
Abstract
Ten cucurbitane-type triterpene glycosides, including five new compounds named charantosides H (1), J (2), K (3), momorcharacoside A (4), goyaglycoside-l (5), and five known compounds (6–10), were isolated from the EtOAc extract of Momordica charantia fruits. The chemical structures of these compounds were identified by 1D and 2D NMR and HRESIMS spectroscopic analyses. Configurations of new compounds were determined by ROESY correlations and comparison of their 13C NMR data with literature reported values. All compounds were evaluated for their inhibition against α-glucosidase, in which compounds 2, 5, 7, 8, 9 showed moderate inhibitory activities with IC50 values ranging from 28.40 to 63.26 μM comparing with the positive control (acarbose, IC50 87.65 ± 6.51 μM).
Graphic Abstract

Similar content being viewed by others
Avoid common mistakes on your manuscript.
1 Introduction
Diabetes mellitus (DM) is a type of metabolic disorder caused by insufficient insulin secretion or insulin utilization disorder, and is marked by persistent hyperglycemia [1]. The latest edition of the International Diabetes Federation (IDF)ʼs Diabetes Atlas estimates that, in 2019, about 463 million adults were living with DM around the world, and 11.3% of global deaths were due to DM [2]. Moreover, it can overwhelm the social and economic welfare of all countries, regardless of their economic level. Therefore, the prevention and treatment of DM is essential. In modern medicine, there are many oral hypoglycemic agents (OHAs) used to treat DM, however, they also have different adverse effects including gastrointestinal upset, lactic acidosis, characteristic hepatocyte injury, dizziness, acute hypoglycemia, and even death [3, 4]. Therefore, screening the new hypoglycemic drugs with high efficiency and low toxicity from the natural products of plants is urgently required. In China, ancient books have recorded many Chinese herbs used in the treatment of DM, among which Momordica charantia (Cucurbitaceae) was very popular and had an incredible hypoglycemic effect. Therefore, M. charantia has great research potential in reducing blood sugar and is a hot spot in modern phytochemical research.
Momordica charantia is a traditional medicinal and edible plant, which has a long history of use in developing countries. Modern phytochemistry research shows that both crude extracts and secondary metabolites (including polysaccharides, triterpenes, saponins, proteins, flavonoids, alkaloids, and steroids, etc.) of M. charantia possess anti-diabetic activity [5,6,7,8,9,10]. Thereinto, cucurbitane-type triterpenoids are the main bioactive ingredients in M. charantia, which can control blood sugar through multiple mechanisms of action (such as PPAR-γ activator, PTP1B inhibitor, and α-glucosidase inhibitor, etc.) [11, 12]. To date, more than 300 kinds of cucurbitane-type triterpenoids have been identified, and some of them showed prominent biological activity [6, 10, 13]. Based on this, we conducted further excavations, hoping to find new cucurbitane-type triterpenoids with good hypoglycemic activity.
2 Results, Discussion and Conclusion
Phytochemical investigation of the fruits of M. charantia resulted in the isolation of five new compounds, charantosides H (1), J (2), K (3), momorcharacoside A (4), and goyaglycoside-l (5), by repeated column chromatography (CC) (Fig. 1). Meanwhile, five known compounds (6–10) were isolated and identified, on the basis of comparison of obtained values with literature values, as (19R,23E)-5β,19-epoxy-19-methoxycucurbita-6,23,25-trien-3β-ol 3-O-β-d-allopyranoside (6) [14], charantoside I (7) [15], charantoside III (8) [15], momordicoside K (9) [16], and 7β,25-dimethoxycucurbita-5(6),23(E)-dien-19-al 3-O-β-d-allopyranoside (10) [17] (Fig. 1), respectively.

