
Abstract
Saldigones A–C (1, 3, 4), three new isoprenylated flavonoids with diverse flavanone, pterocarpan, and isoflavanone architectures, were characterized from the roots of Salvia digitaloides, together with a known isoprenylated flavanone (2). Notably, it’s the first report of isoprenylated flavonoids from Salvia species. The structures of these isolates were elucidated by extensive spectroscopic analysis. All of the compounds were evaluated for their activities on Cav3.1 low voltage-gated Ca2+ channel (LVGCC), of which 2 strongly and dose-dependently inhibited Cav3.1 peak current.
Graphic Abstract

Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
Salvia, one of the largest genera of the widespread family Lamiaceae, is distributed widely in the temperate and warm zones worldwide [1]. The name Salvia was derived from the Latin word “Salvare”, meaning “to heal or to be safe and unharmed”, which rose the folkloric belief of its “magical” remedial function for various indispositions [2, 3]. Phytochemical studies on more than 130 Salvia species have led to an array of secondary metabolites, such as diterpenoids, phenolic acids, triterpenoids, sesquiterpenoids, sesterterpenoids, steroids, as well as flavonoids [1,2,3,4,5,6,7]. Diterpenoids with abietane or clerodane skeletons are considered as the main bioactive and characteristic chemical constituents of this genus [4, 5, 7]. Many diterpenoids from Salvia plants have shown antimicrobial, antioxidant, anti-HIV, antibacterial, antineoplastic, antiplatelet aggregation activities [3,4,5,6,7,8]. Attracted by their diverse chemical structures and extensive biological activities, many scientists have engaged in the phytochemistry, pharmacy, chemical synthesis, and biosynthesis of the chemical constituents of Salvia plants [2, 3, 5, 6].
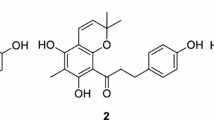
Our group has been committed into the phytochemical studies of genus Salvia since 2000, and discovered a series of novel terpenoids with diverse structures and bioactivities [9,10,11,12,13,14]. As a part of our ongoing research about the plants of Salvia species from southwestern China, unexpectedly, three new isoprenylated flavonoids saldigones A–C (1, 3, 4) (Fig. 1) with various flavanone, pterocarpan, and isoflavanone architectures, were isolated from the roots of S. digitaloides, along with a known isoprenylated flavonone, 5,7,3′-trihydroxy-4′,5′-(2⁗,2⁗-dimethylpyran)-8,2′-di(3-methyl-2-butenyl)-(2S)-flavanone (2), obtained from Dendrolobium Ianceolatum [15]. Terpenoids (accounted for more than 80%) and phenolic acid are the main chemical constituents of the Salvia species, while isoprenylated flavonoids from Salvia were reported for the first time [3,4,5,6]. For the two characterized isoprenylated flavanones (1 and 2), compound 1 owns both methylation and isoprenylation substitutions, and 2 has three isoprenyl groups in which one was further cyclized to form a 2,2-dimethylpyran unit. Moreover, 3 and 4 were characterized as a cis configurated pterocarpan and an isoflavone skeletons with two isoprenyl substituents, respectively.

Structures of compounds 1–4
Previous investigation indicated that some isoprenylated flavonoids led to an enhancement of activities or properties compared to the nonprenylated ones, such as cytotoxicity and bacterial neuraminidase inhibitory activities, but no low voltage-gated Ca2+ channel (LVGCC) activators have been identified [15,16,17,18]. Interestingly, isoprenylated substituents also appear to be important for anchoring certain natural products to the LVGCC [19]. In this study, the biological studies revealed that compound 2 can strongly and dose-dependently inhibited the peak currents of Cav3.1 low voltage-gated Ca2+ channel (LVGCC), an important therapeutic target for absence epilepsy, neuropathic pain, Parkinson’s disease, and insomnia [20,21,22], with an IC50 value of 3.5 μM. Meanwhile, 1, 3, and 4 were inactive at the concentration of 30 μM. In this note, the isolation and structural elucidation of these compounds were described, as well as their inhibitions on Cav3.1 LVGCC.
