
Abstract
Sixteen diterpenoid alkaloids (DAs), including six aconitine-type alkaloids (5 and 9 − 13), seven 7,17-seco-aconitine-type alkaloids (1 − 4, 6 − 8), two napelline-type alkaloids (14 and 15) as well as one veatchine-type alkaloid (16), were isolated from the aerial parts of Aconitum flavum Hand.-Mazz. In which, flavumolines A − D (1 − 4) were four new ones, and flavumoline E (5) was reported as natural compound for the first time. Their chemical structures were elucidated by the analysis of extensive spectroscopic data. The inhibitory activities of these isolates on Cav3.1 low voltage-gated Ca2+ channel, NO production in LPS-activated RAW264.7cells, five human tumor cell lines, as well as acetylcholinesterase (AChE) were tested.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
Aconitum species represent a large genus in the Ranunclaceae family [1]. It is estimated that there are more than 350 species of Aconitum all over the world, which are widely distributed in the northern temperate zone, of which 173 species are endemic to mainland China [1]. Previously chemical and pharmacological studies shown that the diterpenoid alkaloids (DAs) were the main pharmacological constituents of this genus [2]. So far, around 1300 natural DAs, categorized into C20-, C19-, and C18- families depending on the number of contiguous carbon atoms, have been reported [3], [4], [5]. Meanwhile, they have been the targets of medicinal chemists for a broad range of confirmed pharmacological properties including analgesic, antiarrhythmic, anti-inflammatory, hypotensive, neuroprotective and so on [6].
Aconitum flavum Hand.-Mazz, known as a perennial herb, is mainly distributed in Qinghai, Gansu, and other northwest places in China [7]. As one of the most vital Chinese traditional ethnic minority folk medicine, the roots were commonly used for the treatment of traumatic injuries, rheumatic arthritis, and some other inflammations [7]. Previously phytochemical investigations of this plant have resulted in the isolation of more than 20 DAs from the roots, such as aconitine, mesaconitine, and deoxyaconitine, which were not only considered as the predominant toxic components of this folk medicine, but simultaneously, the significant active constituents [8]. In this study, sixteen DAs including four new 7,17-seco-aconitine-type DAs (flavumolines A − D, 1 − 4), one new natural compound (5), together with 11 known analogues (6 − 16) were identified. These compounds could be divided into four different types: six aconitines (5 and 9 − 13), seven 7,17-seco-aconitines (1 − 4 and 6 − 8), two napellines (14 and 15) as well as one veatchine-type alkaloid (16). All of these isolated compounds were evaluated for their inhibitory effects on Cav3.1 low voltage-gated Ca2+ channel, NO production in LPS-activated RAW264.7 cells, five human tumor cell lines, and acetylcholinesterase (AChE). Reported herein, the isolation, structural determination, and biological activity of these compounds were thoroughly described.
2 Results and Discussion
The MeOH extract was subjected to repeated column chromatography to yield four new ones (flavumolines A − D, 1–4), one new natural compound (5), together with 11 known analogues 13-hydroxylfranchetine (6) [9], vilmorisine (7) [10], franchetine (8) [11], 3-acetylaconitine (9) [12], 15-dehydroxyldecludine A (10) [13], pubescensine (11) [14], 3-O-acetyl-20-deethyl-20-formylaconitine (12) [15], 14α-benzoyloxy-N-ethyl-3α,13β,15α-trihydroxy-1α,6α,8β,16β,18-pentamethoxyaconitane (13) [16], 15-acetylsongoramine (14) [17], dehydrolucidusculine (15) [18], and veatchine azomethine (16) [19].