Chemical structures of compounds 1–10 from M. charantia
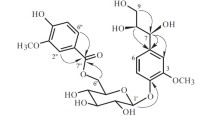
All of the five new compounds were considered as monoglycosides based on the IR absorption bands of a glycosidic function (e.g., 1: νmax 3426, 1084, 1033 cm−1) [15, 18] and an anomeric proton signal of the glycosyl moiety observed in their 1H NMR spectra. After acid hydrolysis, the sugars of 4 and 5 were identified as d-allose and d-glucose by comparing their TLC and specific rotation with the corresponding authentic sample. The predicted structures for these new compounds as depicted below were supported by analysis of the 1H-1H COSY, HMBC, and NOESY data (Figs. 2 and 3), in addition to 13C-DEPT, and HMQC data.

Key 1H-1H COSY and HMBC correlations of 1, 4, and 5

Key ROESY correlations of 1, 2, 4, and 5
Compound 1 was obtained as white amorphous powder. The molecular formula C38H62O9 was deduced from its HRESIMS (positive-ion mode) data (685.4295 [M + Na]+), indicated eight degrees of unsaturation. The 1H NMR spectrum of 1 (Table 1) showed signals assignable to seven methyl groups [δH 1.73, 1.69, 1.47, 1.05, 0.88, 0.86, 0.84], two methoxy groups [δH 3.58, 3.29], three olefinic protons [δH 6.27 (1H, dd, J = 1.8, 9.0 Hz), 5.48 (1H, dd, J = 3.6, 9.6 Hz), 5.22 (1H, d, J = 8.4 Hz)], and an anomeric proton [δH 5.36 (1H, d, J = 7.8 Hz, H-1′)]. The 13C NMR (Table 2) showed 38 carbon signals. The DEPT spectrum exhibited nine methyls, eight methylenes, fifteen methines, and six quaternary carbons. And 13C NMR spectrum showed olefinic carbons appeared at δC 135.1, 134.4, 129.1, and 127.7. The NMR data of 1 were closely similar to those of (19R,23R)-5β,19-epoxy-19,23-dimethoxycucurbita-6,24-dien-3β-ol 3-O-β-d-allopyranoside (charantoside II) [15] except for the signals due to the stereochemistry at C-19. And that ΔδC values [δC (charantoside II) − δC (1)] for the relevant signals were calculated as 1.7 (C-5), − 7.3 (C-8), − 0.7 (C- 9), 2.8 (C-10), 1.6 (C-11) and -1.4 (C-19) from the 13C NMR data of charantoside II and 1, which were feckly consistent with the ΔδC values [δC (19R) − δC (19S)] of 1.8 (C-5), − 7.8 (C-8), − 0.7 (C-9), 2.6 (C-10), 1.8 (C-11) and -2.6 (C-19) calculated from the 13C NMR data of 5β,(19R)- and 5β,(19S)-epoxy-19,23-dimethoxycucurbita-6,24-dien-3β-ol [19]. Therefore, compound 1 has the (S)-configuration at C-19, and the ROESY correlation (Fig. 3) of H-8/H-19 confirmed the above deduction [20]. The actual connection positions were further established on the basis of HMBC correlations (Fig. 2) between H-1′ (δH 5.36) of the sugar moiety and C-3 (δC 84.7) of the aglycon group. And we found that the NMR data of the sugar moiety of 1 was basically consistent with 4, which corroborates the presence of a d-allose. In addition, long-range correlations were also observed at 19-methoxyl protons (δH 3.58)/C-19 (δC 113.8), and 23-methoxyl protons (δH 3.29)/C-23 (δC 74.6). Therefore, the molecular formula of 1, along with the 1D and 2D spectroscopic data illustrated that the structure of 1 could be assigned as (19S,23R)-5β,19-epoxy-19,23-dimethoxycucurbita-6,24-dien-3β-ol 3-O-β-d-allopyranoside, named charantoside H.