2 Result and Discussion
Saldigone A (1) was isolated as colorless gum and its molecular formula was determined as C27H32O6 by HRESIMS (m/z 451.2128 [M – H]−, calcd C27H31O6, 451.2126). The IR spectrum displayed bands for hydroxy (3411 cm−1) and carbonyl (1626 cm−1). The presence of a flavanone skeleton was evident from the characteristic absorption bands at λmax 345 and 297 nm in the UV spectra, the ABX-type spin system (δH 5.26, 1H, dd, J = 12.8, 3.0 Hz, H-2; 2.78, 1H, dd, J = 17.0, 3.0 Hz, H-3α; and 2.97, 1H, dd, J = 17.0, 12.8 Hz, H-3β) in the 1H NMR spectrum (Table 1), conjugated with the oxygenated carbon resonance at δC 78.8 (C-2), a methylene resonance at δC 43.6 (C-3), and a carbonyl resonance at δC 196.7 (C-4) in the 13C NMR spectra [23,24,25]. The 13C NMR and DEPT data presented a total of 27 carbon signals attributable to three signals for C-2, C-3, and C-4 mentioned above, 12 signals for two phenyls, as well as 12 other signals for one methyl (δC 7.1, C-1″), one methoxyl group (δC 61.5, MeO-4′), and two isoprenyls (δC 22.2, C-1‴; 121.9, C-2‴; 133.6, C-3‴; 26.0, C-4‴; 18.1, C-5‴/28.4, C-1⁗; 122.2, C-2⁗; 136.0, C-3⁗; 26.0, C-4⁗; 18.1, C-5⁗). Except for the signals for H-2 and H-3, three hydroxy groups (δH 12.25, 1H, s, OH-5; 6.14, 1H, s, OH-7; 5.64, 1H, s, OH-5′) and one methoxy group (δH 3.79, 3H, s, OMe-4′) can also be easily distinguished in the 1H NMR spectrum.
The locations of these oxygenated substituents as well as the isoprenyl and methyl groups were determined by the HSQC and HMBC experiments (Fig. 2). The cross-peaks of OH-5 with C-5 (δC 160.1), C-6 (δC 104.7), and C-10 (δC 103.0), and the methyl proton (δH 2.02, 3H, s, H-1″) with C-5, C-6, and C-7 (δC 162.2) indicated that the hydroxyl group was attached to C-5 while the methyl was substituted at C-6. The correlations of OH-7 with C-6, C-7, and C-8 (δC 105.3), and a set of isoprenyl protons (δH 3.35, 2H, t, H-1‴) with C-7, C-8, and C-9 (δC 157.3) indicated that the isoprenyl group located at C-8 while the hydroxyl group was attached to C-7 of A-ring. In addition, B ring was established as a 3′-hydroxy-4′-methoxy-5′-isoprenyl substitutions by the HMBC correlations of OH-3′ with C-2′ (δC 111.2), C-3′ (δC 149.3), and C-4′ (δC 145.3), of the methoxyl with C-4′, and of the isoprenyl protons (δH 3.35, 2H, t, H-1⁗) with C-2′, C-3′, and C-4′. The 1H-1H COSY correlation between H-2 and H-3, and HMBC correlations of H-2 with C-4, C-9, C-2′, and C-6′ (δC 119.1), and of H-3 with C-4, C-10 (δC 103.0) and C-1′ (δC 135.8) confirmed the existence of C ring furthermore.

Key HMBC and 1H-1H COSY correlations of 1, 3, 4
Biogenetically, flavanones are formed by cyclization of chalcones mediated by the enzyme chalcone isomerase (CHI) in a highly stereospecific manner through a Michael nucleophilic reaction involving the 2′-OH group and the α, β-unsaturated ketone of the chalcone, leading almost exclusively to the (2S)-isomer [26, 27]. As for compound 1, the circular dichroism (CD) spectra gave the sequential positive and negative Cotton effects near 330 and 280 nm for the n-π* and π-π* electronic transitions (Fig. 3), respectively, which were consistent with the absolute configuration 2S according to the reported data [25, 28,29,30,31]. Therefore, the structure of 1 was determined as (2S)-5,7,3′-trihydroxy-6-methy-8,5′-diisoprenyl-4′-methoxy flavanone named saldigone A.

CD spectra of compounds 1 (red), 3 (black) and 4 (yellow)
The structure of isoprenylated flavonone 2 was elucidated by analysis of 1D, 2D NMR, and CD spectral data confirmed by literature data comparison [15].