Flavumoline A (1) was obtained as a white amorphous powder with the molecule formula as C29H35NO8 determined on the basis of HRESIMS at m/z 548.2259 [M + Na]+ (calcd for C29H35NO8Na, 548.2255). The presence of hydroxyl (3431 cm−1) and carbonyl (1672 cm−1) units was deduced from the IR spectrum. The 13C NMR and DEPT spectra displayed 29 carbons signals, which were divided into five methylenes, nine methines, and five quaternary carbons as well as the signals for a benzoyl and three methoxy groups. Detailly, the characterized signals for a franchetine-type C19-DA core could be distinguished as follows: one representative 6,17-epoxy unit (consisted of two oxygenated methines at δC 83.1, C-17 and δC 76.2, C-6), two quaternary carbons at δC 48.9 (C-4) and 51.0 (C-11), and three methines at δC 46.5 (C-5), 44.3 (C-9), and 47.1 (C-10), together with the typical C-7/C-8 trisubstituted double bond (δC 128.0, d, C-7 and δC 137.4, s, C-8) [3]. The 1H NMR spectrum of 1 also verified resonances assignable to two oxygenated methines at δH 4.62 (d, J = 4.6 Hz, H-17) and δH 4.71 (d, J = 6.1 Hz, H-6), and a singlet at δH 5.78 (d, J = 5.8 Hz, H-7). These evidences, conjugated with the fact that a number of DAs have been reported as the major constituents of this genus, suggested that compound 1 was assigned to be a typical franchetine-type C19-DA [20]. This deduction could be further confirmed by the HMBC correlated signals of H-5 with C-4/C-7/C-10/C-11/C-17/C-18/C-19, H-9 with C-12/C-13/C-14, and H-10 with C-1/C-5/C-9/C-11/C-12/C-17. Furthermore, the 6,17-epoxy unit was confirmed by the correlations from H-17 to C-6/C-11 and H-6 to C-5/C-7/C-8/C-11/C-17. In addition, the correlations of H-3, H-5, H-17, and H-18 with C-19, combined with correlations from N–H (δH 6.29, d, J = 4.7 Hz) to C-4/C-11/C-19/C-17 confirmed the amide moiety between nitrogen atom and C-19 (Fig. 2).
Moreover, the benzoyl group was placed at C-14 according to the HMBC correlation from H-14 (δH 5.07, s) to carbonyl (δC 166.7, s). Three methoxy units were located at C-1, C-16, and C-18 on the basis of the HMBC correlated signals from OCH3-1 (δH 3.41, s) to C-1 (δC 85.2, d), from OCH3-16 (δH 3.50, s) to C-16 (δC 85.8, d), and from OCH3-18 (δH 3.31, s) to C-18 (δC 74.1, t), respectively. Besides, a hydroxyl group should be located at C-13 corresponding to the HMBC correlations from OH to C-12/C-13/C-16.
Biogenetically, the configurations of H-5, 9, 10, could be ascribed to β-orientation whereas H-17 to α-orientation, which could be confirmed by ROESY correlations of H-5/9/10/12β and H-17/12α, respectively [21]. Then, the α-orientation of the benzoyl group at C-14 was determined on the ROESY correlations between H-14 and H-9. And the α-orientation of the OCH3-1 was identificated by the ROESY correlations between H-1/H-10. In addition, the correlations of H-17/H-12α/H-16 and 13-OH/H-14 demonstrated the β-orientation of OCH3-16 and 13-OH (Fig. 3, the dashed lines and solid lines to represent α-orientation and β-orientation, respectively). Therefore, the structure of 1 was determined and named flavumoline A.
The molecular formula of compounds 2 (C30H37NO8) and 3 (C31H41NO8) were established on the analysis of HRESIMS and NMR data. The NMR data of 2 showed the presence of six methylenes, eight methines, three quaternary carbons, and a characteristic trisubstituted intra-annular double bond, in addition to a benzoyl, an aldehyde group and three methoxy groups. All of these spectroscopic data suggested that 2 was an analogue of 13-hydroxylfranchetine (6) [9]. The main difference was the presence of a N-CHO moiety in compound 2 instead of N-ethyl group in 6, which was verified by 1D NMR data and HMBC correlations. The HMBC correlations (Fig. 2) from the singlet for aldehyde group (δH 7.95, s) to C-19 and C-17 suggested that the aldehyde group was placed at N-atom. On the analysis of ROESY data, the correlations of H-14/H-9, H-16/H-12α/H-17, H-1/H-10 confirmed the α-orientation of benzoyl group at C-14, β-orientation of methoxy at C-16, and α-orientation of methoxy at C-1, respectively. Compared with 2, compound 3 possessed a typical N-ethyl moiety, which were elucidated by the HMBC correlations from H-21 to C-17, C-19. A hydroxyl located at C-3 (δC 71.0, d) confirmed by the 1H-1H COSY correlation of H-2/H-3 and HMBC correlations of H-3 with C-2/5/19. Additionally, the ROESY correlations of H-3/H-1/H-5 confirmed α-orientation of 3-OH. Therefore, the structures of flavumoline B (2) and flavumoline C (3) were confirmed as shown above (Fig. 1).