Compound 2 was obtained as white solid. It showed a quasi-molecular ion at 685.4293([M + Na]+) in the HRESIMS (positive-ion mode) spectrum and had the same molecular formula C38H62O9 as 1, which also possessed eight degrees of unsaturation. Detailed analysis of the 1H, 13C NMR, and DEPT spectra (Tables 1 and 2) of compound 2, which showed heavily resemblance in all signals to those of (19S,23R)-5β,19-epoxy-19,23-dimethoxycucurbita-6,24-dien-3β-ol 3-O-β-d-allopyranoside (charantoside H, 1) except that d-allose of 1 were replaced by d-glucose in 2. In the 13C NMR spectrum of compound 2, an anomeric carbon atom (δC 105.2) and a series of oxygenated carbon signals (δC 78.6, 77.8, 76.1, 71.8, and 62.9) were in line with 5, confirmed the presence of a β-d-glucopyranosyl residue [15]. The 1H-1H COSY and HMBC correlations of compound 1 and 2 were similar, but their ROESY spectra (Fig. 3) showed the different correlations between H-3′/H-5′, further proved the type of sugar moiety of 2. Similarly, the absolute configuration of C-19 (S) in 2 was confirmed by ROESY correlation of H-8/H-19. Based on the above corroboration, the structure of compound 2 was identified as (19S,23R)-5β,19-epoxy-19,23-dimethoxycucurbita-6,24-dien-3β-ol 3-O-β-d-glucopyranoside, named charantoside J.
Compound 3 was obtained as white amorphous powder. It revealed a quasi-molecular ion at 685.4296 ([M + Na]+) in the HRESIMS (positive-ion mode) spectrum and had the same molecular formula C38H62O9 as 1. According to the 1H, 13C NMR, and DEPT spectra (Tables 1 and 2) of 3, which were also similar to those of charantoside H (1) except for the signals due to the stereochemistry at C-23. Compound 3 exhibited 1H NMR signals (Table 1) for the side-chain protons at δ 1.03 (3H, d, J = 5.6 Hz, a secondary methyl), 1.69 and 1.73 (each 3H, s, two vinylic methyls), 3.29 (3H, s, an O-methyl), 4.10 (1H, m, an allylic oxymethine), and 5.15 (1H, dt, J = 1.6, 9.6 Hz, an olefinic methine). Detailed comparisons of its 13C NMR data (Table 2) with those of compound 1, the ΔδC values [ΔδC (1) − ΔδC (3)] for the side-chain signals were calculated as − 1.1 (C-20), − 1.0 (C-21), + 0.3 (C-22), − 1.7 (C-23), + 0.5 (C-24), − 1.6 (C-25), − 0.0 (C-26), and − 0.3 (C-27), which were almost in line with the ΔδC values [ΔδC (23R) − ΔδC (23S)] of − 0.9 (C-20), − 0.9 (C-21), + 0.4 (C-22), − 1.6 (C-23), + 0.5 (C-24), − 1.4 (C-25), − 0.1 (C-26), and − 0.4 (C-27) calculated from the 13C NMR data of charantoside II (23R) and charantoside VI (23S) [15]. As a consequence, compound 3 has the (S)-configuration at C-23. The 1H-1H COSY, HMBC, and ROESY correlations of compounds 1 and 3 were similar as well, suggesting that both compounds 1 and 3 have an almost identical planar chemical structure. Analogously, ROESY correlation of H-8/H-19 certified that acetal carbon (C-19) should has the (S)-configuration. Eventually, the structure of compound 3 was identified as (19S,23S)-5β,19-epoxy-19,23-dimethoxycucurbita-6,24-dien-3β-ol 3-O-β-d-allopyranoside, named charantoside K.
