
Abstract
Euphopepluanones F − K (1 − 4), four new jatrophane type diterpenoids were isolated from the seeds of Euphorbia peplus, along with eight known diterpenoids (5 − 12). Their structures were established on the basis of extensive spectroscopic analysis and X-ray crystallographic experiments. The new compounds 1 − 4 were assessed for their activities to induce lysosomal biogenesis through LysoTracker Red staining. Compound 2 significantly induced lysosomal biogenesis. In addition, compound 2 could increase the number of LC3 dots, indicating that it could activate the lysosomal-autophagy pathway.
Graphic Abstract

Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
Plants of the genus Euphorbia are used in traditional medicines for treatments of digestive system disorders, skin ailments, infections, inflammation, and injuries world-wide [1]. The medicinal usage of Euphorbia plants is attributed to the structurally diverse polycyclic diterpenoids produced by these plants, which have reported more than 20 skeletal types [2, 3]. Euphodendroidin D, for example, is a jatrophane type diterpenoid from E. dendroides that show inhibition of the transport activity of P-glycoprotein, an ABC transporter protein related to multidrug resistance by decreasing the intracellular concentration of drugs [4]. Resiniferatoxin, a daphnane diterpenoid from E. resinifera, is known for the alleviation of neuropathic pain and is in phase I human clinical trials in treating severe pain in cancer [5]. In 2012, an ingenane type diterpenoid ingenol 3-angelate from E. peplus was approved by FDA for the treatment of actinic keratosis, a precancerous skin condition [6, 7].
E. peplus Linn., a small annual weed native to Mediterranean coast, was introduced into Yunan province of China [8]. The sap from E. peplus has been used in folk medicine for the treatment of asthma, catarrh and internal tumors [9]. Recently, our group discovered ingenane type diterpenoids 20-deoxyingenol and its analogues from E. peplus, possessing activity to promote lysosome biogenesis, limit amyloid plaque formation in APP/SP1 mice’s brain, which suggested that the potential of these compounds for the treatment of Alzheimer disease [10]. The subsequent phytochemical studies of the plants of the genus Euphorbia led to isolation of several novel diterpenoids with significant bioactivities [11,12,13,14,15,16]. In our continuing efforts to uncover structurally novel diterpenes capable of inducing lysosomal biogenesis, four new jatrophane type diterpenoids euphopepluanones F − K (1 − 4), along with eight known diterpenoids (5 − 12) were obtained from the seed of E. peplus (Fig. 1). Their structures were elucidated based on extensive NMR, X-ray crystallographic and electronic circular dichroism (ECD) experiments. Furthermore, the activity of compounds 1 − 4 in inducing lysosomal biogenesis were tested, in which only 2 displayed significant activity. Herein, we reported the structural elucidation and biological evaluation of these compounds.

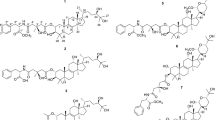
Chemical structures of compounds 1−12
2 Results and Discussion
2.1 Structure Elucidation
Euphopepluanone F (1), was obtained as colorless crystals. The molecular formula of 1 was established as C33H40O12 based on its positive HRESIMS ([M + Na]+, m/z 651.2426, calcd for C33H40O12Na, 651.2412). Its IR spectrum indicated the absorption bands for hydroxyl (3481 cm−1), carbonyl of ester (1743 cm−1), ketone (1723 cm−1), and double bond (1650 cm−1). Its 1D NMR spectra (Table 1) showed typical signals for three acetoxy groups (δC 170.4, 20.5; 170.0, 20.8; and 168.9, 20.7; δH 2.08, 2.05, and 1.67), and a benzoyloxy group (δC 165.6; δH 8.12, 7.57 and 7.45). In addition to these signals, the 13C NMR spectrum displayed four methyls (δC 20.7, 23.1, 24.0 and 25.2), one sp3 methylene (δC 52.3), one sp2 methylene (δC 114.2), five sp3 methines (δC 42.8, 47.7, 65.2, 72.7 and 81.0), two sp2 methines (δC 133.1 and 136.1), three sp3 quaternary carbons (δC 49.6, 79.1 and 85.2) and three sp2 quaternary carbons (δC 138.0, 204.9 and 213.2). These signals account for 19 carbons, indicating the absence of one carbon signal that may be attributed to conformational exchange of the jatrophane type diterpenoid molecule [17, 18]. The spectroscopic data of 1 was similar to (2S*,3S*,4R*,5R*,7S*,8R*,13S*,15R*)-5,7,8-triacetoxy-3-benzoyloxy-15-hydroxyjatropha-6(17),11E-diene-9,14-dione (5) [17] except for the presence of signals responsible for an hydroxy group (δH 2.33) and an additional oxygenated quaternary carbon (δC 79.1), the absence of the proton signal at C-2. These differences suggested that compound 1 harbors an additional hydroxy group attached at C-2, which was confirmed by the HMBC correlations from the hydroxy group to C-1 and C-2 (Fig. 2).