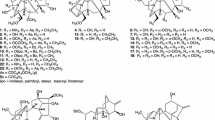
Structures of compounds 1–16 from A. flavum Hand.-Mazz
Flavumoline D (4) was a colorless oily liquid with molecule formula of C31H41NO8 as deduced by analysis of the ion peak at m/z 556.6689 [M + H]+ in the HRESIMS (calcd for C31H42NO8, 556.6702), which indicated 10 degrees of unsaturation. The 13C NMR and DEPT data showed six methylenes, nine methines, four quaternary carbons, as well as the signals for a benzoyl group, three methoxy groups, and a typical N-ethyl group. Side by side, comparison of its NMR data with those of 6, a known franchetine-type DA isolated previously, indicated that the structure of 4 was similar with that of 6 [9], except that the C-7/C-8 double bond was replaced by 7,8-epoxy unit (δC 64.0, C-7 and δC 59.0, C-8). The HMBC correlations of H-5/H-15 with C-7 and H-7/H-9/H-10/H-15 with C-8 confirmed the 7,8-epoxy unit. In addition, the remained three methoxy groups, one hydroxyl, and one benzoyl were elucidated to be attached on C-1, C-16, C-18, C-13, and C-14, respectively, as deduced by the HMBC correlations of OCH3-1 (δH 3.35, s) with C-1 (δC 86.0, d), OCH3-16 (δH 3.42, s) with C-16 (δC 82.6, d), OCH3-18 (δH 3.30, s) with C-18 (δC 79.0, t), hydroxyl group (δH 3.92, s) with C-12/C-13/C-16, and H-14 (δH 5.11, d, J = 3.0 Hz) with the carbonyl (δC 166.7, s) of the benzoyl group (Fig. 2).

Key 1H–1H COSY and HMBC correlations of compounds 1 − 5
The relative configuration of 4 was confirmed by the analysis of ROESY spectrum. At first, the correlation from H-14 to H-9 implied the benzoyl group at C-14 was α-oriented. The configurations of methoxyl groups were confirmed as 1α-OCH3 and 16β-OCH3 on account of the correlations of H-1/H-10 and H-16/H-12α/H-17. Then, the remained hydroxyl group at C-13 was established as β-oriented according to its correlation with 16-OCH3. On the basis of correlations of H-7/H-15α/H-16, the configuration of 7,8-epoxy unit was identified as β-orientation (Fig. 3). Hence, the structure of 4 was established as above.