Compound 4 was obtained as white amorphous powder and assigned a molecular formula of C36H56O7, (HRESIMS m/z 623.3926 [M + Na]+), indicating nine degrees of unsaturation. The absorption at 238 nm in the UV spectrum exhibited a conjugated double bond group. The 1H NMR spectrum of 4 (Table 1) showed signals allocable to seven methyl groups [δH 1.73, 1.72, 1.46, 1.04, 0.87, 0.87, 0.77], five olefinic protons [δH 6.33 (1H, dd, J = 10.8, 15.0 Hz), 6.17 (1H, dd, J = 1.8, 10.2 Hz), 5.91 (1H, d, J = 10.2 Hz), 5.53 (1H, dd, J = 3.6, 9.6 Hz), 5.48 (1H, dd, J = 8.4, 14.4 Hz)], and a β-allopyranoside moiety [δH 5.38 (1H, d, J = 7.8 Hz, H-1′)] [21]. After acid hydrolysis of 4 with HCl/MeOH, d-allose was detected by TLC and specific rotation comparing with the standard. The 13C NMR and DEPT spectrum (Table 2) of 4 revealed signals assignable to the sugar moiety and tetracylic part were very semblable to those of (23E)-5β,19-epoxycucurbita-6,23,25-trien-3β-ol 3-O-β-d-allopyranoside (charantosides IV) [15], while the signals of side chain were significantly disparate. The olefinic carbons at δC 138.9, 124.6, 126.1, 132.1 and their coupling constants in the 1H-NMR spectrum [δH 5.48 (1H, dd, 8.4, 14.4), 6.33 (1H, dd, 10.8, 15.0), 5.91 (1H, d, 10.2)] implied that a conjugated double bond existed in the side chain. This was further confirmed via the 1H-1H COSY correlations of H-21/H-20/H-22/H-23/H-24 and the key HMBC correlations Me-21/C-17, C-20, C-22, H-22/C-21, C-24, H-24/C-22, C-23, H-26/C-24, C-25, C-27, and H-27/C-24, C-25, C-26 (Fig. 2). Therefore, the structure of the side chain was almost identical to 5β,19-epoxy-cucurbita-6,22E,24-trien-3β-ol [22]. Based on the above observation, compound 4 was identified as 5β,19-epoxy-cucurbita-6,22E,24-trien-3β-ol 3-O-β-d-allopyranoside, and named momorcharacoside A.
Compound 5 was obtained as white amorphous powder and assigned a molecular formula of C37H60O9, (HRESIMS m/z 671.4136 [M + Na]+), indicating eight degrees of unsaturation. The 1H NMR spectrum of 5 (Table 1) showed signals assignable to seven methyl groups [δH 1.54, 1.54, 1.54, 0.92, 0.89, 0.82, 0.79], one methoxy groups [δH 3.44], four olefinic protons [δH 6.28 (1H, d, J = 9.6 Hz), 5.92 (1H, overlap), 5.92 (1H, overlap), 5.48 (1H, dd, J = 3.6, 9.6 Hz)], and a β-glucopyanosyl moiety [δH 4.92 (1H, d, J = 7.8 Hz, H-1′)] [18, 21]. The suger moiety were determined to be d-glucose on the basis of acidic hydrolysis and TLC and specific rotation analysis. The 13C NMR (Table 2) showed 37 carbon signals, which were closely similar to those of 19(R)-methoxy-5β,19-epoxycucurbita-6,23-diene-3β,25-diol 3-O-β-d-glucopyranoside (goyaglycoside-a) [23] except for the signals due to the stereochemistry at C-19. Thus, the ΔδC values [δC (goyaglycoside-a) -−δC (5)] for the relevant signals were feckly consistent with those reported in the literature [19]. Therefore, compound 5 has the (S)-configuration at C-19, which was further confirmed by the ROESY correlation (Fig. 3) of H-8/H-19. Based on the above proof, compound 5 was identified as 19(S)-methoxy-5β,19-epoxycucurbita-6,23-dien-3β,25-diol 3-O-β-d-glucopyranoside, named goyaglycoside-l.
