
Abstract
Five previously undescribed guanacastane diterpenoids, namely psathyrellins A–E (1–5), were obtained from cultures of the mushroom Psathyrella candolleana. Their structures with absolute configurations were elucidated by extensive spectroscopic methods. Compounds 1–3 showed antibacterial activity against four strains with MIC values in a range of 16–128 μg/mL.
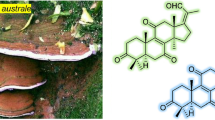
Graphic Abstract

Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
The basidiomycete Psathyrella candolleana (Psathyrellaceae) is a small agaric usually found in the vicinity of recently dead hardwood trees. It has a wide distribution on lawns or pastures in Europe and North America. Previous pharmacological studies indicated that the extracts of P. candolleana showed a marked anticlastogenic effect against DNA damage [1‒3], while chemical investigations on this fungus have demonstrated guanacastane-type diterpenoids [4, 5], which possess a unique 5/7/6 tricyclic backbone that have been found only in several fungal species. These diterpenoids have been detected to possess antibacterial and cytotoxic properties [5‒7]. As our long-term chemical studies on higher fungi [8,9,10,11,12,13], a chemical investigation on this mushroom resulted in the isolation of five new guanacastane diterpenoids, namely psathyrellins A–E (1–5, Fig. 1). Their structures were elucidated by extensive spectroscopic methods, while their absolute configurations were established by the single crystal X-ray diffraction and CD spectra. All compounds were evaluated for their antibacterial activity against four strains. Herein, the isolation, structural elucidation, and the biological activities of these isolates are reported.

Structures of compounds 1–5
2 Results and Discussion
Psathyrellin A (1) was isolated as colorless crystals. Its molecular formula was determined as C20H28O4 on the basis of HRESIMS at m/z 333.20584 (calcd for C20H28O4 [M + H]+, 333.30604), implying seven degrees of unsaturation. The IR absorption bands at 3421, 1710, 1640, 1462 cm−1 revealed the existence of hydroxy, carbonyl, and double bonds. In the 1H NMR spectrum (Table 1), three singlets at δH 1.06, 1.09, 1.85, two doublets at δH 0.79 (3H, d, J = 6.9 Hz), 1.12 (3H, d, J = 6.9 Hz) were readily assigned for five methyl groups. In addition, one doublet at δH 4.46 (1H, dd, J = 14.0, 5.2 Hz) indicated an oxidized methine carbon. In the 13C NMR spectrum, a total of 20 carbon resonances were detected. They were classified into five CH3, four CH2, three CH, and eight non-protonated carbons by the DEPT and HSQC spectra (Table 2). Of them, two carbonyl signals at δC 200.8 (s, C-5) and 205.4 (s, C-13), and four olefinic signals four two double bonds at δC 150.2 (s, C-1), 162.5 (s, C-3), 130.5 (s, C-4), and 149.3 (s, C-14) occupied four degrees of unsaturation, suggesting a tricyclic backbone of 1. All these data, as well as the literature survey [5, 6, 14], suggested that 1 should be a guanacastane diterpenoid characterized with a 5/7/6-fused ring system. Preliminary analysis of 2D NMR data (especially the HMBC data) revealed two α,β-unsaturated keto moieties in rings A and B, respectively (Fig. 2). A HMBC correlation from δH 4.46 (1H, dd, J = 14.0, 5.2 Hz, H-6) to C-5, as well as a 1H–1H COSY correlation between H-6 and H-7, suggested a hydroxy group placed at C-6. After many attempts, a single crystal of 1 was obtained from methanol, while the single crystal X-ray diffraction revealed the absolute configuration of 1 as shown in Fig. 3 (Flack parameter = 0.05(3); CCDC: 2068966).