Key 1H- 1H COSY, HMBC, and ROESY correlations of compound 1
Initially, we try to determine the relative configuration of 1 by a ROESY experiment (Fig. 2). We observed ROESY cross peaks of H-7/H-4, H-7/H-13, OH-2/H-3 and H-8/H3-19. However, the lack of correlation of the hydrogen atom at the five membered ring and the eleven membered ring led the establishment of the gross relative configuration difficult. Furthermore, the disappearance of H-5 signal made the assignment of its relative configuration impossible. Fortunately, after several attempts, crystals of 1 (CCDC: 2,043,474) were obtained, which allowed the assignment of the relative configuration of 1. The absolute configuration of compound 1 was confirmed based on X-ray crystallography with Cu Kα radiation resulted in the Flack parameter = 0.04 (4) (Fig. 3).

ORTEP drawing of compound 1
The molecular formula of euphopepluanone I (2) was determined to be C36H44O12 based on its positive HRESIMS ([M + Na]+, m/z 691.2716, calcd for C36H44O12Na, 691.2725). Comparison of the spectroscopic data of 2 with those of 1 suggested similar structure but with different esterification patterns. The former harbored an angeloxy group (δH 6.16, 2.01, 1.87) instead of the acetoxy group at C-7. HMBC correlations of the carbonyl carbons (δC 166.9) and oxymethine protons (δH 6.09) placed the angeloxy group at C-7. The relative configuration of 2 was the same as that of 1 by their similar ROESY cross-peaks.
The positive HRESIMS data of euphopepluanone J (3) showed an [M + Na]+ ion at m/z 693.2512 (calculated for 693.2518), corresponding to the molecular formular C35H42O13. The mass spectrum indicate compound 3 was 42 mass units more than compound 1, suggesting that one of hydroxyl group in 1 was acetylated in compound 3. On comparing its NMR data with those of 1 (Table 1), 3 displayed additional signals responsible for an acetoxy group (δH 2.19; δC 169.8, 22.3), and the absence of the OH-2 signal. Thus, the OH-2 in 1 was inferred as being replaced by an acetoxy group in 3, which was supported by the HMBC correlations from H-2 to the carbonyl carbon. Therefore, the structure of 3 was delineated as shown.
Euphopepluanone K (4) possessed a molecular formula of C27H34O8 as deduced from its positive HRESIMS ([M + Na]+, m/z m/z 509.2151, calcd for C27H34O8Na, 509.2146). The spectra data of 4 closely related to those of 5, except for the absence of acetate signals at C-5, C-7, and C-8, suggesting the replacement of them with hydroxyl groups. Indeed, the resonances of C-4, C-6 and C-9 were up-shielded (ΔδC + 2.7, + 6.8, + 6.4 ppm), and C-5 and C-8 were down-shielded (ΔδC -0.7, -1.6) in 4, further supporting the presence of hydroxy groups at C-5, C-7, and C-8, instead of acetoxy groups in compound 5. The relative configuration of 4 was assigned as that of 1 by the nearly identical ROSEY data of these two compounds. Furthermore, the similar electronic circular dichroism (ECD) spectra of compounds 1, 2, 3 and 4 (Fig. 4) indicated they share the same absolute configurations.

