
Abstract
Phytochemical investigation on the whole plants of Ypsilandra yunnanensis was carried out for the first time and led to the isolation of five new cholestanol glycosides, ypsiyunnosides A–E (1–5), and one known analogue. Their structures were determined mainly by detailed spectroscopic analysis, including extensive 1D and 2D NMR, MS and UV, as well as chemical methods. Among them, compound 1 possessed a rare 6/6/6/5/5 fused-rings cholestanol sketelon, which was identified as (23R,25R)-3β,16α,26-triol-16,23-cyclocholest-5,17(20)-dien-22-one. Their induced platelet aggregation activities and cytotoxicities were evaluated.
Graphical Abstract
Five new cholestanol glycosides, ypsiyunnosides A–E (1–5), were isolated from the whole plants of Ypsilandra yunnanensis. Compound 1 possessed a rare 6/6/6/5/5 fused-rings cholestanol sketelon. Their structures were elucidated by a combination of 1D and 2D NMR, MS and chemical analysis.

Similar content being viewed by others
Avoid common mistakes on your manuscript.
1 Introduction
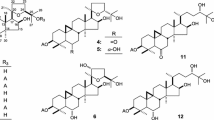
Ypsilandra yunnanensis W. W. Sm. et J. F. Jeffr., belonging to the family of Liliaceae, is an erect, perennial herb and distributed mainly in the northwest of Yunnan Province and the southeast of the Tibet Autonomous Region of China. It usually grows in grass slope, forest-shrub edge, or under the bushes at the altitude between 3300 and 4000 m [1]. Previously, we have performed a systemic phytochemical investigation of the whole plants of Y. thibetica and obtained a series of spirostanol-, furostanol-, and 23-spirocholestanol glycosides with cytotoxic, antifungal, hemostatic, and anti-HIV activities [2–9]. However, the phytochemicals and the biological activity of Y. yunnanensis have not been reported so far. The HPLC analysis revealed that the secondary metabolites of Y. thibetica and Y. yunnanensis were very different, especially in the water-soluble part of the crude extract. As part of our continuing search for structurally diverse and bioactive steroidal glycosides from the Ypsilandra plants, the chemical constituents of Y. yunnanensis were investigated. The result led to the isolation of five new cholestane glycosides (1–5) and one known analogue, parispseudoside C (6) [10] as shown in Fig. 1 from the 70 % EtOH extract of the whole title plant. Their structures were determined by the analysis of spectroscopic data (HR-ESI-MS, 1D and 2D NMR) and chemical methods. This paper presents herein the isolation, structural elucidation, and the induced platelet aggregation activities and cytotoxicities of these compounds.

Chemical structures of compounds 1–5
2 Results and Discussion
Ypsiyunnoside A (1) was obtained as a white amorphous powder, \(\left[ \alpha \right]_{\text{D}}^{23}\) – 139.6 (c 0.05, MeOH); UV (MeOH) λ max (log ε) 237 (3.03) and 197 (2.76) nm. Its molecular formula was determined as C57H90O26 on the basis of a positive-ion at m/z 1213.5616 [M + Na]+ (calcd. 1213.5618) in its HR-ESI-MS and 13C NMR data (Table 2), corresponding to 13° of unsaturation. The IR spectrum of 1 exhibited absorption bands for hydroxyl (3440 cm−1) and α,β-unsaturated ketone (1701 and 1658 cm−1) functionalities, which was confirmed by the UV absorption at λ max (MeOH) 237 nm. The 1H NMR spectrum of the aglycone moiety of 1 (Table 1) displayed signals for four characteristic steroidal methyls at δ H 1.08 (s, Me-19), 1.46 (s, Me-18), 1.83 (s, Me-21), and 1.12 (d, J = 6.5 Hz, Me-27) and an olefinic proton at δ H 5.35 (br s). The 13C NMR and DEPT spectra (Table 2) showed a total of 57 carbon signals, which were classified as 7 methyls, 11 methylenes, 32 methines, and 7 quaternary carbons. Among them, 27 carbon signals were assigned to the aglycone including four methyls at δ C 8.6 (q, Me-21), 15.7 (q, Me-18), 17.1 (q, Me-27) and 19.5 (q, Me-19), three oxygenated carbons at δ C 76.6 (t, C-26), 78.1 (d, C-3) and 83.0 (s, C-16), and four olefinic carbons at δ C 121.7 (d, C-6), 128.3 (s, C-20), 141.1 (s, C-5) and 182.1 (s, C-17), as well as one carbonyl carbon at δ C 212.3 (s, C-22). Furthermore, the 1H NMR and 13C NMR spectra of 1 exhibited five anomeric protons at δ H 4.84 (d, J = 7.7 Hz, H-1′′′′′), 4.97 (d, J = 7.4 Hz, H-1′), 5.85 (br s, H-1′′′), 6.30 (br s, H-1′′′′) and 6.41 (br s, H-1′′) (Table 1), corresponding to five carbons at δ C 105.3 (d, C-1′′′′′), 100.5 (d, C-1′), 102.4 (d, C-1′′′), 103.3 (d, C-1′′′′) and 102.2 (d, C-1′′) (Table 2), respectively. Acid hydrolysis of 1 gave d-glucose and l-rhamnose, which were determined by GC chromatographic analysis of their l-cysteine methyl ester-TMS derivates. The above spectroscopic information hinted that compound 1 was a cholestanol pentaglycoside.