Key 1H–1H COSY and HMBC correlations of 1 and 3

ORTEP diagrams of 1 (left) and 2 (right)
Psathyrellin B (2) was isolated as colorless crystals. Its molecular formula was determined as C20H26O4 by HRESIMS data at m/z 331.19031 [M + H]+ (calcd for C20H27O4+, 331.19039). The UV data at 282 nm suggested a conjugated system in 2. The 1D and 2D NMR spectra revealed similar patterns to those of 1 except that one more double bond in 2. In the HMBC spectrum, correlations from δH 1.11 (3H, s, H3-17) to δC 142.8 (s, C-1) and 158.6 (s, C-12), from δH 6.99 (1H, br s, H-2) to C-1 and δC 158.6 (s, C-3), and from δH 1.74 (3H, s, H3-15) to C-3 and δC 131.5 (s, C-4) suggested that three double bonds were distributed at C-12/C-13, C-1/C-2, and C-3/C-4, respectively. Detailed analysis of 2D NMR data suggested that the other parts of 2 were the same to those of 1. A single crystal X-ray diffraction not only proved the planar structure, but also determined the absolute configuration of 1 as shown in Fig. 3 (Flack parameter = − 0.12(14); CCDC: 2068967).
Psathyrellin C (3) was isolated as a yellow oil. The molecular formula was determined as C20H28O5 on the basis of HRESIMS data at m/z 349.20093 [M + H]+ (calcd for C20H29O5+, 349.20095). All NMR data suggested that 3 was structurally similar to that of 2 (Fig. 2). In compound 3, C-15 was oxidized into a hydroxymethylene at δC 59.4. In addition, C-5 was reduced into a hydroxymethine at δC 75.0. These information were supported by the HMBC and 1H–1H COSY data. In the ROESY spectrum, correlation of H-6 with H-9 suggested that OH at C-6 should be α oriented. Based on this information, the coupling constant of J5,6 = 7.9 Hz indicated that the OH at C-5 should be β oriented. The CD spectrum of 3 revealed similar Cotton effects with those of 2 (Fig. 4), indicating the absolute configuration of 3 to be the same to that of 2.

CD curves of compounds 2–5
Psathyrellin D (4) was isolated as a yellow oil. The molecular formula was determined as C22H28O6 on the basis of HRESIMS data at m/z 411.17749 [M + Na]+ (calcd for C22H28O6Na+, 411.17781). All spectroscopic data of 4 were similar to those of 2 excepted two additional carbons at δC 20.8 (q) and 172.8 (s) in 4 that were easily assigned as an O-acetyl moiety. The HMBC correlations from δH 4.40 and 4.30 to δC 172.8 (s) and from δH 1.27 (3H, d, J = 6.9 Hz, H3-20) to δC 32.3 (d, C-18), 67.1 (t, C-19) suggested that the O-acetyl moiety should be placed at C-19. Detailed analysis of 2D NMR data suggested that the other parts of 4 were the same to those of 2. The CD spectrum almost showed the same curve to that of 2, indicating the absolute configuration of the main backbone in 4 to be the same to that of 2 (Fig. 4). However, the stereochemistry of C-18 could not be established currently.
Psathyrellin E (5) was isolated as a yellow oil. The molecular formula was determined as C22H28O5 on the basis of HRESIMS data at m/z 373.20084 [M + H]+ (calcd for C22H29O5+, 373.20095). According to analysis of 1D and 2D NMR data, compound 5 was easily identified as a 19-O-acetyl derivative of 2, which was also similar to 4. The only difference was that the hydroxymethine of C-6 in 4 was reduced into a methylene at δC 34.5 (t, C-6) in 5, as supported by the HMBC correlation from H-6 to C-5 and the 1H–1H COSY data between H-6 and H-7. The CD spectrum almost indicated the same curve to that of 2, indicating the absolute configuration of the backbone in 5 to be the same to that of 2 (Fig. 4). However, the stereochemistry of C-18 could not be established currently.
Compounds 1–5 were evaluated for their antibacterial activities against Escherichia coli, Staphylococcus aureus, Salmonella enterica, and Pseudomonas aeruginosa. As a result, compounds 1–3 showed antibacterial activity with MIC values in a range of 16–128 μg/mL (Table 3).
