
Abstract
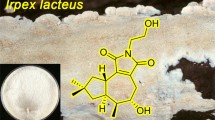
Six previously undescribed 5,6-seco-tremulane analogues, together with two known ones, were isolated from the culture broth of the medicinal fungus Irpex lacteus HFG1102. The structures of the new compounds were elucidated via extensive spectroscopic methods, including NMR and HRMS spectroscopic analyses.
Graphical Abstract

Similar content being viewed by others
Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
1 Introduction
Higher fungi are valuable and irreplaceable resources of natural product-derived lead compounds for drug research and development [1]. The fungus Irpex lacteus has been used as a medicinal fungus for decades in China for the treatment of inflammation, bacterial/fungal infection, and urinary retention [2]. The polysaccharide products of the submerged culture of I. lacteus have been developed and approved by SFDA as a clinically used drug for curing chronic glomerulonephritis [2]. However, although this fungus has been widely used as an industrial source to obtain polysaccharide products, its secondary metabolites remained unknown.
The 5,6-seco-tremulane skeleton, which was regarded as the resultant of Baeyer–Villiger oxidation of tremulane, was first and mainly reported from the basidiomycete Conocybe siliginea [3, 4]. To the best of our knowledge, the distribution of this type of sesquiterpenes only reported from three species, the basidiomycete Flavodon flavus BCC 17421 [5], and the endophytic fungi Colletotrichum capsici [6] and Ceriporia lacerate [7]. Notably, this type of sesquiterpenes always presented in inseparable mixtures. This study, as part of our ongoing search for promising lead compounds from higher fungi, concentrated on the secondary metabolites of the culture broth of the medicinal fungus I. lacteus, led to the isolation of six undescribed 5,6-seco-tremulane congeners (1–6) and two known tautomeric isomers (7, 8) (Fig. 1). Herein, we report the isolation, structure elucidation of the new compounds.

Structures of compounds 1–8
2 Results and Discussion
Compounds 1 and 2 were obtained as an inseparable colourless mixture with the ratio of approximately 1:0.9 deduced from the intensity of 1H NMR resonance. The 1H and 13C NMR spectra of the mixture displayed paired signals, indicating the similar structures of 1 and 2. Nevertheless, comprehensive analysis of the 1D and 2D NMR spectra expedited the structural elucidation of 1 and 2 unambiguously based on the relatively less overlapped signals. Elucidating of the HSQC, HMBC, and 1H–1H COSY spectra facilitated the assignments of two class of signals corresponding to 1 and 2.
The signals assigned for compound 1 consisted of a methyl singlet, six methylenes, four methines (including a ketal carbon), and four quaternary carbons (two olefinic, one carbonyl) (Tables 1, 2). The 1H–1H COSY of H-13/H-6/H-7/H-8, and H-4/H-3/H-12 enabled the connection of C-13–C-6–C-7–C-8 and C-12–C-3–C-4, respectively. The HMBC correlations from H3-14 (δH 1.09) and H-15 (δH 3.22, 3.19) to C-9 (δC 44.6), C-10 (δC 42.7), and C-8 (δC 44.4), along with from H-10 (δH 2.31, 2.00) to C-1 (δC 139.4), C-2 (δC 133.2), and C-7 (δC 49.3) enabled the completion of a five-membered ring constructed by C-1–C-7–C-8–C-9–C-10 and an exocyclic double bond located at C-1 (Part A) (Fig. 2). Moreover, the HMBC correlations from oxygen-bearing methylene H-11 (δH 4.49, 4.39) to C-1, C-2, C-3 (δC 42.4), and C-12 (δC 110.6), and from H-12 (δH 6.09) and H-4 (δH 2.92, 2.49) to the carbonyl C-5 (δC 178.3) led to construction of tetrahydrofuro[2,3-b]furan-2(3H)-one moiety which connected with part A through the double bond C-1–C-2 (Fig. 2). The above evidences led to the establishment of the planar structure of 1, which also in accordance with the molecular formula deduced from HRESIMS result which gave a sodium adduct ion peak at m/z 287.1258 [M+Na]+ (calcd for C15H20O4Na, 287.1254).