Comparison of the 1H and 13C NMR spectra of the aglycone of 1 with those of parispseudoside C (6) [10] led to find the absence of one carbonyl group, one methylene and the appearance of an oxygen-bearing quaternary carbon (δ C 83.0) and one methine (δ H 2.44; δ C 57.6) in 1. In addition, one carbonyl group, two double bonds and five monosaccharides in compound 1 accounted for 8 degrees of unsaturation, and the remaining five degrees of unsaturation required that the aglycone of 1 be pentacyclic. Therefore, it was supposed that compound 1 was a 23,16-aldol condensation product of parispseudoside C (6), which was verified by the 1H–1H COSY and HMBC spectra. The 1H–1H COSY correlations (Fig. 2) of δ H 4.03 (Ha-26) and 3.72 (Hb-26) with δ H 2.51 (H-25), of δ H 2.51 (H-25) with δ H 2.39 (Ha-24) and 1.98 (Hb-24), of δ H 2.51 (H-25) with δ H 1.12 (Me-27), and of δ H 1.98 (Hb-24) with δ H 2.44 (H-23), together with the HMBC correlation (Fig. 2) of δ H 2.51 (H-25) with δ C 57.6 (C-23) indicated that the methine was assigned to C-23. The observed HMBC correlations (Fig. 2) from Me-21 to C-17, C-20, and C-22, from H-23 to C-16, and C-22, from H2-24 to C-16 and C-22 supported that the formation of a cyclopentane with α,β-unsaturated ketone group by 23,16-aldol condensation (Fig. 3). The ROESY correlations (Fig. 4) of Me-18/H-8, H-8/H β -15 (δ H 1.98), and H β -15/H-23 suggested that H-23 was β-oriented. Furthermore, the obvious correlation from H β -15 to H-23 hinted that OH-16 was α-oriented. The 25R-configuration of 1 was deduced from the geminal proton resonances of Ha-26 and Hb-26, which showed Δab (δ Ha – δ Hb) of 0.31, less than 0.48 [11].

The IH–1H COSY correlations and selected key HMBC correlations of 1

Possible biosynthetic pathway for the aglycone of 1

Key ROESY correlations for the aglycone of 1
The β-configurations for the two glucopyranosyls were deduced by large J 1H-2H values (3 J 1,2 = 7.4–7.7 Hz) of their anomeric protons, while the anomeric configuration of the three rhamnopyranosyls were assigned as α-configuration from the 13C NMR data of C-3′′, C-5′′, C-3′′′, C-5′′′, C-3′′′′, and C-5′′′′ with those of the corresponding carbons of methyl α- and β-rhamnopyranoside [12, 13]. The sequence of the sugars and the linkage sites to the aglycone were in good agreement with those of 6, which was supported by their almost identical NMR data and the HMBC correlations from H-1′ (δ H 4.97) of 3-Glu to C-3 (δ C 78.1) of aglycone, from H-1″ (δ H 6.41) of 2′-Rha to C-2′ (δ C 78.5) of 3-Glu, from H-1′′′ (δ H 5.85) of 4′-Rha to C-4′ (δ C 77.8) of 3-Glc, from H-1′′′′ (δ H 6.30) of 4′′′ -Rha to C-4′′′ (δ C 80.5) of 4′-Rha, and from H-1′′′′′ (δ H 4.84) of 26-Glu to C-26 (δ C 76.6) of aglycone. Thus, the structure of ypsiyunnoside A (1) was elucidated as 26-O-β-d-glucopyranosyl-(23R,25R)-3β,16α,26-triol-16,23-cyclocholest-5,17(20)-dien-22-one-3-O-α-l-rhamnopyranosyl-(1 → 4)-α-l-rhamnopyranosyl-(1 → 4)-[α-l-rhamnopyranosyl-(1 → 2)]-β-d-glucopyranoside.