In this study, five new and five known compounds were isolated from M. charantia, all of which were cucurbitane-type triterpene glycosides. All compounds were evaluated for their α-glucosidase inhibitory activities with acarbose as a positive control. Compounds 2, 5, 7, 8, and 9 showed moderate inhibitory activities with IC50 values of 63.26 ± 3.04, 59.13 ± 4.67, 35.08 ± 4.15, 36.38 ± 3.03, 28.40 ± 2.08 μM, respectively. The IC50 value of positive control (acarbose) was 87.65 ± 6.51 μM (Table 3). Interestingly, all the active compounds contained β-d-glucopyranosyl, suggesting that the presence of glucose groups may affect the activity of triterpenes. However, further studies are needed to determine the structure–activity relationship of the cucurbitacene-type triterpenes. The results of this study also showed that cucurbitane-type triterpene glycosides might be the key ingredient in the hypoglycemic effect of M. charantia, some of them had significant blood sugar lowering effect.
3 Experimental Section
3.1 General Experimental Procedures
UV spectra were recorded on a UV-2401PC spectrometer (Shimadzu, Kyoto, Japan). Optical rotations were measured in methanol on JASCO P-1020 digital polarimeter (Jasco, Tokyo, Japan). IR spectra were scanned on a Bruker Tensor-27 Fourier transform infrared spectrometer with KBr pellets (Bruker, German). High-resolution (HR) ESI mass spectra data were measured on a Waters API QSTAR Pulsar spectrometer. 1D and 2D NMR spectra were obtained in pyridine-d5 on Bruker Ascend-400, 600 and 800 MHz NMR spectrometers with tetramethylsilane (TMS) as internal standard (Bruker, Zurich, Switzerland). Column chromatography (CC) was performed on macroporous resin (D-101, Tianjin, China), Lichroprep RP-18 (Merck, German), sephadex LH-20 (Pharmacia, USA), silica gel (200–300 mesh, Qingdao, China), and Semi-preparative HPLC was performed on an Agilent 1260 liquid chromatography system equipped with a ZORBAX SB-C18 column (5 μm, 9.4 × 250 mm, 3.0 mL/min) and a DAD detector. Fractions were detected by TLC, and spots were visualized by spraying with 10% H2SO4 in EtOH, followed by heating. α-glucosidase inhibitory activity was evaluated on the basis of the ability of the compounds to decrease glucosidase activity and then inhibit the breaking of glycosidic bonds in p-nitrophenyl-α-d-glucopyranoside (PNPG). Water was purchased from wahaha group co. LTD. Acetonitrile (chromatographic grade) was purchased from OCEANPAK (Sweden). Common organic solvents are industrial grade, used after re-distillation. PNPG was obtained from Sigma Chemical Co. (St. Louis, Mo, USA). α-glucosidase was purchased from Shanghai yuanye biotechnology Co., Ltd. Potassium phosphate buffer solution (PPBS) was obtained from Shanghai Yidian Scientific Instrument Co., Ltd. 96-well plates was purchased from Nest biotechnology co., LTD.
3.2 Plant Material
Dried slices of M. charantia were purchased from the Luosiwan Chinese Herbal Medicine Market in Kunming, Yunnan Province, China, in February 2017. The material was identified by associate Prof. Jian-Chao Cheng from Kunming Institute of Botany (KIB), Chinese Academy of Science (CAS). A specimen was deposited in the State Key Laboratory of Phytochemistry and Plant Resource in West China, Kunming Institute of Botany, Kunming, China.