ECD spectra of compounds 1−4
Eight known diterpenoids were characterized as hydroxyjatropha-6(17),11E-diene-9,14-dione (5) [17], 3,5,7,15-tetraacetoxy-9-nicotinoyloxy-14-oxojatropha-6(17),11-diene (6) [19], pepluanin D (7) [20], ingenol-20-angelate (8) [21], ingenol-3-angelate (9) [22], 5-O-benzoyl-20-deoxyingenol (10) [23], 5,8,9,15-tetraacetoxy-3-benzoyloxy-11,16-dihydroxypepluane (11) [24], and 5,8,9,11,15-pentaacetoxy-3-benzoyloxy-16-hydroxypepluane (12) [25] by comparing their spectroscopic data with those in the literatures.
2.2 Bioactivity Evaluation
To assess the activity to enhance lysosomal biogenesis of the new compounds 1–4, LysoTracker Red staining method was used. All the four new compounds increased the LysoTracker staining intensity. The cells were treated for 3 h with compounds 1 − 4 at 20 μM, and these compounds increased the LysoTracker staining intensity by 141.3%, 151.7%, 136.4% and 130.1%, respectively (Fig. 5a). Hep-14 was used as positive control [10]. It was further tested whether the lysosome biogenesis activities of these compounds are time- and concentration-dependent. As shown in Fig. 5b, HeLa cells were treated for 1, 3 and 6 h with 10, 20 and 40 μM of compound 2 as indicated. Induction of lysosomes was observed in a time- and concentration-dependent manner, with the greatest increase at 40 μM when the cells were treated for 6 h. Many lysosomal genes were upregulated during lysosome biogenesis. To confirm that compound 2 induce lysosomal biogenesis, the expression levels of a set of lysosomal genes were checked, including lysosomal-associated membrane protein 1 (LAMP1), cathepsin B (CTSB), cathepsin A (CTSA), lysosomal sulfatase (ARSB), and ATPase H+ transporting V0 subunit E1 (ATP6 V0E1). As shown in Fig. 5c, all these genes were upregulated at mRNA levels 3 h after treatment with compound 2. These data further demonstrated that compound 2 can induce lysosomal biogenesis. Then, we checked the level of LC3 dots, the marker for the activation of autophagy, induced by compound 2. The number of LC3 dots increased with the treatment of compound 2 in a dose-dependent manner (Fig. 5d, e). These results indicate that compound 2 could activate the lysosomal-autophagy pathway.

Compound 2 activates the lysosomal-autophagy pathway. a Compounds 1−4 induced lysosomal biogenesis. The number of lysosomes was stained with LysoTracker and mean fluorescence intensity (MFI) of LysoTracker was quantified. The bar graph showed the fold change of MFI of LysoTracker. Hep-14 served as positive control. b Compound 2 induced lysosomal biogenesis in a dose- and time-dependent manner. Cells were treated with compound 2 as indicated dose and time. The fold change of MFI of LysoTracker was analyzed. c Compound 2 induce the expression of lysosomal genes. HeLa cells were treated with compound 2 (20 μM, 3 h) and subjected to qRT-PCR analysis. The mRNA levels of lysosomal related genes were measured, and actin was used as an internal control. d, e Compound 2 increased the LC3 dots in a dose-dependent manner. The cells with CFP-LC3 were treated with compound 2 for 12 h with indicated concentration and the LC3 dots were imaged with confocal microscope. The representative images were shown in d and the quantification of LC3 dots were plotted in e. All experiments were carried out in triplicates and bar graph represents mean ± SD. p < 0.05 were considered statistically significant. **p < 0.01, ***p < 0.001
3 Experimental Section
3.1 General Experimental Procedures
Optical rotations were measured with a Jasco P-1020 automatic polarimeter. CD spectra were obtained on the Applied Photophysics circular dichroism spectrometer (Applied Photophysics, Leatherhead, Surrey, UK). High-resolution MS data were measured on an Agilent 1290 UPLC/6540 Q-TOF mass spectrometer in positive mode. IR spectra were determined on a NICOLET iS107 Mid-infrared spectrometer. NMR spectra were measured on Bruker AVANCE III 500 MHz and AV 600 MHz NMR spectrometers with TMS as the internal standard. An Agilent 1260 series instrument equipped with a SunFire-C18 column (5 μm, 10 mm × 250 mm) and XSelect HSS T3(5 μm, 10 mm × 150 mm) were used for high-performance liquid chromatography (HPLC). Silica gel (100 − 200, 200 − 300, 300 − 400) mesh (Qingdao Marine Chemical, Inc), NH MB 100–40/75 Silica gel (FUJI SILYSIA CHEMICAL LTD), Lichroprep RP-18 (40 − 63 μm, Fuji), and Sephadex LH-20 (20 − 150 μm, Pharmacia) was used for CC.
