.uk/.
Crystal data for compound 1: C15H20O4, M = 264.31, a = 8.1236 (3) Å, b = 10.9775 (5) Å, c = 15.0127 (6) Å, α = 90°, β = 95.6520 (10)°, γ = 90°, V = 1332.28 (9) Å3, T = 100. (2) K, space group P1211, Z = 4, μ (Cu Kα) = 0.774 mm−1, 33,394 measured reflections, 5199 independent reflections (Rint = 0.0763). The final R1 values were 0.0397 [I > 2σ (I)]. The final wR (F2) values were 0.1190 [I > 2σ (I)]. The final R1 values were 0.0447 (all data). The final wR (F2) values were 0.1207 (all data). The goodness of fit on F2 was 1.115. Flack parameter = 0.11 (6). CCDC 2,142,473.
Crystal data for compound 7: C15H20O4, M = 264.31, a = 5.6882 (2) Å, b = 15.5718 (4) Å, c = 7.6450 (2) Å, α = 90°, β = 93.2180 (10)°, γ = 90°, V = 676.09 (3) Å3, T = 100. (2) K, space group P1211, Z = 2, μ (Cu Kα) = 0.762 mm−1, 14,454 measured reflections, 2570 independent reflections (Rint = 0.0451). The final R1 values were 0.0379 [I > 2σ (I)]. The final wR (F2) values were 0.0984 [I > 2σ (I)]. The final R1 values were 0.0379 (all data). The final wR (F2) values were 0.0985 (all data). The goodness of fit on F2 was 1.070. Flack parameter = 0.04 (8). CCDC 2,142,472.
Crystal data for compound 8: C15H22O4·2 (H2O), M = 302.36, a = 10.1051 (5) Å, b = 5.9207 (3) Å, c = 13.4788 (6) Å, α = 90°, β = 96.4640 (10)°, γ = 90°, V = 801.30 (7) Å3, T = 100. (2) K, space group P1211, Z = 2, μ (Cu Kα) = 0.796 mm−1, 13,564 measured reflections, 3137 independent reflections (Rint = 0.0336). The final R1 values were 0.0292 [I > 2σ (I)]. The final wR (F2) values were 0.0752 [I > 2σ (I)]. The final R1 values were 0.0294 (all data). The final wR (F2) values were 0.0755 (all data). The goodness of fit on F2 was 1.081. Flack parameter = 0.06 (4). CCDC 2,142,471.
Crystal data for compound 9: C15H22O4, M = 266.32, a = 8.2909 (4) Å, b = 8.2909 (4) Å, c = 40.8360 (19) Å, α = 90°, β = 90°, γ = 90°, V = 2807.0 (3) Å3, T = 100. (2) K, space group P41212, Z = 8, μ (Cu Kα) = 0.735 mm−1, 44,745 measured reflections, 2766 independent reflections (Rint = 0.0520). The final R1 values were 0.0273 [I > 2σ (I)]. The final wR (F2) values were 0.0705 [I > 2σ (I)]. The final R1 values were 0.0273 (all data). The final wR (F2) values were 0.0706 (all data). The goodness of fit on F2 was 1.091. Flack parameter = 0.05 (2). CCDC 2,142,474.
References
Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68:394–424.
Fattovich G, Stroffolini T, Zagni I, Donato F. Hepatocellular carcinoma in cirrhosis: incidence and risk factors. Gastroenterology. 2004;127:35–50.
Fitzmaurice C, Akinyemiju TF, Al Lami FH, Alam T, Alizadeh-Navaei R, Allen C, Alsharif U, Alvis-Guzman N, Amini E, Anderson BO, Aremu O, Artaman A, Asgedom SW, Assadi R, Atey TM, Avila-Burgos L, Awasthi A, Ba Saleem HO, Barac A, Bennett JR, Bensenor IM, Bhakta N, Brenner H, Cahuana-Hurtado L, Castaneda-Orjuela CA, Catala-Lopez F, Choi JYJ, Christopher DJ, Chung SC, Curado MP, Dandona L, Dandona R, et al. Global, regional, and national cancer incidence, mortality, years oflife lost, years lived with disability, and disability- adjusted life-years for 29 cancer groups, 1990 to 2016: a systematic analysis for the global burden of disease study. JAMA Oncol. 2018;4:1553–68.
Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, Bray F. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2021;71:209–49.
Llovet JM, Kelley RK, Villanueva A, Singal AG, Pikarsky E, Roayaie S, Lencioni R, Koike K, ZucmanRossi J, Finn RS. Hepatocellular carcinoma. Nat Rev Dis Primers. 2021;7:6.
Anwanwan D, Singh SK, Singh S, Saikam V, Singh R. Challenges in liver cancer and possible treatment approaches. Biochim Biophys Acta Rev Cancer. 2020;1873:188314.
Llovet JM, Ricci S, Mazzaferro V, Hilgard P, Gane E, Blanc JF, de Oliveira AC, Santoro A, Raoul JL, Forner A, Schwartz M, Porta C, Zeuzem S, Bolondi L, Greten TF, Galle PR, Seitz JF, Borbath I, Haussinger D, Giannaris T, Shan M, Moscovici M, Voliotis D, Bruix J. Sorafenib in advanced hepatocellular carcinoma. N Engl J Med. 2008;359:378–90.
Dai Q, Zhang C, Yuan Z, Sun Q, Jiang Y. Current discovery strategies for hepatocellular carcinoma therapeutics. Expert Opin Drug Discov. 2020;15:243–58.