Key 1H–1H COSY and HMBC correlations of compounds 1–6
A ROESY experiment determined the relative configuration of compound 1. The ROESY cross peaks of H-7/H-8β/H-15 led to the assignment of H-7 and 15-CH2OH as β orientation. The significant ROESY correlation between H-3 and H-12, H-10 and H-11 suggested that H-3 and H-12 were at the same side, and the C-1–C-2 double bond were Z configuration. Therefore, the structure of compound 1 was established as 11,12-epoxy-15-hydroxy-5,6-seco-tremula-1,6(13)-dien-5,12-olide (Fig. 1).
The other set of signals for compound 2 showed resemblance with those of compound 1, but with the presence of an additional methoxy group (δH 3.65, δC 52.3). Careful analysis of the 2D NMR led to the conclusion that the only difference between 1 and 2 was the lactone group in 1 opened in 2 and the carbonyl group was methyl esterified, which was confirmed by the HMBC correlation from the methoxy (δH 3.65) to the carbonyl C-5 (δC 174.3) as well as the absence of HMBC correlation from H-12 to C-5. The relative configuration of 2 was identical with that of 1 by interpretation of the ROESY spectrum. The HRESIMS result of the mixture also gave a molecular formula of C16H24O5 (m/z 319.1529 [M+Na]+, calcd for C16H24O5Na, 319.1516) which further reinforced the above assignment. Thus, compound 2 was deduced as methyl 12β,15-dihydroxy-5,6-seco-tremula-1,6(13)-dien-5-oate (Fig. 1).
Compounds 3 and 4 were isolated as an inseparable mixture with the ratio of 1:0.6. The 1D NMR data of the mixture showed high similarities with those of the 1/2 mixture, implying that compounds 3 and 4 were analogues of 1 and 2. Elucidating the 2D NMR spectra of the mixture promoted the completion of 3 and 4 which possessing the same planar structures with those of 1 and 2, respectively. Considering of the above information, a conclusion could be drawn which compounds 3 and 4 differed from 1 and 2 in spatial configuration. Analysis of the ROESY spectrum of 3/4, in combination with ChemBio3D simulations confirmed the speculation. The ROESY spectrum displayed cross peaks of H-14 (δH 3.41, 3.40 for 3; 3.40, 3.39 for 4)/H-8 (δH 1.67, 1.55 for 3; 1.64, 1.54 for 4), H-14/H-10 (δH 2.22, 1.99 for 3; 2.16, 1.88 for 4). As shown in Fig. 3, the minimized energy optimized molecular models of 3 and 4 suggested that only when the 14-CH2OH adopted the equatorial direction, the above ROESY correlation could be seen. Therefore, compounds 3 and 4 were C-9 epimers of compounds 1 and 2, respectively. This conclusion was also evidenced by the large discrimination of 13C NMR chemical shifts of C-14 and C-15 between 1 and 3, 2 and 4. Thus, compounds 3 and 4 were identified as shown in Fig. 1, and were given the respective names 11,12-epoxy-14-hydroxy-5,6-seco-tremula-1,6(13)-dien-5,12-olide, and methyl 12β,14-dihydroxy-5,6-seco-tremula-1,6(13)-dien-5-oate.

Key ROESY correlations of compounds 1–4
Compound 5 was isolated as colourless oil. The HRESIMS result presented a molecular formula of C15H24O4 as indicated by the sodium adduct ion peak at m/z 291.1572 [M+Na]+ (calcd for C15H24O4Na, 291.1567). The 1D NMR data of 5 (Tables 1, 2) were classified into three methyls, five methylenes (two oxygenated), three methines (one oxygenated), and four quaternary carbons (two olefinic ones, and a carbonyl). These data closely related to the co-isolate conocenolide A (7) [3], suggesting compound 5 was a congener of 7. The main discrepancy between 5 and 7 was that the absence of the C-6–C-13 double bond while the presence of an oxygen-bearing methine and a methyl doublet in 5 compared to those of 7, suggested that the C-6 in 7 was oxygenated in 5, which was verified by the 1H–1H COSY correlations of H-6/H-7, H-6/H-13 (Fig. 2). Unfortunately, the inadequate sample of 5 (0.8 mg) precluded the determination of the absolute configuration of 6-OH. Therefore, compound 5 was identified as 6,11-dihydroxy-5,6-seco-tremul-1-en-5,12-olide (Fig. 1).