Ypsiyunnoside B (2) was isolated as a white amorphous powder, \(\left[ \alpha \right]_{\text{D}}^{24}\) – 148.7 (c 0.05, MeOH); UV (MeOH) λ max (log ε) 217 (2.78) and 203 (2.92) nm. It had a molecular formula of C57H90O26 as deduced by HR-ESI-MS ion peak at m/z 1213.5616 [M + Na]+ (calcd. 1213.5618) and 13C NMR data (Table 2). A careful comparison of the NMR and MS data of 2 (Tables 1 and 2) with those of 6 revealed that they were similar, except for the presence of a methine (δ H 3.71; δ C 63.5) and a terminal double bond [δ H 5.90 and 6.56 (each 1H); δ C 129.7 (t) and δ C 143.2 (s)] and the absence of a methyl and a Δ17(20) double bond in 2. These spectroscopic characteristics suggested that the aglycone of 2 was (25R)-3β,26-diol-cholest-5,20-dien-16,22-dione. The HMBC correlations of Me-18 with C-17 (δ C 63.5), of H-17 with C-12, C-13, C-14, C-16, C-18, C-20, C-21, and C-22, and of H2-21 (δ H 5.90 and 6.56) with C-17 (δ C 63.5) and C-22 (δ C 202.0) evidenced this suggestion. According to the consistency of the 1H and 13C NMR data (Tables 1 and 2) of the sugar moieties in 1, 2, and 6, these three compounds were ascertained to have the same sugar sequence and linkage position. Consequently, the structure of ypsiyunnoside B (2) was determined to be 26-O-β-d-glucopyranosyl-(25R)-3β,26-diol-cholest-5,20-dien-16,22-dione-3-O-α-l-rhamnopyranosyl-(1 → 4)-α-l-rhamnopyranosyl-(1 → 4)-[α-l-rhamnopyranosyl-(1 → 2)]-β-d-glucopyranoside.
Ypsiyunnoside C (3) was isolated as white amorphous powder, \(\left[ \alpha \right]_{\text{D}}^{23}\) – 109.2 (c 0.05, MeOH); UV (MeOH) λ max (log ε) 245 (2.69) and 204 (2.85) nm. Its molecular formula was established as C57H90O27 by the positive HR-ESI-MS at m/z 1229.5563 [M + Na]+ (calcd. 1229.5567) and 13C NMR data (Table 2). Inspection of the 1H and 13C NMR spectroscopic data of 3 (Tables 1 and 2) with those of 6 revealed their considerable structural similarity. The major differences were observed for the replacement of a methene in 6 by an oxygenated methine (δ H 4.04; δ C 73.2). The oxymethine was placed at C-7 on the basis of the 1H–1H COSY correlation of δ H 5.69 (H-6) with δ H 4.04 (H-7). The relative configuration of OH-7 was determined to be β-oriented by the chemical shift of C-7 (δ C 73.2), while the signals for C-7 would be at δ C 64.7 of the 7α-isomer [7]. Other parts were identical to those of 6 based on 2D NMR experiments. Therefore, the structure of ypsiyunnoside C (3) was characterized as 26-O-β-d-glucopyranosyl-(25R)-3β,7β,26-triol-cholest-5,17(20)-dien-16,22-dione-3-O-α-l-rhamnopyranosyl-(1 → 4)-α-l-rhamnopyranosyl-(1 → 4)-[α-l-rhamnopyranosyl-(1 → 2)]-β-d-glucopyranoside.