Saldigone B (3) was isolated as a white powder and was assigned the molecular formula C26H30O5, based on HRESIMS (m/z 421.2021 [M – H]−, calcd. C26H29O5, 421.2020). The IR spectrum displayed bands for hydroxy (3427 cm−1). The UV spectrum showed absorption bands at λmax 286 nm. In the 1H NMR spectrum (Table 2), the characteristic set of signals at δH 3.35 (1H, br t, J = 6.6 Hz, H-6a), δH 4.19 (1H, dd, J = 11.0, 4.9 Hz, H-6α), δH 3.57(1H, br t, J = 11.0 Hz, H-6β), and δH 5.65 (1H, d, J = 6.6 Hz, H-11a), together with the corresponding carbon signals at δC 39.2, δC 66.7, δC 76.7 for the typical C3 unit (C-6a, C-6, and C-11a) in the 13C NMR displayed that 3 possessed a pterocarpan skeleton [18, 32, 33]. Its 13C and DEPT NMR spectra displayed 26 carbon resonances attributable to the C3 unit mentioned above, 12 signals for two phenyls, as well as 11 other signals for one methoxyl (δC 63.2, MeO-1), and two isoprenyls groups (δC 23.4, C-1′; 122.7, C-2′; 134.8, C-3′; 18.2, C-4′; 18.1, C-5′/22.5, C-1″; 122.5, C-2″; 133.6, C-3″; 26.0, C-4″; 26.0, C-5″). The pterocarpan framework can be further evidenced by the 1H-1H COSY cross peaks of H-6/H-6a/H-11a, couple with the HMBC correlations from H-6 to C-4a (δC 152.9), C-11a, and C-6b (δC 119.8) as well as from H-11a to C-1 (δC 157.1), C-4a, C-11b (δC 106.7), and C-6b (Fig. 2).
According to the molecular formula (C26H30O5), there should be two OH groups in the structure except for the signals for the two isoprenyls, one methoxy, and 9 signals (including H-6, 2H; 6a, 1H; H-7, 1H, d, J = 7.7 Hz; H-8, 1H, d, J = 6.33; H-10, 1H, s; H-11a, 1H, and two oxygen atoms) for the pterocarpan skeleton. One of the two OH groups were located on C-3 (δC 155.6) by the HMBC correlations of OH-3 (δH 5.66, s) with C-2 (δC 114.2), C-3, and C-4 (δC 112.1). Then, the remaining OH group was attached on C-9 evidenced by the side-by-side comparison of the NMR spectral data with those of eryvarin J [32], as well as the HMBC correlations of H-7/8/10 with distinguished oxygenated quaternary carbon signal for C-9 at δC 157.1.
Structurally, there are two chiral centers (C-6a, 11a) in the pterocarpans skeleton whose absolute configurations are exclusively cis (6aR/11aR or 6aS/11aS) over the trans in nature [23, 28]. As reviewed previously, the positive 1Lb-band (260–310 nm) and negative 1La-band (220–240 nm) indicated the (6aR, 11aR)-cis configuration of pterocaroans [22, 28]. As for compound 3, the diagnostic positive Cotton effect at 287 nm (Δε + 8.35) and negative Cotton effect at 236 nm (Δε − 25.07) observed in the CD spectrum (Fig. 3) conformed the cis configuration of 3 as well as both the R absolute configurations of C-6a and C-11a [19, 22, 23]. Then, 3 was characterized as (6aR,11aR)-3,9-dihydroxy-2,4-diisoprenyl-1-methoxy pterocarpan named saldigone B.
Saldigone C (4) was isolated as pale-yellow gum and assigned a molecular formula of C26H30O6, based on HRESIMS (m/z 437.1968 [M – H]−, calcd. C26H29O6, 437.1970). The IR spectrum displayed bands for hydroxy (3429 cm−1) and carbonyl (1618 cm−1). It was deduced to be an isoflavanone based on the characteristic absorption bands in the UV spectrum (λmax 254 and 296 nm) together with three typical aliphatic proton signals (δH 4.47, 1H, br t, J = 11.1 Hz, H-2α; 4.41, 1H, dd, J = 11.1, 5.5 Hz, H-2β; 4.27, 1H, dd, J = 11.1, 5.5 Hz, H-3) in the 1H NMR spectrum (Table 3). This deduction can be further confirmed by the corresponding oxygenated methylene resonance (δC 70.7, C-2), a methine resonance (δC 46.9, C-3), and a carbonyl resonance at (δC 198.2, C-4) exclusively for an isoflavanone skeleton observed in the 13C NMR spectra [34, 35]. Furthermore, the 13C NMR and DEPT data displayed that the presence of totally 26 carbon signals owing to the signals for C-2, C-3, and C-4 described above, 12 signals for two phenyls, as well as 11 other signals for one methoxyl (δC 55.8, MeO-2′) and two isoprenyls (δC 21.5, C-1″; 122.1, C-2″; 134.9, C-3″; 18.1, C-4″; 26.1, C-5″/21.9, C-1‴; 122.4, C-2‴; 134.2, C-3‴; 18.1, C-4‴; 26.1, C-5‴).