Compound 6 was obtained as colourless oil. The 1H and 13C NMR spectra exhibited signals which ascribable to three methyls, four methylenes (one oxygenated), four methines (two oxygenated), and four quaternary carbons (a tetrasubstituted double bond, a carbonyl). These data were reminiscent of those of compounds 1 and 3 (Tables 1, 2). Compared the NMR data of 6 with those of 1 revealed that the large differences located at C-6, C-13, and C-15. Detailed analysis of the 2D NMR spectra of 6 indicated that the C-6–C-13 double bond was oxygenated, while C-15 remained unoxygenated. This postulation was validated by the 1H–1H COSY correlations of H-6/H3-13, and H-6/H-7, and HMBC correlation from H3-14 to H3-15 (Fig. 2). Due to scarcity of samples, 6-OH remained unassigned. The above assignments were consistent with the HRESIMS data, which gave a sodium adduct ion peak at m/z 289.1416 [M +Na]+, suggesting a molecular formula of C15H22O4 (calcd for C15H22O4Na, 289.1410). Thus, the structure of 6 was established as shown in Fig. 1 and was named as 11,12-epoxy-6-hydroxy-5,6-seco-tremul-1-en-5,12-olide.
Conocenolides A (7) and B (8) [3] were two abundant and separable but tautomeric isomers accompanied in this study. Their structures were identified compared with the literature data.
In conclusion, six hitherto unknown 5,6-seco-tremulanes were isolated from the culture broth of the medicinal fungus I. lacteus. All these 5,6-seco-tremulane skeletons were proposed to be biosynthesized via Baeyer–Villiger oxidation, rearrangement, elimination, oxidation, and esterification of the corresponding 5-oxo-tremulane. This study represents the first report of 5,6-seco-tremulanes from the fungus I. lacteus, and also expands the distribution of 5,6-seco-tremulane-type sesquiterpenoids as fungal metabolites.
3 Experimental
3.1 General Experimental Procedures
Optical rotations were obtained on a JASCO P-1020 digital polarimeter (Horiba, Kyoto, Japan). UV spectra were recorded on a Shimadzu UV-2401PC UV–Visible recording spectrophotometer (Shimadzu, Kyoto, Japan). A Tenor 27 spectrophotometer (Bruker Optics GmbH, Ettlingen, Germany) was used for scanning IR spectroscopy using KBr pellets. 1D and 2D NMR spectra were obtained on a Bruker Avance III 600 MHz spectrometer (Bruker Corporation, Karlsruhe, Germany). HRESIMS were recorded on an Agilent 6200 Q-TOF MS system (Agilent Technologies, Santa Clara, CA, USA). Sephadex LH-20 (Amersham Biosciences, Uppsala, Sweden) and silica gel (Qingdao Haiyang Chemical Co., Ltd., Qingdao, China) were used for column chromatography (CC). Medium Pressure Liquid Chromatography (MPLC) was performed on a Büchi Sepacore System equipped with pump manager C-615, pump modules C-605 and fraction collector C-660 (Büchi Labortechnik AG, Flawil, Switzerland), and columns packed with Chromatorex C-18 (dimensions 450 mm × i.d. 14 mm, particle size: 40–75 μm, Fuji Silysia Chemical Ltd., Kasugai, Japan). Preparative high performance liquid chromatography (prep-HPLC) were performed on an Agilent 1260 liquid chromatography system equipped with a Zorbax SB-C18 column (particle size 5 μm, dimensions 150 mm × i.d. 9.4 mm, flow rate 7 ml min−1) and a DAD detector (Agilent Technologies, Santa Clara, CA, USA).