Ypsiyunnoside D (4), obtained as white amorphous powder, gave the molecular formula of C51H80O21 from its HR-ESI-MS ion peak at m/z 1051.5083 [M + Na]+ (calcd. 1051.5090) and 13C NMR data (Table 2). The 1H and 13C NMR spectroscopic data of 4 (Tables 1 and 2) were similar to those of 6, differing only in the disappearance of glucopyranosyl signals and the upfield shift of C-26 (δ C 75.1 ppm → 67.9 ppm). This indicated that no sugar moiety was attached to C-26. By the detailed analysis of 1D and 2D-NMR data (Tables 1 and 2) of the sugar moieties in compounds 4 and 6, the two cholestanol glycosides were considered to have the same tetraglycoside and the linkage position at C-3. On the basis of the above evidence, the structure of ypsiyunnoside D (4) was elucidated as (25R)-3β,26-diol-cholest-5,17(20)-dien-16,22-dione-3-O-α-l-rhamnopyranosyl-(1 → 4)-α-l-rhamnopyranosyl-(1 → 4)-[α-l-rhamnopyranosyl-(1 → 2)]-β-d-glucopyranoside.
Ypsiyunnoside E (5) was obtained as white amorphous powder with \(\left[ \alpha \right]_{\text{D}}^{23}\) – 84.8 (c 0.05, MeOH) and UV (MeOH) λ max (log ε) 242 (2.78) and 203 (2.88) nm. It had a molecular formula of C33H50O9 based on the positive-ion HR-ESI-MS (m/z 613.3357 [M + Na]+; calcd. 613.3353) and 13C NMR data (Table 2). The 1H and 13C NMR spectroscopic data of 5 showed its aglycone resembling that of compounds 4 and 6. However, the 1H NMR spectrum (Table 1) showed only one anomeric proton signal at δ H 4.82 (d, J = 7.8 Hz), which was identified as a β-d-glucopyranosyl by the aforementioned method. The HMBC correlation of H-1′ (δ H 4.82) of the glucopyranosyl with C-26 (δ C 74.9) revealed that the glucopyranosyl was linked to C-26. In conclusion, the structure of ypsiyunnoside E (5) was established as 26-O-β-d-glucopyranosyl-(25R)-3β,26-diol-cholest-5,17(20)-dien-16,22-dione.
Considering the traditional use of Ypsilandra plants as hemostatic medicine for Yi minority [14] and the cytotoxic activity of steroidal glycosides previously obtained from Y. thibetica [3, 6], compounds 1–6 were evaluated for their induced platelet aggregation activities and cytotoxicities against HEK293 and HepG2 human cancer cell lines. Unfortunately, the results showed that none of them had any obviously induced platelet aggregation activity at the concentration of 300 μg/mL and cytotoxic activity against the two human cancer cell lines (HepG2 and HEK293, IC50 > 20 μM).
3 Experimental
3.1 Plant Material
The whole plants of Y. yunnanensis were collected in September 2012 from Gongshan county, Yunnan Province, China, and identified by Dr. Rong Li (Institute of Botany, Chinese Academy of Sciences). A voucher specimen (No. HY0019) has been deposited at the State Key Laboratory of Phytochemistry and Plant Resources in West China, Kunming Institute of Botany, Chinese Academy of Sciences.
3.2 Extraction and Isolation
The air-dried and powdered whole plants of Y. yunnanensis (5.0 kg) were extracted three times with 70 % EtOH (20 L × 3) under reflux for a total of 6 h (3 × 2 h) and the combined extract was concentrated under reduced pressure to give an aqueous solution (4.3 L). The solution was partitioned with n-BuOH (5 L × 4). After evaporation of the n-BuOH soluble fraction, the total saponin (905 g) was obtained. The saponin was subjected to CC (silica gel, 200–300 mesh; gradient CHCl3-MeOH 10:1–1:1, v/v) to afford 19 fractions (Fr. 1–19). The water-soluble part (Fr. 18 and Fr.19) showed different peaks from the extract of Y. thibetica on the HPLC chromatograms. Thus, the two fractions were further purified by different separation methods. Fr. 18 (208 g) was separated by CC (silica gel, 200–300 mesh; gradient CHCl3-MeOH, 9:1 → 1:1 v/v) to give five subfractions. Fr. 18-1 was subjected to Sephadex LH-20 column (MeOH) and semi-prep. HPLC (MeCN-H2O, 10:90 → 25:75, 0–10 min; MeCN-H2O 25:75 → 35:65, 10–30 min) to yield 5 (5.6 mg). Fr. 18-4 was subjected to an RP-18 column (MeOH-H2O 10:90 → 40:60 v/v) and further purified by semi-prep. HPLC (MeCN-H2O 15:85 → 50:50, 35 min) to afford 3 (3.2 mg), 4 (3.8 mg), and 6 (2.5 mg). Fr. 19 (130 g) was subjected to a MCI gel CC and then RP-18 column (MeOH-H2O, 10:90 → 50:50 v/v) to give four fractions. Fr. 19-2 was repeatedly subjected to Sephadex LH-20 CC (MeOH) to give 2 (11.9 mg). Fr.19-3 was subjected to CC (silica gel, 200–300 mesh; gradient CHCl3-MeOH, 9:1 → 1:1 v/v) and followed by semi-prep. HPLC (MeCN-H2O 20:80 → 30:70, 30 min) to yield 1 (75 mg).