Key ROESY correlations of compounds 1 − 5
Flavumoline E (5), was isolated as white amorphous powder. Its molecular formula was established as C33H41NO10 on the basis of HRESIMS at m/z 612.2799 [M + H]+ (calcd for C33H42NO10, 612.2803), in combination with NMR spectroscopic data. The IR spectrum of 5 showed absorptions due to the hydroxyl (3487 cm−1) and carbonyl (1676 cm−1) functionalities. The 13C NMR and DEPT spectra of 5 displayed 33 carbon resonances including five quaternary carbons (one carbonyl at δC 200.5, s, C-3, two oxygenated carbons at δC 92.1, C-8 and δC 74.3, C-13), eleven methines, three methylenes, and 14 other signals attributable to a benzoyl, a typical N-ethyl group, an acetoxyl, and three methoxy groups. By carefully analyzing the characteristic resonances of a N-ethyl group at δC 49.2, t (C-21) and δC 13.2, q (C-22), a nitrogen-bearing methine and methylene at δC 60.8, d (C-17) and δC 51.0, t (C-19), an α,β-unsaturated ketone moiety at δH 6.27, 6.46 (each 1H, d, J = 10.3 Hz) together with the C-atom signals at δC 132.3, 147.3, and 200.5 were clearly observed. The 1H NMR spectrum also verified resonances assignable to a nitrogen-bearing methine and methylene at δH 2.76 (s, H-17), δH 2.62 (d, J = 11.4 Hz, H-19a) and 2.38 (overlap, H-19b), as well as a representative N-ethyl group at δH 2.39, 2.69 (m, H-21) and δH 1.00 (t, J = 7.1 Hz, H-22). These evidences suggested that 5 should be a typical aconitine-type C19-DA [22]. This deduction could be further confirmed by the HMBC correlations of H-17 with C-5/C-6/C-8/C-10/C-11/C-19, H-19 with C-3/C-4/C-5/C-17/C-18/C-21, H-5 with C-4/C-7/C-10/C-11/C-17/C-18/C-19, H-9 with C-8/C-12/C-13/C-14/C-15, H-10 with C-8/C-9/C-12/C-13/C-17, toghther with the connection of C-17, C-19, and C-21 to the same nitrogen atom evidenced by the HMBC correlations from H-19 to C-17 and C-21, from H-21 to C-17 and C-19 (Fig. 2).
According to the HMBC spectrum, the correlations between one OH group (δH 4.01, s) and C-12/C-13/C-16, another OH group (δH 4.47, d, J = 3.0 Hz) and C-13/C-15/C-16, OAc (δH 1.45, s) and C-8, H-14 (δH 4.98, d, J = 5.0 Hz) and carbonyl of benzoyl group, identified the location of these substituent groups at C-13, C-15, C-8, and C-14, respectively. Additionally, the substitutions of 6-OCH3, 16-OCH3, and 18-OCH3 were identified by the correlations from OCH3-6 to C-6, OCH3-16 to C-16, OCH3-18 to C-18. The HMBC correlated resonances between H-1 and C-10/C-17, H-2 and C-4/C-11, H-18a,b/H-19a,b and C-3, demonstrated that the C = C bond was located between C-1 and C-2 and the carbonyl group at C-3 (200.5, s).
Biosynthetically, the configurations of H-5, 9, 10, 8-OAc, 13-OH could be ascribed to β-orientation and H-17 to α-orientation, respectively, due to the caged core of aconitine-type C19-DA [23]. Then, the configurations of two methoxy groups and one benzoyl at C-6, 16, and 14 were confirmed as α, β, and α, respectively, according to the correlations between H-6/H-5, H-16/H-12α/H-17 and H-14/H-9 (Fig. 3). However, the configuration of 15-OH has not been confirmed yet because of the simultaneous correlations of H-15 with H-7/H-16/16-OCH3 and 15-OH with H-7/H-16/16-OCH3. Eventually, the structure of 5 was identified as shown in Fig. 1, which was reported as a natural compound for the first time according to the literature on synthesis [24].
Since diterpenoid alkaloids in Aconitum were reported to commonly treat traumatic injury, arrhythmia, and rheumatism, in which ion channels or inflammation were involved in the pathophysiological process and inhibitors of ion channels or NO release were considered as potential agents for the treatment of these diseases [25], these isolated compounds were evaluated for their inhibitory effects on T-type ion channels using the whole-cell recording patch clamp method, NO production in LPS-activated RAW264.7 cells using Griess assay [26], on five human tumor cell lines [27], as well as acetylcholinesterase (AChE) [28]. As a result, compound 8 (30 μM) exhibited 64.5% inhibitory rate on Cav3.1 low voltage-gated Ca2+ channel. Compounds 3, 4, 5, 6, 7, and 11 showed potential inhibitory effects on NO production ranging from 20% to 32% at 50 μM. Additionally, compound 5 showed potential inhibitory effects on four human tumor cell lines HL-60, A-549, SMMC-7721, MCF-7 with IC50 value as 16.88, 33.11, 23.97, 24.21 μM in vitro, and no compound showed inhibitory effect on AChE.