3 Experimental Section
3.1 General Experimental Procedures
Optical rotations were measured on a Rudolph Autopol IV polarimeter. UV spectra were obtained on a UH5300 UV–VIS Double Beam Spectrophotometer. IR spectra were obtained by using a Shimadzu Fourier Transform Infrared spectrometer with KBr pellets. NMR spectra were acquired with a Bruker Avance III 600 instrument. CD spectra were recorded with an Applied Photophysics Chirascan-Plus spectrometer. High resolution electrospray ionization mass spectra (HRESIMS) were recorded on a LC–MS system consisting of a Q Exactive™ Orbitrap mass spectrometer with an ESI ion source used in ultra-high resolution mode (140,000, at m/z 200) and a Dionex UltiMate 3000 RSLC UPLC system. Silica gel (200–300 mesh and 500–800 mesh), RP-18 gel (40–75 µm) and Sephadex LH-20 were used for column chromatography (CC). Preparative HPLC was performed on an Agilent 1260 liquid chromatography system with a Zorbax SB-C18 (5 µm, 9.4 × 150 mm) column, a Daicel chiral column (AS-H, 5 µm, 4.6 × 250 mm) and a DAD detector.
3.2 Fungal Material and Cultivation Conditions
Fruiting bodies of P. candolleana were collected at Jingdong of Yunnan Province, China in 2003. They were identified by Prof. Zhu-Liang Yang of Kunming Institute of Botany, Chinese Academy of Sciences. The voucher specimen (NO.CGBWSHF00118.2) was deposited at School of Pharmaceutical Sciences, South-Central University for Nationalities. The strains were cultured in PDA and stored at − 4 °C. Culture medium was composed of glucose (5%), pork pepton (0.15%), yeast (0.5%), KH2PO4 (0.05%) and MgSO4 (0.05%). Initial pH was adjusted to 6.0, the fermentation was first carried out on an erlenmeyer flask for 6 days till the mycelium biomass reached to the maximum. Then it was transferred to rice medium at 24 °C in dark culture for 40 days. The rice medium in each 250 mL-Erlenmeyer flask was composed of rice (50 g) and water (50 mL). A total of 180 bottles were used in this study.
3.3 Extraction and Isolation
The rice fermentation (9 kg) was extracted four times with EtOAc. The organic layer was evaporated to give a crude extract (90 g). The extract was subjected to silica gel CC (200–300 mesh) eluted with a gradient solvent system of petroleum ether (PE)/Me2CO (from 20:1 to 1:1) to afford eight fractions A–H. Fraction C (3.8 g) was isolated by CC over silica gel using PE/Me2CO (6/1) to give subfractions C1–C6. Compound 1 was deposited from fraction C4 as colorless crystals (6 mg; purity > 95%). Fraction E (4 g) was first isolated by silica gel CC (200–300 mesh) eluted with PE/Me2CO (5/1) to give five subfractions E1-E5. Fraction E2 (800 mg) was further isolated by CC using RP-C18 silica gel (MeOH/H2O from 6/4 to 9/1) to give subfractions E2a-E2e. HPLC preparation (MeCN/H2O from 7/3 to 8/2 in 20 min) on fraction E2d (82 mg) afforded compounds 3 (1.8 mg, retention time (tR) = 12.1 min; purity 90%), 4 (2.8 mg, tR = 12.8 min; purity 90%), and 2 (4.3 mg, tR = 14.6 min; purity > 95%). Fraction E2e (70 mg) was separated by CC over Sephadex LH-20 (MeOH) to give a mixture. The mixture was subjected to HPLC (MeCN/H2O from 7/3 to 8/2 in 20 min) to give compound 5 (2.6 mg, tR = 15.1 min; purity 90%).
3.4 Spectroscopic Data of Compounds
3.4.1 Psathyrellin A (1)
Colorless crystals (MeOH); [α] 15D – 208.1 (c 0.22, MeOH); UV (MeOH) λmax (log ε) 192 (3.36), 244 (3.18) nm; IR (KBr) νmax 3421, 3349, 2829, 1710, 1702, 1640, 1462, 1038 cm−1; 1H (600 MHz) and 13C NMR (150 MHz) data (methanol-d4), see Table 1; HRESIMS m/z 333.20584 [M + H]+ (calcd for C20H29O4+, 333.20604).