3.3 Extraction and Isolation
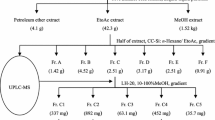
The dried fruits of M. charantia (40.0 kg) were sliced and extracted with MeOH. The solution was concentrated under reduced pressure to obtain a crude extract (25 kg), which was then successfully partitioned with petroleum ether (PE), EtOAc (EA), and n-butanol, respectively. The EtOAc fraction (2.0 kg) was subjected to the D101 macroporous resin, eluting with gradient system of MeOH/H2O (30:70, 50:50, 70:30, 90:10, 100:1) to afford five fractions. The fraction (MeOH/H2O 90:10, 87.0 g) was chromatographed on a silica gel column, eluting with gradient system of CHCl3/MeOH (100:1–1:1) to give four fractions (Fr.1–Fr.4). Fr.2 (16.8 g) was applied to ODS column, eluting with MeOH/H2O to give six sub fractions (Fr.2.1–Fr.2.6). Fr.2.5 (4.2 g) was separated over silica gel column (PE/EA) followed by semi-preparative HPLC (CH3CN/H2O), to yield compounds 1 (1.0 mg), 2 (1.0 mg), 3 (2.5 mg), and 10 (4.0 mg), respectively. Fr.2.6 (2.1 g) was successively purified by open column CC (CHCl3/MeOH) and semi-preparative HPLC (CH3CN/H2O), respectively, to afford compounds 4 (28.0 mg), 6 (6.0 mg), 7 (5.0 mg), 8 (1 mg), and 9 (30.0 mg). Similarly, Fr.3 (643 mg) was purified by RP-HPLC with CH3CN /H2O as eluent to obtain compounds 5 (10.0 mg).
3.3.1 Charantoside H (1)
White amorphous powder; [α]19D − 49.62 (c 0.11, MeOH); UV (MeOH) λmax (log ε) 196 (4.22) nm; IR (KBr) νmax 3426, 3027, 2926, 2875, 2815, 1736, 1634, 1465, 1447, 1380, 1320, 1292, 1260, 1218, 1182, 1154, 1108, 1084, 1049, 1033, 986, 953, 926, 882, 843, 801, 775, 747, 721, 696, 557, 529, 513, 467, 440, 411, 402 cm−1; For 1H NMR and 13C NMR (pyridine-d5) spectroscopic data, see Tables 1 and Table 2; HRESIMS m/z 685.4295 [M + Na]+ (calcd for C38H62O9Na, 685.4286).
3.3.2 Charantoside J (2)
White solid; [α]20D − 88.78 (c 0.11, MeOH); UV (MeOH) λmax (log ε) 196 (4.33) nm; IR (KBr) νmax 3424, 3026, 2928, 2875, 2815, 1736, 1636, 1465, 1446, 1383, 1320, 1292, 1260, 1218, 1183, 1153, 1110, 1086, 1050, 1033, 987, 952, 919, 817, 801, 775, 747, 721, 695, 618, 582, 557, 529, 513, 466, 411, 402 cm−1; For 1H NMR and 13C NMR (pyridine-d5) spectroscopic data, see Table 1 and Table 2; HRESIMS m/z 685.4293 [M + Na]+ (calcd for C38H62O9Na, 685.4286).
3.3.3 Charantoside K (3)
White amorphous powder; [α] − 35.77 (c 0.13, MeOH); UV (MeOH) λmax (log ε) 196 (4.15) nm; IR (KBr) ν23Dmax 3428, 3028, 2925, 2874, 1736, 1630, 1465, 1448, 1377, 1307, 1287, 1260, 1212, 1197, 1180, 1155, 1108, 1082, 1049, 1033, 985, 953, 941, 926, 882, 843, 803, 778, 755, 731, 691, 550, 522, 496, 467, 449, 440, 411, 402 cm−1; For 1H NMR and 13C NMR (pyridine-d5) spectroscopic data, see Tables 1 and 2; HRESIMS m/z 685.4296 [M + Na]+ (calcd for C38H62O9Na, 685.4286).