3 Experimental
3.1 General Experimental Procedures
Optical rotations were measured on a Jasco P-1020 polarimeter. UV spectra were detected on a Shmadzu UV-2401PC spectrometer. IR spectra were determined on a Bruker FT-IR Tensor-27 infrared spectrophotometer with KBr disks. All 1D and 2D NMR spectra were recorded on Bruker DRX-600 spectrometers using TMS as an internal standard. Unless otherwise specified, chemical shifts (δ) were expressed in ppm with reference to the solvent signals. ESIMS and HRESIMS analysis were carried out on Waters Xevo TQS and Aglient G6230 TOF mass spectrometers, respectively. Semi-preparative HPLC was performed on a Waters 2695 HPLC with a 5C18-MS-II column [4.6 × 150 mm]. Silica gel (100–200, 200–300 mesh, Qingdao Marine Chemical Co., Ltd., People’s Republic of China), and MCI gel (75–150 μm, Mitsubishi Chemical Corporation, Tokyo, Japan) were used for column chromatography. Fractions were monitored by TLC (GF 254, Qingdao Marine Chemical Co., Ltd.), and spots were visualized under a UV lamp at 254 nm or by spraying the Dragendorff’ reagent and heating silica gel plates sprayed with 10% H2SO4 in EtOH.
3.2 Plant Material
The plants of A. flavum were collected in June 2019 from the Sanjiangyuan Nature Reserve of Guoluo, Qinghai Province, People’s Republic of China. Plant identity was verified by prof. Li-Xin Wei. A voucher specimen (No. 201901H19) was deposited in the Kunming Institute of Botany.
3.3 Extraction and Isolation
Air-dried and powdered plant material (aerial parts) of A. flavum (45 kg) was extracted three times (3 × 150 L) with MeOH at room temperature and then concentrated to 3.88 kg under reduce pressure. The crude extract was suspended in 1% HCl followed by basification with 10% aqueous NH4OH (pH 9 − 10) and subsequently, extracted with ethyl acetate to afford crude alkaloids (808 g). The total alkaloids fraction was separated on a silica gel column (CHCl3/CH3OH, 100:1 − 1:1) to yield eight fractions (Fr. 1 − 8). Then Fr. 1 (35 g) was separated on a silica gel column (PE/EtOAc/DEA, 50:1:1 − 3:1:1) to yield six sub-fractions (Fr. 1a − 1f). Fr. 1a (2.3 g) was separated on a silica gel column (PE/EtOAc, 80:1–3:1) and then semi-preparative HPLC (Waters 2695 C18, i.d. 150 × 10 mm, 5 μm, 3.0 ml/min, UV 210 nm) using MeOH/H2O (85/15, v/v) as the mobile phase afforded 6 (57 mg), 7 (10 mg) and 8 (23 mg). Fr. 1b (2.7 g) was separated on a silica gel column (PE/EtOAc, 60:1–3:1) followed by preparative HPLC using MeOH/H2O (75/25, v/v) to yield 4 (128 mg), 14 (15 mg), 15 (9.7 mg) and 16 (6 mg). Fr. 1c (3 g) was separated on a silica gel column (PE/EtOAc, 50:1–3:1), followed by semi-preparative HPLC using MeOH/H2O (70/30, v/v) as the mobile phase to afford 10 (8 mg), 11 (3 mg) and then yield 9 (230 mg) by recrystallizaiton. Fr. 1d (7 g) was separated on a silica gel column (PE/EtOAc, 50:1–1:1) yielded 12 (50 mg) and then on semi-preparative HPLC using MeOH/H2O (70/30, v/v) as the mobile phase to afford 13 (10 mg). Fr. 1e (5.5 g) yielded 2 (7.3 mg) and 3 (20.1 mg) by preparative HPLC using MeOH/H2O (70/25, v/v) as mobile phase, subsequently, afforded 5 (4.7 mg) and 1 (2 mg) isolated by Sephadex II column using acetone.