Su L-H, Ma Y-B, Geng C-A, Li T-Z, Huang X-Y, Hu J, Zhang X, Tang S, Shen C, Gao Z, Zhang X-M, Chen J-J. Artematrovirenins A-P, guaiane-type sesquiterpenoids with cytotoxicities against two hepatoma cell lines from Artemisia atrovirens. Bioorg Chem. 2021;114:105072.
Tu Y-Y. The discovery of artemisinin (qinghaosu) and gifts from Chinese medicine. Nat Med. 2011;17:1217–20.
Wang S, Li J, Sun J, Zeng K-W, Cui J-R, Jiang Y, Tu P-F. NO inhibitory guaianolide-derived terpenoids from Artemisia argyi. Fitoterapia. 2013;85:169–75.
Geng C-A, Huang X-Y, Chen X-L, Ma Y-B, Rong G-Q, Zhao Y, Zhang X-M, Chen J-J. Three new anti-HBV active constituents from the traditional Chinese herb of Yin-Chen (Artemisia scoparia). J Ethnopharmacol. 2015;176:109–17.
Bora KS, Sharma A. The genus Artemisia: a comprehensive review. Pharm Biol. 2011;49:101–9.
Wang J, Xu C, Wong Y-K, Li Y-J, Liao F-L, Jiang T-L, Tu Y-Y. Artemisinin, the magic drug discovered from traditional Chinese medicine. Engineering. 2019;5:32–9.
Yao W, Wang F, Wang H. Immunomodulation of artemisinin and its derivatives. Sci Bull. 2016;61:1399–406.
Lone SH, Bhat KA, Khuroo MA. Arglabin: from isolation to antitumor evaluation. Chem Biol Interact. 2015;240:180–98.
Lee SH, Lee MY, Kang HM, Han DC, Son KH, Yang DC, Sung ND, Lee CW, Kim HM, Kwon BM. Anti-tumor activity of the farnesyl-protein transferase inhibitors arteminolides, isolated from Artemisa. Bioorg Med Chem. 2003;11:4545–9.
Su L-H, Zhang X-T, Ma Y-B, Geng C-A, Huang X-Y, Hu J, Li T-Z, Tang S, Shen C, Gao Z, Zhang X-M, Chen J-J. New guaiane-type sesquiterpenoid dimers from Artemisia atrovirens and their antihepatoma activity. Acta Pharm Sin B. 2021;11:1648–66.
Su L-H, Li T-Z, Ma Y-B, Geng C-A, Huang X-Y, Zhang X, Gao Z, Chen J-J. Artematrovirenolides A-D and Artematrolides S-Z, Sesquiterpenoid dimers with cytotoxicity against three hepatoma cell lines from Artemisia atrovirens. Chin J Chem. 2022;40:104–14.
Su L-H, Li T-Z, Geng C-A, Ma Y-B, Huang X-Y, Wang J-P, Zhang X-M, Chen J-J. Trimeric and dimeric sesquiterpenoids from Artemisia atrovirens and their cytotoxicities. Org Chem Front. 2021;8:1249–56.
Su L-H, Geng C-A, Li T-Z, Ma Y-B, Huang X-Y, Zhang X-M, Chen J-J. Artatrovirenols A and B: Two cagelike sesquiterpenoids from Artemisia atrovirens. J Org Chem. 2020;85:13466–71.
Zhang X-T, Hu J, Su L-H, Geng C-A, Chen J-J. Artematrolide A inhibited cervical cancer cell proliferation via ROS/ERK/ mTOR pathway and metabolic shift. Phytomedicine. 2021;91:153707.
Li T-Z, Yang X-T, Wang J-P, Geng C-A, Ma Y-B, Su L-H, Zhang X-M, Chen J-J. Biomimetic synthesis of lavandiolides H, I, and K and Artematrolide F via diels-alder reaction. Org Lett. 2021;23:8380–4.
Zan K, Chen X-Q, Zhao M-B, Jiang Y, Tu P-F. Cytotoxic sesquiterpene lactones from Artemisia myriantha. Phytochem Lett. 2020;37:33–6.
Tang S, Zhang X-T, Ma Y-B, Huang X-Y, Geng C-A, Li T-Z, Zhang X-M, Shen C, Su L-H, Gao Z, Chen J-J. Artemyrianolides A-S, Cytotoxic Sesquiterpenoids from Artemisia myriantha. J Nat Prod. 2020;83:2618–30.
Zan K, Chen X-Q, Zhao M-B, Tu P-F. Sesquiterpenoids from aerial parts of Artemisia myriantha. China J Chin Mater Med. 2016;41:2833–7.
Zan K, Chen X-Q, Tu P-F. Guaianolides from aerial parts of Artemisia myriantha. China J Chin Mater Med. 2018;43:2295–9.
Acknowledgements
This work was supported by the Key Program of the National Natural Science Foundation of China (22137008) and the Yunnan Wanren Project (YNWR-KJLJ-2019-002).
Author information
Authors and Affiliations
Contributions
All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare that there is no conflict of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1.
Supporting Information.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zhang, X., Ma, YB., He, XF. et al. Artemyrianosins A–J, cytotoxic germacrane-type sesquiterpene lactones from Artemisia myriantha. Nat. Prod. Bioprospect. 12, 16 (2022). https://doi.org/10.1007/s13659-022-00340-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s13659-022-00340-5