Side-by-side comparison of the 1D NMR data of 4 with those of eryzerin B indicated their structures were closely similar to each other except for the existence for an additional hydroxyl group (δH 12.53) in 4 as evidenced by the MS and 1D NMR [35]. And the location of this OH group on C-5 was deduced by its HMBC correlations with C-5 (δC 159.9), C-6 (δC 107.3), and C-10 (δC 103.4). In addition, the HMBC correlations of the isoprenyl protons at δH 3.33 (1H, d, J = 7.1 Hz, H-1″) and δH 3.27 (1H, d, J = 7.1 Hz, 1‴) with C-5/6/7(δC 162.2) and C-7/8 (δC 106.3)/9 (δC 158.4), respectively, as well as of methoxy protons (δH 3.74, 3H, s, MeO-2′) to C-2′ (δC 158.8) indicated that the isoprenyls and one methoxyl groups were attached to C-6, C-8, and C-2′, respectively.
To analogy the data for similarly substituted isoflavanone systems, the positive Cotton effect near 310 nm and the negative Cotton effect near 280 nm (Fig. 3) indicated that the absolute configuration at C-3 was R for 4 [25, 28, 35]. Thus, 4 was characterized as (3R)-5,7,4′-thihydroxy-2′-methoxy-6,8-diisoprenyl isoflavanone named saldigone C.
As mentioned above, isoprenyl substituents appear to be important for anchoring some natural products to the low voltage-gated Ca2+ channel (LVGCC) [19]. The inhibitors of Cav3.1 LVGCC are promising agents for drug development against a number of neurological disorders [20, 21]. In this study, compound 2 was also tested to exhibit strong inhibitory activity on Cav3.1. In HEK 293 cells expressing human Cav3.1, 10 μM 2 notably antagonized currents that elicited by depolarizing voltage steps (− 80 to + 60 mV) (Fig. 4A). It took approximate 120 s to arrive the steady-state inhibition after the perfusion of 2 and this effect was hard to reverse upon washout (Fig. 4B), suggesting that the binding sites of 2 may be buried deeply inside the channel. Dose–response studies showed that 2 inhibited Cav3.1 peak currents with an IC50 value of 3.5 μM (Fig. 4C and Table S1). While, the Hill coefficient of 2 is 0.9, indicating a negative cooperativity. Mibefradil, the positive control, indicates a stronger activity than 2, with an IC50 value of 1.12 μM (Fig. 4D and Table S1). Moreover, compounds 1, 3 and 4 showed negligible effects on Cav3.1 at the higher concentration of 30 μM (Fig. S1).

Inhibitory effect of Compound 2 on Cav3.1. A Normalized current–voltage (I–V) curves of Cav3.1 in the absence or presence of 10 μM 2. Cav3.1 Currents were evoked from a holding potential (HP) of − 100 mV by 150 ms depolarization from − 80 to + 60 mV in 10 mV increment at 4 s intervals. Data are represented as mean ± SD (n = 3). B Time course of peak current inhibition by 10 μM 2. Ordinate axis, peak current during exposure to 2, which normalized by the peak current after drug exposure. C Representative Cav3.1 peak current trace that elicited by 150 ms depolarization to − 40 mV at 4 s intervals from a HP of − 100 mV in the absence (Bath) and presence of various concentrations of 2. D Dose–response curve of inhibiton of the peak current by 2. Solid curve represents fit to the Hill equation. Data are represented as mean ± SD (n = 3)
In conclusion, saldigones A–C (1, 3, 4), three new isoprenylated flavonoid constituents featured with diverse flavanone, pterocarpan, and isoflavanone architectures, were characterized from S. digitanoides, along with a known isoprenylated flavonone (2). To our knowledge, it’s the first report of isoprenylated flavonoid type metabolites from the Salvia plants despite many flavonoids have been isolated from this genus [3,4,5,6,7,8]. In detail, compounds 1 and 2 are isoprenylated flavonoids with methylation and isoprenylation substitutions, while 3 and 4 are two isoprenylated flavonoids with different pterocarpan and isoflavanone skeletons, respectively. In addition, compound 2 was proved to be a potent inhibitor of Cav3.1 LVGCC, indicating that isoprenylated flavonoids are deserved further in-depth exploration in genus Salvia. Totally, these findings could enrich diversities of secondary metabolites of the genus Salvia, and increased universality of biological activities and properties.