3.3.1 Flavumoline A (1)
White powder; [α] 19D −4.9 (c 0.175, MeOH); IR (KBr) νmax: 3431, 2975, 2933, 2828, 1718, 1672, 1282, 1099 cm−1; For 13C NMR data (see Table 1) and 1H NMR data (see Table 2). ESIMS m/z 526 [M + H]+; HRESIMS m/z: 548.2259 [M + Na]+ (calcd for C29H35NO8Na, 548.2255).
3.3.2 Flavumoline B (2)
White powder; [α] 18D −12.5 (c 0.100, MeOH); IR (KBr) νmax: 3447, 2938, 2882, 2828, 1717, 1670, 1280, 1099 cm−1; For 13C NMR data (see Table 1) and 1H NMR data (see Table 2). ESIMS m/z 540 [M + H]+; HRESIMS m/z: 562.2408 [M + Na]+ (calcd for C30H37NO8Na, 562.2411).
3.3.3 Flavumoline C (3)
White powder; [α] 18D −9.7 (c 0.103, MeOH); IR (KBr) νmax: 3534, 3402, 2974, 2824, 1717, 1280, 1103 cm−1; For 13C NMR data (see Table 1) and 1H NMR data (see Table 2). ESIMS m/z 556 [M + H]+; HRESIMS m/z: 556.2905 [M + H]+ (calcd for C31H42NO8Na, 556.2905).
3.3.4 Flavumoline D (4)
Colorless oily liquid; [α] 19D 19 −3.9 (c 0.101, MeOH); IR (KBr) νmax: 3437, 2973, 2932, 2826, 1719, 1279, 1101 cm−1; For 13C NMR data (see Table 1) and 1H NMR data (see Table 2). ESIMS m/z 556 [M + H]+; HRESIMS m/z: 556.6689 [M + H]+ (calcd for C31H42NO8, 556.6702).
3.3.5 Flavumoline E (5)
White powder; [α] 18D + 4.4 (c 0.122, MeOH); IR (KBr) νmax: 3487, 2973, 2936, 1721, 1676, 1280 cm−1; For 13C NMR data (see Table 1) and 1H NMR data (see Table 2). ESIMS m/z 612 [M + H]+; HRESIMS m/z 612.2799 [M + H]+ (calcd for C33H42NO10, 612.2803).
4 Bioassays
4.1 T-Type Ion Channel Inhibitory Activity Assay
All experiments were performed at room temperature (~ 22 °C). Pipettes were fabricated from borosilicate glass (World Precision Instru-ments) using a micropipette puller (P-1000, Sutter Instrument), and were fire-polished to resistances of 2 ~ 4 M for whole-cell recording. Whole-cell currents were elicited by 150 ms depolarization to − 40 mV at 4 s intervals from a holding potential (HP) of − 100 mV. Currents were amplified by Axopatch 200B and digitized by Digidata 1440A (Molecular Devices). Currents were low-pass filtered at 2 kHz and sampled at 10 kHz. pCLAMP 10 software (Molecular Devices) was used for data acquisition and analysis. The extracellular solutions contained (in mM) 142 CsCl, 1 MgCl2, 2 CaCl2, 10 Glucose and 10 HEPES (pH 7.4 adjusted with CsOH). The intracellular solutions contained (in mM) 127 Cs-methanesulphonate, 2MgCl2, 2Na2ATP, 10 HEPES and 11 EGTA (pH 7.4 adjusted with CsOH). The tested compounds (30 μM) were added.
4.2 Anti-Inflammatory Activity Assay
The murine macrophage cell line RAW264.7 was obtained from Cell Bank of Chinese Academy of Sciences. RAW264.7 cells were seeded in 96-well cell culture plates (1.5 × 105 cells/well) and treated with serial dilutions of the compounds with a maximum concentration of 50 μM in triplicate, followed by stimulation with 1 μg/mL LPS (Sigma) for 18 h. NO production in the supernatant was assessed by Griess reagents (Sigma). The absorbance at 570 nm was measured with a microplate reader (Thermo, Waltham, MA, USA). NG-Methyl-L-arginine acetate salt (L-NMMA, Sigma) was used as a positive control [29]. The viability of RAW264.7 cells was evaluated by the MTS assay simultaneously to exclude the interference of the cytotoxicity of the test compounds.