3.2 Plant Material
In August 2018, the seeds of E. peplus were collected from Kunming Botanical Garden, Yunnan Province, People’s Republic of China. A voucher specimen (No. kep-09–13) identified by Prof. Hu Shi-Jun (Southwest Forestry University) was deposited in the herbarium of the Kunming Institute of Botany, Chinese Academy of Sciences.
3.3 Extraction and Isolation
The air-dried the seeds of E. peplus (24 kg) were powdered and extracted with methanol thrice at room temperature. The extract was suspended in water and extracted with petroleum ether, and ethyl acetate. The ethyl acetate extract (800 g) was subjected to a silica gel column using petroleum ether/ethyl acetate (100:0 to 0:100, v/v) as eluent to obtain 10 fractions, F1 − F10, in which diterpenes are mainly concentrated in F7 and F8.
Fraction F7 (58 g) was subjected to MCI gel developed with MeOH-H2O (40:60–100:0) to give 12 fractions. F7.7 (5.5 g) was submitted to silica gel column chromatography and eluted with ether/ethyl acetate gradient (20:1–1:1) to afford 7 fractions F7.7.1-F7.7.7. The fraction F7.7.3 (2.3 g) was isolated by amino-silica gel column (ether/ethyl acetate, 20:1–1:1) giving 8 fractions. Fraction F7.7.3.6 (99 mg) was purified by semi-preparative HPLC to yield compound 4 (2.4 mg). The fraction 7.7.4 (2.1 g) was separated by Sephadex LH-20 (MeOH/MeCl3 50:50) to give four fractions, in which F7.7.4.2 (134 mg) was applied to preparative HPLC leading to compounds 3 (13.2 mg) and 5 (5.8 mg). F7.8 (6.3 g) was separated by silica gel column chromatography using ether/ethyl acetate gradient (20:1–1:1) to afford 8 fractions. Fraction F7.8.2 (1.5 g) and F7.8.3 (0.6 g) were chromatographed on Sephadex LH-20 (MeOH/MeCl3 50:50) to afford five and seven fractions, respectively. F7.8.2.5 (29 mg) and F7.8.3.2 (42 mg) were each submitted to semi-preparative HPLC to give compounds 10 (4.8 mg) and 2 (6.7 mg), respectively.