3.4.2 Psathyrellin B (2)
Colorless crystals (MeOH); [α] 15D – 239.2 (c 0.18, MeOH); UV (H2O) λmax (log ε) 190 (3.34), 252 (3.20), 282 (2.86) nm; ECD (MeOH) λmax (Δε) 223 (+49), 251 (+45), 287 (–39), 339 (–27) nm; IR (KBr) νmax 3432, 3346, 2910, 1711, 1706, 1642, 1446, 1036 cm−1; 1H (600 MHz) and 13C NMR (150 MHz) data (methanol-d4), see Table 1; HRESIMS m/z 331.19031 [M + H]+ (calcd for C20H27O4+, 331.19039).
3.4.3 Psathyrellin C (3)
Yellow oil; [α] 15D – 182.9 (c 0.12, MeOH); ECD (MeOH) λmax (Δε) 209 (–33), 224 (+26), 287 (–29), 341 (–8) nm; 1H (600 MHz) and 13C NMR (150 MHz) data (methanol-d4), see Table 1; HRESIMS m/z 349.20093 [M + H]+ (calcd for C20H29O5+, 349.20095).
3.4.4 Psathyrellin D (4)
Yellow oil; [α] 15D – 221.9 (c 0.14, MeOH); ECD (MeOH) λmax (Δε) 222 (+20), 243 (+22), 287 (–15), 338 (–12) nm; 1H (600 MHz) and 13C NMR (150 MHz) data (methanol-d4), see Table 1; HRESIMS m/z 411.17749 [M + Na]+ (calcd for C22H28O6Na+, 411.17781).
3.4.5 Psathyrellin E (5)
Yellow oil; [α] 15D – 292.4 (c 0.12, MeOH); ECD (MeOH) λmax (Δε) 221 (+27), 242 (+31), 290 (–24), 338 (–18) nm; 1H (600 MHz) and 13C NMR (150 MHz) data (methanol-d4), see Table 1; HRESIMS m/z 373.20084 [M + H]+ (calcd for C22H29O5+, 373.20095).
3.4.6 X-Ray Crystallographic Data for Psathyrellin A (1)
C20H28O4, M = 332.42, a = 14.0862(3) Å, b = 7.26370(10) Å, c = 17.3636(3) Å, α = 90°, β = 99.5460(10)°, γ = 90°, V = 1752.01(5) Å3, T = 100.(2) K, space group P1211, Z = 4, μ(Cu Kα) = 0.692 mm−1, 33652 reflections measured, 6741 independent reflections (Rint = 0.0259). The final R1 values were 0.0306 [I > 2σ(I)]. The final wR(F2) values were 0.0799 [I > 2σ(I)]. The final R1 values were 0.0307 (all data). The final wR(F2) values were 0.0800 (all data). The goodness of fit on F2 was 1.056. Flack parameter = 0.05(3). CCDC: 2068966 (www.ccdc.cam.ac.uk).
3.4.7 X-Ray Crystallographic Data for Psathyrellin B (2)
C20H26O4, M = 330.41, a = 8.0026(8) Å, b = 8.3729(9) Å, c = 14.4078(15) Å, α = 90.00°, β = 105.783(3)°, γ = 90.00°, V = 929.00(17) Å3, T = 150.(2) K, space group P1211, Z = 2, μ(CuKα) = 1.54178 mm−1, 12059 reflections measured, 3552 independent reflections (Rint = 0.0456). The final R1 values were 0.0559 [I > 2σ(I)]. The final R1 values were 0.0415 (all data). The final wR(F2) values were 0.1156 (all data). The goodness of fit on F2 was 1.092. Flack parameter = − 0.12(14). CCDC: 2068967 (www.ccdc.cam.ac.uk).