4.3 Cytotoxicity Assay
The human tumor cell lines HL-60, SMMC-7721, A-549, MCF-7, and SW-480 were obtained from ATCC (Manassas, VA, USA). Cells were cultured in RMPI-1640 or DMEM medium (Biological Industries, Kibbutz Beit-Haemek, Israel) supplemented with 10% fetal bovine serum (Biological Industries) at 37 °C in a humidified atmosphere with 5% CO2. The cytotoxicity assay was evaluated by the 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium, inner salt (MTS) (Promega, Madison, WI, USA) assay [30]. Briefly, cells were seeded into each well of a 96-well cell culture plate. After 12 h of incubation at 37 °C, the test compound (40 μM) was added. After incubated for 48 h, cells were subjected to the MTS assay. Compounds with a growth inhibition rate of 50% were further evaluated at concentrations of 0.064, 0.32, 1.6, 8, and 40 μM in triplicate, with cisplatin and paclitaxel (Sigma, St. Louis, MO, USA) as positive controls. The IC50 value of each compound was calculated with Reed and Muench’s method [31].
4.4 AChE Inhibitory Activity Assay
Acetylcholinesterase (AChE) inhibitory activity of the compounds isolated was assayed by the spectrophotometric method developed by Ellman et al.[32] with slightly modification. S-Acetylthiocholine iodide, S-butyrylthiocholine iodide, 5,5′-dithio-bis-(2-nitrobenzoic) acid (DTNB, Ellmans’ reagent), acetylcholinesterase derived from human erythrocytes were purchased from Sigma Chemical. Compounds were dissolved in DMSO. The reaction mixture (totally 200 μL) containing phosphate buffer (pH 8.0), test compound (50 μM), and acetyl cholinesterase (0.02U/mL), was incubated for 20 min (37 °C). Then, the reaction was initiated by the addition of 40 μL of solution containing DTNB (0.625 mM) and acetylthiocholine iodide (0.625 mM) for AChE inhibitory activity assay, respectively. The hydrolysis of acetylthiocholine was monitored at 405 nm every 30 s for one hour. Tacrine was used as positive control with final concentration of 0.333 μM. All the reactions were performed in triplicate. The percentage inhibition was calculated as follows: (%) inhibition = (E—S)/E × 100 (E is the activity of the enzyme without test compound and S is the activity of enzyme with test compound).
References
G.Y. Jiang, L.L. Qin, F. Gao, S. Huang, X.L. Zhou, Fitoterapia 141, 104738 (2020)
R.P. Zhang, Y.J. Lin, H.F. Yu, S.Y. Chen, J. Zhou, Nat. Prod. Bioprospect. 10, 105–108 (2020)
F.P. Wang, Q.H. Chen, X.Y. Liu, Nat. Prod. Rep. 27, 529–570 (2010)
Z.Q. Mu, H. Gao, Z.Y. Huang, Org. Lett. 14, 2758–2761 (2012)
J.F. Zhang, L. Chen, S. Huang, J. Nat. Prod. 80, 3136–3142 (2017)
F.Z. Chen, D.L. Chen, Q.H. Chen, F.P. Wang, J. Nat. Prod. 72, 18–23 (2009)
T. Wang, D.H. Xu, H.Y. Xu, P. Sheng, Lishizhen. Med. Mater. Med. Res. 19, 2162–2163 (2008). ((in Chinese))
J. Wang, X.H. Meng, T. Chai, J.L. Yang, Y.P. Shi, Nat. Prod. Bioprospect. 9, 419–423 (2019)