3.3.4 Momorcharacoside A (4)
White amorphous powder; [α]24D − 53.88 (c 0.29, MeOH); UV (MeOH) λmax (log ε) 238 (4.02), 196 (4.07), 211(3.73) nm; IR (KBr) νmax 3391, 3124, 2949, 2873, 2387, 2318, 1637, 1592, 1469, 1397, 1377, 1348, 1310, 1085, 1034, 1000, 777, 749, 684, 661, 628, 586, 531, 493, 410 cm−1; For 1H NMR and 13C NMR (pyridine-d5) spectroscopic data, see Tables 1 and 2; HRESIMS m/z 623.3926 [M + Na]+ (calcd for C36H56O7Na, 623.3918).
3.3.5 Goyaglycoside-l (5)
White amorphous powder; [α]20D − 71.96 (c 0.14, MeOH); UV (MeOH) λmax (log ε) 196 (4.21) nm; IR (KBr) νmax 3427, 3027, 2970, 2947, 2927, 2873, 1735, 1632, 1464, 1449, 1377, 1312, 1288, 1256, 1199, 1158, 1138, 1112, 1078, 1050, 980, 950, 941, 925, 844, 803, 778, 753, 733, 695, 579, 549, 529, 491, 466, 450, 439, 429, 412 cm−1; For 1H NMR and 13C NMR (pyridine-d5) spectroscopic data, see Tables 1 and 2; HRESIMS m/z 671.4136 [M + Na]+ (calcd for C37H60O9Na, 671.4130).
3.4 Acid Hydrolysis of Compounds 4 and 5 for Sugar Analysis
Compounds 4 and 5 (5 mg each), were separately dissolved in 2 M HCl/CH3OH (1:1, 5 mL) and heated at 80 °C for 4 h in a water bath. CHCl3/H2O 1:1 (5 mL × 3) was used for extraction. The aqueous phase was neutralized with Na2CO3. Each H2O layer was concentrated in vacuo to give a monosaccharide, which was identified by TLC [BuOH/acetic ether/H2O (4:1:5 upper layer)] and specific rotation compared with the authentic samples, All: Rf = 0.47, [α]21D = + 24.4; Glc: Rf = 0.51, [α]21D = + 51.9.
3.5 α-Glucosidase Inhibitory Activity
The α-glucosidase inhibition assay was performed according to the method adapted from the literature with slight modifications [24]. α-glucosidase can cut glycosidic bonds in the PNPG to produce 4-nitrophenol (yellow), then measured its absorbance can determine the activity of enzyme. The test samples and ursolic acid (positive control) were dissolved in dimethylsulfoxide (DMSO), and then diluted with PPBS (pH 6.86) to the required concentration. The α-glucosidase (1.0 U/mL) and substrate (PNPG, 2.5 mM) were dissolved in PPBS. Sample wells included 60 μL of PPBS, 10 μL of test substances, 30 μL of enzyme stock solution, and incubated at 37 °C for 10 min. After the pre-incubation phase, 40 μL of PNPG solution was added and the mixture was incubated for another 20 min at 37 °C. Finally, 80 μL Na2CO3 (0.2 M) solution was added to the sample wells to stop the reaction. The absorbance of the reaction mixture was recorded at 405 nm using a microplate reader. All samples were measured in triplicate. The inhibition rate (%) was calculated by the following formula: Inhibition (%) = [1 − (Asample/Acontrol)] × 100.