3 Experimental
3.1 General Experimental Procedures
Silica gel (100–200 and 200–300 mesh, Qingdao Marine Chemical Co., Ltd., Qingdao, China), MCI gel (75–150 μm, Mitsubishi Chemical Corporation, Tokyo, Japan) were used for column chromatography. Fractions were monitored by TLC (GF 254, Qingdao Marine Chemical Co., Ltd. Qingdao, China), and spots were visualized by heating silica gel plates immersed in 8% H2SO4 in ethanol. Semi-preparative HPLC was performed on Waters 2695 HPLC with a COSMOSIL C18 -MS-II (4.6ID × 250 mm) packed column. 1D and 2D NMR spectra were recorded on a Bruker DRX-600 spectrometer using TMS as an internal standard. Unless otherwise specified, chemical shifts (δ) are expressed in ppm with reference to the solvent signals. ESIMS and HRESIMS data were acquired on Agilent G6200 TOF mass spectrometer. Optical rotations were measured on a Jasco P-1020 polarimeter. UV spectra were recorded on a Shimadzu UV-2401PC spectrometer. IR spectra were recorded on a Bruker FT-IR Tensor-27 infrared spectrophotometer with KBr disks.
3.2 Plant Material
Roots of S. digitaloides were collected in Haba, Yunnan Province, People’s Republic of China, in August, 2011. The plant material was identified by Dr. Chun-Lei Xiang, Kunming Institute of Botany, Chinese Academy of Sciences. A voucher specimen was deposited in Kunming Institute of Botany, Chinese Academy of Sciences with identification number 20110788.
3.3 Extraction and Isolation
The air-dried roots of S. digitaloides (25.0 kg) were extracted with acetone for three times at room temperature (each 100 L, 3 days). The combined extracts were concentrated, the crude extract (730 g) was subjected to a silica gel column chromatography successively eluted with CHCl3, EtOAc and MeOH. The EtOAc fraction (180 g) was chromatographed on a silica gel column, eluted with petroleum ether/EtOAc (from 500:1 to 0:1, v/v), to yield five fractions (Fr. A–E) on the basis of TLC analysis. Fraction C (130.0 g) was separated through MPLC on an MCI gel column (MeOH-H2O from 60:40 to 100:0) to obtain four fractions (Fr. C1 – C4). Fraction C1 (20.0 g) was then chromatographed on a silica gel column, eluted with petroleum ether/acetone (from 200:1 to 0:1, v/v), to yield six fractions (Fr. C1-1 – C1-6).
Fraction C1-6 (4.0 g) was subjected to column chromatography over a silica gel column eluted with petroleum ether/CHCl3/EtOAc (9/0.7/0.3) to yield seven fractions (C1-6-1 – C1-6-7). Fraction C1-6–2 (900 mg) was chromatographed on Sephadex LH-20 (acetone) and further purified by semi-preparative HPLC (MeCN – H3O+, 72:28) to afford compounds 1 (2.9 mg, tR = 11.0 min), 2 (2.7 mg, tR = 12.9 min), 3 (7.5 mg, tR = 16.1 min) and 4 (2.2 mg, tR = 17.3 min).
3.3.1 Saldigone A (1)
Colorless gum; [α] 22D − 28.3 (c 0.08, MeOH); UV (MeOH) λmax (log ε) 345 (3.63), 297 (4.27), 195 (5.01), 328 (3.57), 254 (3.55) nm; CD (MeOH; c 1.19 × 10–4): Δε 350 (+ 1.74), 293 (− 14.15), 252 (+ 0.47), 242 (− 1.77), 227 (+ 3.57), 215 (− 1.74), 205 (+ 11.52) nm; IR (KBr) vmax 3411, 2922, 1626, 1607, 1374, 1186, 845, 554 cm−1; 1H and 13C NMR data, see Table 1; ESIMS m/z 451 [M – H]−; HRESIMS m/z 451.2128 [M – H]− (calcd for C27H31O6, 451.2126).