F. Zhang, S.L. Peng, F. Luo, L.S. Ding, Chin. Chem. Lett. 16, 1043–1046 (2005)
B. Zhao, J.Y. Zhao, ShSh. Sagdullaev, H.A. Aisa, Chem. Nat. Compd. 54, 828–830 (2018)
D.H. Chen, W.L. Song, Acta Chim. Sinica 41, 843–846 (1983)
Y.Q. He, B.H. Yao, Z.Y. Ma, J. Phar. Anal. 1, 57–59 (2011)
T.P. Yin, L. Cai, G. Lei, J.W. Dong, Chin. J. Org. Chem. 33, 2528–2532 (2013)
L. Chen, L.H. Shan, J.F. Zhang, W.L. Xu, M.Y. Wu, S. Huang, X.L. Zhou, Nat. Prod. Commun. 12, 2063–2065 (2015)
Y. Shen, A.X. Zuo, Z.Y. Jiang, X.M. Zhang, H.L. Wang, J.J. Chen, Helv. Chim. Acta. 93, 863–868 (2010)
X.X. Zong, X.J. Yan, J.L. Wu, Z.Q. Liu, H. Zhou, N. Li, L. Liu, J. Nat. Prod. 82, 980–989 (2019)
F. Gao, Y.Y. Li, D. Wang, X. Huang, Q. Liu, Molecules 17, 5187–5194 (2012)
H. Yamashita, K. Takeda, M. Haraguchi, Y. Abe, N. Kuwahara, S. Suzuki, A. Terui, T. Masaka, N. Munakata, M. Uchida, M. Nunokawa, K. Kaneda, M. Goto, K.H. Lee, K. Wada, J. Nat. Med. 72, 230–237 (2018)
S. William Pelletier, D.M. Locke, J. Am. Chem. Soc. 87, 761–776 (1965)
Y. Si, X. Ding, T.A. Adelakuna, Y. Zhang, X.J. Hao, Fitoterapia 147, 104738 (2020)
H. Cheng, F.H. Zeng, D. Ma, M.L. Jiang, L. Xu, F.P. Wang, Org. Lett. 16, 2299–2301 (2014)
F.L. Wen, Y.Y. Jiang, H. Tang, D.L. Chen, F.P. Wang, Nat Prod Commun. 12, 329–330 (2017)
F. Shaheen, M. Zeeshan, M. Ahmad, S. Anjum, S. Ali, H.K. Fun, M.I. Choudhary, A. Rahman, J. Nat. Prod. 69, 823–825 (2006)
X.K. She, X.X. Jian, D.L. Chen, Q.H. Chen, F.P. Wang, J. Asian. Nat. Prod. Res. 14, 665–677 (2012)
F. Wang, Z.G. Yue, P. Xie, L. Zhang, Z. Li, B. Song, Z.S. Tang, X.M. Song, Molecules 21, 1175 (2016)
V.M. Dirsch, H. Stuppner, A.M. Vollmar, Planta Med. 64, 423–426 (1998)
K. Wada, H. Yamashita, Molecules 24, 2317 (2019)
H. Ahmad, S. Ahmad, M. Ali, A. Latif, H. Naz, N. Rahman, F. Shaheen, A. Wadood, H.U. Khan, M. Ahmad, Bioorg. Chem. 78, 427–435 (2018)
D.W. Reif, S.A. Mc Creedy, Arch, Biochem. Biophys. 1, 170–176 (1995)
A.H. Cory, Cancer Commun. 3, 207–212 (1991)
L.J. Reed, H. Muench, Am. J. Trop. Med. Hyg. 27, 493–497 (1938)
G.L. Ellman, K.D. Courtney, V.J. Andres, R.M. Featherstone, Biochem. Pharmacol. 7, 88–95 (1961)
Acknowledgements
The work was financially supported by the Second Tibetan Plateau Scientific Expedition and Research (STEP) program (2019QZKK0502), State Key Laboratory of Phytochemistry and Plant Resources in West China (P2017-KF02 and P2019-ZZ05), and the Natural Sciences Foundation of Yunnan Province (2019FA003).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
All authors declare no conflict of interest.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zhang, N., Xia, F., Li, SY. et al. Diterpenoid Alkaloids from the Aerial Parts of Aconitum flavum Hand.-Mazz. Nat. Prod. Bioprospect. 11, 421–429 (2021). https://doi.org/10.1007/s13659-021-00302-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13659-021-00302-3