Fraction F8 (66 g) was subjected to MCI gel with MeOH-H2O (40:60–100:0) to give 13 fractions, in which compound 1 (378.3 mg) was obtained from fraction 7 as colorless crystals. Fraction F8.7 (12 g) was submitted to silica gel column with ether/ethyl acetate gradient (20:1- 1:1) leading to 6 fractions, in which fraction F8.7.1 (218 mg) was further purified by silica gel column with gradient of ether/ethyl acetate (10:1–1:1) to give five fractions F8.7.1.1-F8.7.1.5, and then the fraction F8.7.1.5 (33.7 mg) was subjected to preparative HPLC to yield compound 7 (22.3 mg). Fraction F8.7.3 (3.1 g) was chromatographed on Sephadex LH-20 (MeOH/MeCl3 50:50) giving three fractions. The resulted F8.7.3.1 (400 mg) was purified by silica gel column and eluted with ether/ethyl acetate (10:1–5:1) to afford six fractions, and 11 (42.7 mg) was obtained from fraction F8.7.3.1.2 by preparative HPLC. Fraction F8.7.3.2 (2.3 g) was chromatographed on silica gel column with ether/ethyl acetate (1:5) leading to seven fractions. The obtained fraction F8.7.3.2.1 (141 mg) was purified by preparative HPLC to yield compound 8 (51.9 mg). The fraction F8.8 (7.9 g) was submitted to Sephadex LH-20 (MeOH/MeCl3 50:50) to give six fractions, in which fraction F8.8.1 (6.1 g) was separated by silica gel column with gradient of ether/ethyl acetate (10:1–5:1) to afford 10 fractions. The fractions F8.8.1.4 (43 mg) and F8.8.1.6 (50 mg) were each purified by preparative HPLC producing compounds 12 (24.4 mg) and 6 (9.1 mg), respectively. Fraction F8.8.3 (695 mg) was chromatographed on with gradient of ether/ethyl acetate (10:1–5:1) to give eight fractions, in which F8.8.3.6 (51 mg) was further separated by preparative HPLC leading to compound 9 (16.8 mg).
Euphopepluanone F (1): a colorless massive crystal (MeOH/H2O, 20/1); mp 216–220 °C; [α] 25D + 64.9 (c 0.11, MeOH); UV (MeOH) λmax (log ε) 195 (4.52), 229 (3.96) nm; IR (KBr) vmax 3481, 2977, 1743, 1723, 1650, 1454, 1374, 1277, 1224 cm −1; 1H and 13C NMR data, see Table 1; ( +)-HRESIMS m/z [M + Na]+ 651.2426 (calcd for C33H40O12Na, 651.2412).
Euphopepluanone I (2): a white amorphous powder; [α] 25D + 41.2 (c 0.13, MeOH); UV (MeOH) λmax (log ε) 195 (4.48), 226 (4.05) nm; IR (KBr) vmax 3436, 2975, 2934, 1748, 1726, 1644, 1453, 1374, 1273 cm −1; 1H and 13C NMR data, see Table 1; ( +)-HRESIMS m/z [M + Na]+ 691.2716 (calcd for C36H44O12Na, 691.2725).
Euphopepluanone J (3): a white amorphous powder; [α] 25D + 16.3 (c 0.08, MeOH); UV (MeOH) λmax (log ε) 195 (4.54), 229 (3.91) nm; IR (KBr) vmax 3447, 2981, 1747, 1728, 1633, 1453, 1372, 1239 cm −1; 1H and 13C NMR data, see Table 1; ( +)-HRESIMS m/z [M + Na]+ 693.2512 (calcd for C35H42O13Na, 693.2518).
Euphopepluanone K (4): a white amorphous powder; [α] 25D + 59.3 (c 0.11, MeOH); UV (MeOH) λmax (log ε) 195 (4.45), 226 (3.92) nm; IR (KBr) vmax 3436, 2970, 2933, 1710, 1631, 1452, 1380, 1278 cm −1; 1H and 13C NMR data, see Table 1; ( +)-HRESIMS m/z [M + Na]+ 509.2151 (calcd for C27H34O8Na, 509.2146).
3.4 X-ray Crystallographic Analyses
Crystallographic Data for Compound 1. C33H40O12, M = 628.65, a = 10.0011(3) Å, b = 17.0114(4) Å, c = 10.2133(3) Å, α = 90°, β = 114.3100(10)°, γ = 90°, V = 1583.54(8) Å3, T = 100(2) K, space group P21, Z = 2, μ(CuKα) = 0.839 mm−1, 17,488 reflections measured, 5524 independent reflections (Rint = 0.0390). The final R1 values were 0.0330 (I > 2σ(I)). The final wR(F2) values were 0.0852 (I > 2σ(I)). The final R1 values were 0.0330 (all data). The final wR(F2) values were 0.0853 (all data). The goodness of fit on F2 was 1.065. Flack parameter = 0.04(4). These data can be obtained free of charge from The Cambride Crystallographic Data Centre via http://www.ccdc.cam.ac.uk/data_request/cif.