3.3.2 Saldigone B (3)
White powder; [α] 20D − 187.3 (c 0.09, MeOH); UV (MeOH) λmax (log ε) 286 (4.06), 208 (5.06), 257 (3.56), 203 (5.04) nm; CD (MeOH; c 1.1 × 10–4): Δε 287 (+ 8.35), 236 (− 25.07), 229 (− 22.58), 215 (− 49.89), 203 (+ 19.75) nm; IR (KBr) vmax 3427, 2924, 1608, 1496, 1354, 1264, 1142, 1124, 1099, 959, 839, 445 cm−1; 1H and 13C NMR data, see Table 1; ESIMS m/z 421 [M – H]−; HRESIMS m/z 421.2021 [M – H]− (calcd for C26H29O5, 421.2020).
3.3.3 Saldigone C (4)
Pale yellow gum; [α] 20D + 10.9 (c 0.08, MeOH); UV (MeOH) λmax (log ε) 194 (4.79), 196 (4.81), 254 (3.56), 296 (4,08), 420 (2.35) nm; CD (MeOH; c 2.2 × 10–4): Δε 367 (− 0.48), 311 (+ 4.24), 285 (− 3.29), 215 (+ 6.69), 199 (− 10.45) nm; IR (KBr) vmax 3429, 2924, 1618, 1468, 1178, 989, 567, 486 cm−1; 1H and 13C NMR data, see Table 1; ESIMS m/z 437 [M – H]−; HRESIMS m/z 437.1968 [M – H]− (calcd for C26H29O6, 437.1970).
3.3.4 Cell Preparation and Expression
Human embryonic kidney (HEK) 293 cells were grown in DMEM (HyClone) plus 10% newborn calf serum (Gibco) and penicillin (100 U/ml)/streptomycin (0.1 mg/ml) (Biological Industries). HEK 293 cells were transiently transfected with pCDNA3.1-T type and pCDNA3.1-EGFP plasmids together using LipoD293™ (SignaGen Laboratories) and used in 48 h.
Change history
12 May 2021
A Correction to this paper has been published: https://doi.org/10.1007/s13659-021-00308-x
References
E.M. Guajardo Touche, E.G. Loprz, A.P. Reyes, H. Sánchez, F. Honecker, H. Achenbach, Phytochemistry 45, 387–390 (1997)
A. Ulubelen, H. Birman, S. Öksüz, G. Topçu, U. Kolak, A. Barla, W. Voelter, Planta Med. 68, 818–821 (2002)
Y.B. Wu, Z.Y. Ni, Q.W. Shi, M. Dong, H. Kiyota, Y.C. Gu, B. Cong, Chem. Rev. 112, 5967–6026 (2012)
M. Akaberi, S. Mehri, M. Iranshahi, Fitoterapia 100, 118–132 (2015)
J.P. Wang, J.P. Xu, X. Gong, M. Yang, C.H. Zhang, M.H. Li, Molecules 24(1), 155 (2019)
J.P. Xu, K.H. Wei, G.J. Zhang, L.J. Lei, D.W. Yang, M.H. Li, J. Ethnopharmacol. 225, 18–30 (2018)
A. Kabouche, Z. Kabouche, Stud. Nat. Prod. Chem. 35, 753–833 (2008)
G.P.P. Kamatou, N.P. Makunga, W.P.N. Ramogola, A.M. Viljoen, Ethnopharmacology 119, 664–672 (2008)
G. Xu, A.J. Hou, R.R. Wang, G.Y. Liang, Y.T. Zheng, Z.Y. Liu, X.L. Li, Y. Zhao, S.X. Huang, L.Y. Peng, Q.S. Zhao, Org. Lett. 8, 4453–4456 (2006)
G. Xu, A.J. Hou, Y.T. Zheng, Y. Zhao, X.L. Li, L.Y. Peng, Q.S. Zhao, Org. Lett. 9, 291–293 (2007)
G. Xu, F. Zhao, X.W. Yang, J. Zhou, Y.L. Xin, X.L. Shen, Y.J. Hu, Q.S. Zhao, Nat. Prod. Bioprospect. 1, 81–86 (2011)
G. Xu, X.W. Yang, C.Y. Wu, X.N. Li, J. Su, X. Deng, Y. Li, H.B. Qin, L.X. Yang, Q.S. Zhao, Chem. Commun. 48, 4438–4440 (2012)
F. Xia, C.Y. Wu, X.W. Yang, X. Li, G. Xu, Nat. Prod. Bioprospect. 5, 307–312 (2015)