3.2 Fungal Material
The fungus Irpex lacteus was collected from Changbai Mountain Nature Reserve in 2012. The strain of I. lacteus in this study was isolated from the fresh fruiting bodies and kept on potato, dextrose, and agar (PDA) culture medium. A voucher specimen (No. CGBWSHFG1102) was deposited in the Herbarium of Kunming Institute of Botany, Chinese Academy of Sciences. The culture medium to ferment this fungus consisted of glucose (5%), peptone from porcine meat (0.15%), yeast powder (0.5%), KH2PO4 (0.05%) and MgSO4 (0.05%). Sixty Erlenmeyer flasks (500 ml) each containing 350 ml of above-mentioned culture medium were inoculated with I. lacteus strains, respectively. Fermentation were carried out on rotatory shakers at 25 °C and 150 rpm for 25 days in dark environment.
3.3 Extraction and Isolation
The culture broth (20 l) of I. lacteus HFG1102 was filtered and concentrated to 3 l followed by partitioned between EtOAc and water for four times to give an EtOAc layer. Meanwhile, the mycelia were extracted by EtOH (95%) for three times. The EtOAc layer together with the mycelium extract were concentrated under reduced pressure to afford a crude extract (wt 16.0 g). This residue was separated by MPLC (MeOH/H2O, 0–100%) to give sixteen main fractions (A–P).
Subfraction I was separated by Sephadex LH-20 CC (MeOH) to afford three subfractions I1–I3. Subfraction I3 was further purified by prep-HPLC (MeCN/H2O, 30–50%, 25 min) to give two isolated but interchangeable compounds 7 (tR = 16.5 min) and 8 (tR = 17.8 min) with a totally yield 22 mg.
Subfraction J was separated by Sephadex LH-20 CC (MeOH) to remove colourants, and further separated by Sephadex LH-20 (acetone) to afford four subfractions J1–J4. Subfraction J2 was purified by prep-HPLC (MeCN/H2O, 26–46%, 25 min) to give 1/2 (tR = 16.5 min) with a yield 2.0 mg. Subfraction J3 was purified by prep-HPLC (MeCN/H2O, 26–46%, 25 min) to give 3/4 (tR = 18.2 min) with a yield 1.8 mg.
Subfraction K was separated by Sephadex LH-20 CC (MeOH) to remove colourants, and further separated by Sephadex LH-20 (acetone) to afford five subfractions K1–K5. Subfraction K1 was purified by prep-HPLC (MeCN/H2O, 25–45%, 25 min) to give 5 (tR = 17.5 min) with a yield 1.0 mg. Subfraction K3 was purified by prep-HPLC (MeCN/H2O, 26–46%, 25 min) to give 6 (tR = 18.9 min) with a yield 0.8 mg.
3.4 Spectroscopic Data of Compounds
3.4.1 11,12-Epoxy-15-hydroxy-5,6-seco-tremula-1,6(13)-dien-5,12-olide (1) and methyl 12β,15-dihydroxy-5,6-seco-tremula-1,6(13)-dien-5-oate (2)
Colourless oil; \([\alpha ]_{{\text{D}}}^{{26}}\) − 56.9 (c 0.09, MeOH); UV (c 0.0306 mg ml−1, MeOH) λmax nm (Abs): 205 (0.7693), 232 (0.3060); IR (KBr) νmax 3425, 2927, 2858, 1632, 1384, 1031 cm−1; 1H NMR (600 MHz, CD3OD) data, see Table 1; 13C NMR (150 MHz, CD3OD) data, see Table 2; HRESIMS 1: C15H20O4 m/z 287.1258 [M+Na]+ (calcd for C15H20O4Na, 287.1254); 2: C16H24O5 m/z 319.1529 [M+Na]+ (calcd for C16H24O5Na, 319.1516).