3.5 Antibacterial Assay
The tested bacteria strains Escherichia coli ATCC25922, Staphylococcus aureus subsp. aureus ATCC29213, Salmonella enterica subsp. enterica ATCC14028, Pseudomonas aeruginosa ATCC27853 were purchased from China General Microbiological Culture Collection Center, (CGMCC). All these strains were cultured in Mueller Hinton broth (MHB) (Guangdong Huankai Microbial Sci. &Tech. Co., Ltd.) at 37 °C overnight with shaking (200 rpm). A sample of each culture was then diluted 40-fold in fresh MHB broth and incubated with shaking (200 rpm) at 37 °C for 2.5 h. The resultant mid-log phase cultures were diluted to a concentration of 5 × 105 CFU/mL, then 50 mL was added to each well of the compound-containing plates. The minimum inhibition concentration (MIC) was determined by measuring bacterial growth after 24 h on performing 1:2 serial dilutions of each compound ranging from 1 to 128 μg/mL. Chloramphenicol was used as a positive control.
References
M. Kuo, Psathyrella candolleana. http://www.mushroomexpert.com/psathyrella_candolleana.html
M.N. Al-Habib, J.C. Holliday, D. Tura, J. Med. Mushrooms 16, 617–622 (2014)
M.N. Al-Habib, J.C. Holliday, M.S. Aladahmy, Int. J. Med. Mushrooms 20, 749–759 (2018)
X. Yin, T. Feng, Z.H. Li, Y. Leng, J.K. Liu, Nat. Prod. Bioprospect. 4, 149–155 (2014)
Y.P. Liu, Q. Dai, W.X. Wang, J. He, Z.H. Li, T. Feng, J.K. Liu, J. Nat. Prod. 83, 1725–1729 (2020)
Y. Feng, F.X. Ren, S.B. Niu, L. Wang, L. Li, X.Z. Liu, Y.S. Che, J. Nat. Prod. 77, 873–881 (2014)
Y.P. Liu, Q. Dai, C.J. Pu, M. Wang, Z.H. Li, J.K. Liu, T. Feng, Nat. Prod. Comm. 14, 1–4 (2019)
H.X. Yang, H.L. Ai, T. Feng, W.X. Wang, B. Wu, Y.S. Zheng, H. Sun, J. He, Z.H. Li, J.K. Liu, Org. Lett. 20, 8069–8072 (2018)
J. He, M.S. Yang, W.X. Wang, Z.H. Li, W.A.M. Elkhateeb, T.C. Wen, H.L. Ai, T. Feng, RSC Adv. 9, 128–131 (2019)
W.X. Wang, Z.H. Li, T. Feng, J. Li, H. Sun, R. Huang, Q.X. Yuan, H.L. Ai, J.K. Liu, Org. Lett. 20, 7758–7761 (2018)
T. Feng, J. He, H.L. Ai, R. Huang, Z.H. Li, J.K. Liu, Nat. Prod. Bioprospect. 5, 205–208 (2015)
M. Wang, Z.H. Li, M. Isaka, J.K. Liu, T. Feng, Nat. Prod. Bioprospect. 11, 215–222 (2021)
C. Shi, Y.L. Peng, J. He, Z.H. Li, J.K. Liu, T. Feng, Nat. Prod. Bioprospect. 11, 235–241 (2021)
H. Zhang, M.H. Yang, Y. Li, X.B. Cheng, Y.H. Pei, L.Y. Kong, Fitoterapia 133, 219–244 (2019)
Acknowledgements
The work is financially supported by the National Key Research and Development Program of China (Grant No. 2017YFC1704007). The authors thank Analytical & Measuring Centre, South-Central University for Nationalities for the spectra measurements.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare no conflict of interest.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Wu, H., Yang, HX., Li, ZH. et al. Psathyrellins A–E, Antibacterial Guanacastane Diterpenoids from Mushroom Psathyrella candolleana. Nat. Prod. Bioprospect. 11, 447–452 (2021). https://doi.org/10.1007/s13659-021-00316-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13659-021-00316-x