F. Xia, W.Y. Li, X.W. Yang, J. Yang, X. Li, Y. Nian, G. Xu, Org. Lett. 21, 5670–5674 (2019)
S. Kanokmedhakul, K. Kanokmedhakul, K. Nambuddee, P. Kongsaeree, J. Nat. Prod. 67, 968–972 (2004)
W. Wätjen, R. Rohrig, A. Kulawik, C.M. Passreiter, J. Nat. Prod. 71, 735–738 (2008)
X. Li, D. Wang, M.Y. Xia, Z.H. Wang, W.N. Wang, Z. Cui, Chem. Pharm. Bull. 57, 302–306 (2009)
H.S. Woo, D.W. Kim, J.H. Lee, J.Y. Kim, J.E. Kang, K.H. Park, Bioorg. Med. Chem. Lett. 21, 6100–6103 (2011)
Y.S. Ye, W.Y. Li, S.Z. Du, J. Yang, Y. Nian, G. Xu, J. Med. Chem. 63, 1709–1716 (2020)
G.W. Zamponi, Nat. Rev. Drug Discov. 15, 19–34 (2016)
E. Cheong, H.S. Shin, Physiol. Rev. 93, 961–992 (2013)
K.H. Choi, Expert Opin. Drug Dis. 8, 919–931 (2013)
M. Ichimaru, M. Moriyasu, Y. Nishiyama, A. Kato, S.G. Mathenge, F.D. Juma, J.N. Nganga, J. Nat. Prod. 59, 1113–1116 (1996)
L. Cui, D.T. Ndinteh, M. Na, P.T. Thuong, D. Njamen, J.T. Mbafor, Z.T. Fomum, J.S. Ahn, J. Nat. Prod. 70, 1039–1042 (2007)
M.A. Zaki, N.P.D. Nanayakkara, M.H. Hetta, M.R. Jacob, S.I. Khan, R. Mohammed, M.A. Ibrahim, V. Samoylenko, C. Coleman, F.R. Fronczek, D. Ferreira, I. Muhammad, J. Nat. Prod. 79, 2341–2349 (2016)
J.M. Jez, M.E. Bowman, R.A. Dixon, J.P. Noel, Nat. Struct. Mol. Biol. 7, 786–791 (2000)
C.S. Funari, T.G. Passalacqua, D. Rinaldo, D.H.S. Silva, Phytochemistry 72, 2052–2061 (2011)
M.A. Versiani, T. Diyabalanage, R. Ratnayake, C.J. Henrich, S.E. Bates, J.B. McMahon, K.R. Gustafson, J. Nat. Prod. 74, 262–266 (2011)
D. Slade, D. Ferreira, J.P.J. Marais, Phytochemistry 66, 2177–2215 (2005)
W. Gaffield, Tetrahedron 26, 4093–4108 (1970)
Q.T. Han, Y. Ren, G.S. Li, K.L. Xiang, S.J. Dai, Phytochemistry 152, 91–96 (2018)
T. Rukachaisirikul, P. Innok, A. Suksamrarn, J. Nat. Prod. 71, 156–158 (2008)
Y.W. Chin, L.K. Mdee, Z.H. Mbwambo, Q. Mi, H.B. Chai, G.M. Cragg, S.M. Swanson, A.D. Kinghorn, J. Nat. Prod. 69, 1649–1652 (2006)
H. Tanaka, T. Oh-Uchi, H. Etoh, M. Sako, F. Asai, Phytochemistry 64, 753–758 (2003)
J.R. Villinski, C. Bergeron, J.C. Cannistra, J.B. Gloer, S. Gafner, J. Nat. Prod. 77, 521–526 (2014)
Acknowledgements
The work was financially supported by the Foundation of Supported by National Natural Science Foundation of China (32070392 and 32070393) and the Second Tibetan Plateau Scientific Expedition and Research (STEP) program (2019QZKK0502-0303).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare no competing financial interest.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zhao, JJ., Li, SY., Xia, F. et al. Isoprenylated Flavonoids as Cav3.1 Low Voltage-Gated Ca2+ Channel Inhibitors from Salvia digitaloides. Nat. Prod. Bioprospect. 11, 671–678 (2021). https://doi.org/10.1007/s13659-021-00307-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13659-021-00307-y