3.5 Cell Culture
The activity to enhance lysosomal biogenesis of compounds 1–4 was evaluated using HeLa cell line, which was cultured at 37 °C with 5% CO2 in Dulbecco’s modified Eagle’s medium supplemented with 10% fetal bovine serum (HyClone), 100,000 U/mL penicillin and 100 mg/mL streptomycin. HeLa cell was purchased from ATCC.
3.6 Screening for Compounds That Induce Lysosomal Biogenesis
Briefly, HeLa cells with 85% cell density in 96-well plates were treated with individual compounds at 20 μM in triplicate. Three hours later, cells were grown in fresh medium containing LysoTracker Red DND-99 (0.2 μM) for 30 min. Then, medium was changed to LysoTracker-free medium and images were taken with ArrayScan Infinity (Cellomics, ArrayScan VTI HCS). Positive compounds were subjected to validation by treating HeLa cells with different concentrations (10, 20 and 40 μM) and at 1, 3 and 6 h in triplicate and staining with LysoTracker Red DND-99.
3.7 Confocal Microscopy
CFP-LC3 expressing HeLa cells were treated with indicated compounds and images were collected by confocal microscopy. For live-cell imaging, cells grown on glass-bottom dishes were observed directly. All samples were examined with an inverted Olympus FV1000 confocal microscope. Images were analyzed with FV10-ASW 4.0a Viewer.
3.8 Quantitative Real-Time PCR with Reverse Transcription (qRT-PCR)
Total RNA was isolated from HeLa cells by using TRIzol Reagent (Invitrogen) according to the manufacturer’s recommendation. A reverse-transcription kit (Promega) was used to reverse transcribe RNA (1 µg) in a 20 µL reaction mixture. A real-time PCR system (7900HT Fast; Applied Biosystems) was used to quantify gene expression in triplicate. Amplification of the sequence of interest was normalized with the reference endogenous gene actin.
3.9 Statistics and Reproducibility
Data analyses were carried out using Prism 5, and Student’s t tests were employed for statistical analyses with a level of significance of p < 0.05.
References
M. Ernst, O.M. Grace, C.H. Saslis-Lagoudakis, N. Nilsson, H.T. Simonsen, N. Rønsted, J. Ethnopharmacol. 176, 90–101 (2015)
Q.W. Shi, X.H. Su, H. Kiyota, Chem. Rev. 108, 4295–4327 (2008)
A. Vasas, J. Hohmann, Chem. Rev. 114, 8579–8612 (2014)
G. Corea, E. Fattorusso, V. Lanzotti, O. Taglialatela-Scafati, G. Appendino, M. Ballero, P.-N. Simon, C. Dumontet, A.D. Pietro, J. Med. Chem. 46, 3395–3402 (2003)
J. Heiss, M. Iadarola, F. Cantor, A. Oughourli, R. Smith, A. Mannes, J. Pain 15, S67 (2014)
A. Vasas, D. Rédei, D. Csupor, J. Molnár, J. Hohmann, Eur. J. Org. Chem. 2012, 5115–5130 (2012)
B. Berman, Clin. Cosmet. Investig. Dermatol. 20, 111–122 (2012)
Editorial Committee of Flora of China of Chinese Academy of Science, Flora of Chia, vol. 44 (Science Press, Beijing, 1997), pp. 111