3.4.2 11,12-Epoxy-14-hydroxy-5,6-seco-tremula-1,6(13)-dien-5,12-olide (3) and methyl 12β,14-dihydroxy-5,6-seco-tremula-1,6(13)-dien-5-oate (4)
Colourless oil; \([\alpha ]_{{\text{D}}}^{{26}}\) − 77.5 (c 0.10, MeOH); UV (c 0.015 mg ml−1, MeOH) λmax nm (Abs): 205 (0.4599), 238 (0.1222); IR (KBr) νmax 3425, 2927, 2860, 1728, 1631, 1032 cm−1; 1H NMR (600 MHz, CD3OD) data, see Table 1; 13C NMR (150 MHz, CD3OD) data, see Table 2; HRESIMS 3: C15H20O4 m/z 287.1250 [M+Na]+ (calcd for C15H20O4Na, 287.1254); 4: C16H24O5 m/z 319.1513 [M+Na]+ (calcd for C16H24O5Na, 319.1516).
3.4.3 6,11-Dihydroxy-5,6-seco-tremul-1-en-5,12-olide (5)
Colourless oil; C15H24O4; \([\alpha ]_{{\text{D}}}^{{26}}\) + 14.6 (c 0.07, MeOH); UV (MeOH) λmax nm (log ε): 206 (4.13), 254 (2.97); IR (KBr) νmax 3422, 2952, 2930, 2868, 1750, 1632, 1383, 1193, 1022 cm−1; 1H NMR (600 MHz, CD3OD) data, see Table 1; 13C NMR (150 MHz, CD3OD) data, see Table 2; HRESIMS m/z 291.1572 [M +Na]+ (calcd for C15H24O4Na, 291.1567).
3.4.4 11,12-Epoxy-6-hydroxy-5,6-seco-tremul-1-en-5,12-olide (6)
Colourless oil; C15H22O4; \([\alpha ]_{{\text{D}}}^{{26}}\) + 47.8 (c 0.07, MeOH); UV (MeOH) λmax nm (log ε): 205 (3.46), 245 (2.18); IR (KBr) νmax 3432, 2927, 2859, 1631, 1385, 1031 cm−1; 1H NMR (800 MHz, CDCl3) data, see Table 1; 13C NMR (200 MHz, CDCl3) data, see Table 2; HRESIMS m/z 289.1416 [M+Na]+ (calcd for C15H22O4Na, 289.1410).
References
H.P. Chen, J.K. Liu, Prog. Chem. Org. Nat. Prod. 106, 1–201 (2017)
X.M. Dong, X.H. Song, K.B. Liu, C.H. Dong, Mycosystema 36, 28–34 (2017)
D.Z. Liu, F. Wang, J.K. Liu, J. Nat. Prod. 70, 1503–1506 (2007)
X.Y. Yang, T. Feng, J.H. Ding, X. Yin, H. Guo, Z.H. Li, J.K. Liu, Nat. Prod. Bioprospect. 3, 48–51 (2013)
M. Isaka, S. Palasarn, S. Supothina, K. Srichomthong, R. Choeyklin, Helv. Chim. Acta 99, 232–236 (2016)
F. Wang, H. Ma, Z. Hu, J. Jiang, H. Zhu, L. Cheng, Q. Yang, H. Zhang, G. Zhang, Y. Zhang, Nat. Prod. Res. 31, 1849–1854 (2017)
Y.M. Ying, C.P. Tong, J.W. Wang, W.G. Shan, Z.J. Zhan, J. Chem. Res. 38, 304–305 (2014)
Acknowledgements
This work was financially supported by National Natural Science Foundation of China (81561148013), the Key Projects of Technological Innovation of Hubei Province (No. 2016ACA138), and the Fundamental Research Funds for the Central University, South-Central University for Nationalities (CZT18014, CZT18013).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare no conflict of interest.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Chen, HP., Zhao, ZZ., Li, ZH. et al. Seco-tremulane Sesquiterpenoids from the Cultures of the Medicinal Fungus Irpex lacteus HFG1102. Nat. Prod. Bioprospect. 8, 113–119 (2018). https://doi.org/10.1007/s13659-018-0157-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13659-018-0157-y