References
G.H. Wu, Y.X. Zang, Y.X. Liu, Chinese Health Management Dictionary (2001), 468 pp
IDF diabetes atlas (2019), https://www.diabetesatlas.org/. Accessed 6 Feb 2020
E.K. Stuermer, M. Besser, N. Terberger, V. Koester, H.S. Bachmann, A.L. Severing, Naunyn-Schmiedebergs Arch. Pharmacol. 392, 371–380 (2019)
E.L. Peter, F.M. Kasali, S. Deyno, A. Mtewa, P.B. Nagendrappa, C.U. Tolo, P.E. Ogwang, D. Sesaazi, J. Ethnopharmacol. 231, 311–324 (2019)
C. Zhang, M. Huang, R. Hong, H. Chen, Int. J. Biol. Macromol. 122, 619–627 (2019)
S.R. Shivanagoudra, W.H. Perera, J.L. Perez, G. Athrey, Y.X. Sun, G.K. Jayaprakasha, B.S. Patil, Bioorg. Chem. 87, 31–42 (2019)
S. Jiang, L. Xu, Y. Xu, Y.S. Guo, L. Wei, X.T. Li, W. Song, Electron. J. Biotechnol. 43, 41–47 (2020)
G. Chhabra, A. Dixit, Bioinformation 9, 766–770 (2013)
B. Shan, J.H. Xie, J.H. Zhu, Y. Peng, Food Bioprod. Process. 90, 579–587 (2012)
S. Jia, M. Shen, F. Zhang, J. Xie, Int. J. Mol. Sci. 18, 2555 (2017)
P.C. Hsiao, C.C. Liaw, S.Y. Hwang, H.L. Cheng, L.J. Zhang, C.C. Shen, F.L. Hsu, Y.H. Kuo, J. Agric. Food Chem. 61, 2979–2986 (2013)
S.Y. Lee, S.H. Eom, Y.K. Kim, N.I. Park, S.U. Park, J. Med. Plants Res. 3, 1264–1269 (2009)
J. Yue, Y. Sun, J. Xu, X. Zhang, Y. Zhao, J. Nat. Med. 74, 41 (2020)
J. Qi, X. Cao, Y. Sun, L. Cheng, Patent No. CN107556362A (2018)
T. Akihisa, N. Higo, H. Tokuda, M. Ukiya, H. Akazawa, Y. Tochigi, Y. Kimura, T. Suzuki, H. Nishino, J. Nat. Prod. 70, 1233–1239 (2007)
H. Okabe, Y. Miyahara, T. Yamauchi, Tetrahedron. Lett. 23, 77–80 (1982)
Y. Liu, Z. Ali, I.A. Khan, Planta Med. 74, 1291–1294 (2008)
S. Nakamura, T. Murakami, J. Nakamura, H. Kobayashi, H. Matsuda, M. Yoshikawa, Chem. Pharm. Bull. 54, 1545–1550 (2006)
K. Zeng, Y.N. He, D. Yang, J.Q. Cao, X.C. Xia, S.J. Zhang, X.L. Bi, Y.Q. Zhao, Eur. J. Med. Chem. 81, 176–180 (2014)
C.C. Liaw, H.C. Huang, P.C. Hsiao, L.J. Zhang, Z.H. Lin, S.Y. Hwang, F.L. Hsu, Y.H. Kuo, Planta Med. 81, 62–70 (2015)
J. Yue, Y. Sun, J. Xu, J. Cao, G. Chen, H. Zhang, X. Zhang, Y. Zhao, Phytochemistry 157, 21–27 (2019)
J.Q. Cao, B.Y. Zhang, Y.Q. Zhao, Chin. Herb. Med. 5, 234–236 (2013)
T. Murakami, A. Emoto, H. Matsuda, M. Yoshikawa, Chem. Pharm. Bull. 49, 54–63 (2001)
J.L. Perez, G.K. Jayaprakasha, B.S. Patil, Food Chem. 288, 178–186 (2019)
Acknowledgements
The work was supported by a program of the National Natural Science Foundation of China (Nos. 31872675 and 81373288) and the cooperation program between Chinese Academy of Sciences and Guangdong Province (2013B09110011).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
All authors declare no conflict of interest.
Additional information
Dedicated to Professor Han-Dong Sun on the occasion of his 80th birthday.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Gao, Y., Chen, JC., Peng, XR. et al. Cucurbitane-Type Triterpene Glycosides from Momordica charantia and Their α-Glucosidase Inhibitory Activities. Nat. Prod. Bioprospect. 10, 153–161 (2020). https://doi.org/10.1007/s13659-020-00241-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13659-020-00241-5