S.M. Ogbourne, P.G. Parsons, Fitoterapia 98, 36–44 (2014)
Y. Li, M. Xu, X. Ding, C. Yan, Z. Song, L. Chen, X. Huang, X. Wang, Y. Jian, G. Tang, C. Tang, Y. Di, S. Mu, X. Liu, K. Liu, T. Li, Y. Wang, L. Miao, W. Guo, X. Hao, C. Yang, Nat. Cell. Biol. 18, 1065–1077 (2016)
N.D. Zhao, X. Ding, Y. Song, D.Q. Yang, H.L. Yu, T.A. Adelakun, W.D. Qian, Y. Zhang, Y.T. Di, F. Gao, X.J. Hao, S.L. Li, J. Nat. Prod. 81, 1209–1218 (2018)
Y.L. Ma, X.H. Tang, W.J. Yuan, X. Ding, Y.T. Di, X.J. Hao, Nat. Prod. Bioprospect. 8, 131–135 (2018)
Y.N. Chen, X. Ding, Q.Y. Lu, D.M. Li, B.T. Li, S. Liu, L. Yang, Y. Zhang, Y.T. Di, X. Fang, X.J. Hao, Bioorg. Chem. 105, 104464 (2020)
T.A. Adelakun, X. Ding, R.M. Ombati, N.D. Zhao, O.O. Obodozie-Ofoegbu, Y.T. Di, Y. Zhang, X.J. Hao, Nat. Prod. Res. 34, 3027–3035 (2020)
Y.N. Chen, Q.Y. Lu, D.M. Li, Y.Y. Li, X.X. Pu, B.T. Li, X.H. Tang, H.Y. Tang, S. Liu, L. Yang, Y. Zhang, Y.T. Di, X. Fang, X.J. Hao, Nat. Prod. Res. (2020). https://doi.org/10.1080/14786419.2020.1749609
W.J. Yuan, W.F. Gao, J.Y. Zhao, Y. Zhang, D.Z. Chen, S.L. Li, Y.T. Di, X.J. Hao, Fitoterapia 144, 104583 (2020)
J. Hohmann, D. Rédei, P. Forgo, J. Molnár, G. Dombi, T. Zorig, J. Nat. Prod. 66, 976–979 (2003)
A. Hasan, G.-Y. Liu, R. Hu, H.A. Aisa, J. Nat. Prod. 82, 724–734 (2019)
J. Hohmann, A. Vasas, G. Gunther, G. Dombi, G. Blazso, G. Falkay, L. Mathe, G. Jerkovich, Phytochemistry 51, 673–677 (1999)
G. Corea, E. Fattorusso, V. Lanzotti, R. Motti, P.-N. Simon, C. Dumontet, A.D. Pietro, J. Med. Chem. 47, 988–992 (2004)
L.J. Lin, A.D. Kinghorn, J. Agric. Food. Chem. 31, 396–400 (1983)
J. Hohmann, F. Evanics, L. Berta, T. Bartok, Planta Med. 66, 291–294 (2000)
L. Zhang, L. Gao, Z. Li, X. Yan, A. Ding, Int. J. Mol. Sci. 13, 11247–11259 (2012)
J. Hohmann, G. Günther, A. Vasas, A. Kálmán, G. Argay, J. Nat. Prod. 62, 107–109 (1999)
J. Jakupovic, T. Morgenstern, M. Bittner, M. Silva, Phytochemistry 47, 1601–1609 (1998)
Acknowledgements
This research was supported by the National Natural Science Foundation of China under Grant (Numbers 21432010; 31872666; 82073740); National Key R&D Program of China under Grant (Number 2018YFA0900600); Technological leading talent project of Yunnan (2015HA020); Special Fund for Talent Introduction of Kunming Institute of Botany, CAS (to Xin Fang); and Key R&D Program of Yunnan under Grant (2019ZF011-2).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
All authors declare that they have no conflict of interest.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Chen, YN., Ding, X., Li, DM. et al. Jatrophane Diterpenoids from the Seeds of Euphorbia peplus with Potential Bioactivities in Lysosomal-Autophagy Pathway. Nat. Prod. Bioprospect. 11, 357–364 (2021). https://doi.org/10.1007/s13659-021-00301-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13659-021-00301-4