3.3 Ypsiyunnoside A (1)
White amorphous powder, \(\left[ \alpha \right]_{\text{D}}^{23}\) – 139.6 (c 0.05, MeOH); UV (MeOH) λ max (log ε) 237 (3.03) and 197 (2.76) nm; IR (KBr) ν max 3440, 2935, 1701, 1658, 1452, 1383, 1041, 984, 911, 884, 838 cm−1; 1H NMR data see Table 1; 13C NMR data see Table 2; ESIMS m/z 1213 [M + Na]+; HRESIMS m/z 1213.5616 (calcd for C57H90O26Na [M + Na]+, 1213.5618).
3.4 Ypsiyunnoside B (2)
White amorphous powder, \(\left[ \alpha \right]_{\text{D}}^{24}\) – 148.7 (c 0.05, MeOH); UV (MeOH) λ max (log ε) 217 (2.78) and 203 (2.92) nm; IR(KBr) ν max 3441, 2934, 1734, 1632, 1452, 1383, 1043, 985, 910, 882, 836 cm−1; 1H NMR data see Table 1; 13C NMR data see Table 2; ESIMS m/z 1213 [M + Na]+; HRESIMS m/z 1213.5616 (calcd for C57H90O26Na [M + Na]+, 1213.5618).
3.5 Ypsiyunnoside C (3)
White amorphous powder, \(\left[ \alpha \right]_{\text{D}}^{23}\) – 109.2 (c 0.05, MeOH); UV (MeOH) λ max (log ε) 245 (2.69) and 204 (2.85) nm; IR(KBr) ν max 3441, 2931, 1631, 1453, 1384, 1043, 984, 912, 882, 837 cm−1; 1H NMR data see Table 1; 13C NMR data see Table 2; ESIMS m/z 1229 [M + Na]+; HRESIMS m/z 1229.5563 (calcd for C57H90O27Na [M + Na]+, 1229.5567).
3.6 Ypsiyunnoside D (4)
White amorphous powder with \(\left[ \alpha \right]_{\text{D}}^{23}\) – 142.8 (c 0.05, MeOH); UV (MeOH) λ max (log ε) 246 (2.78) and 202 (2.87) nm; IR(KBr) ν max 3445, 2931, 1715, 1632, 1451, 1383, 1048, 984, 913, 880, 837 cm−1; 1H NMR data see Table 1; 13C NMR data see Table 2; ESIMS m/z 1027 [M – H]–; HRESIMS m/z 1051.5083 (calcd for C51H80O21Na [M + Na]+, 1051.5090).
3.7 Ypsiyunnoside E (5)
White amorphous powder with \(\left[ \alpha \right]_{\text{D}}^{23}\) – 84.8 (c 0.05, MeOH); UV (MeOH) λ max (log ε) 242 (2.78) and 203 (2.88) nm; IR(KBr) ν max 3445, 2928, 1709, 1634, 1515,1451, 1383, 1040, 922, 898, 877 cm−1; 1H NMR data see Table 1; 13C NMR data see Table 2; ESIMS m/z 613 [M + Na]+; HRESIMS m/z 613.3357 (calcd for C33H50O9Na [M + Na]+, 613.3353).
3.8 Acid Hydrolysis and GC Analysis
Compounds 1–5 (1–2 mg, each) were refluxed with 2 M HCl (1:1 v/v, 2 mL) on water bath for 2 h. After cooling, the reaction mixture was neutralized with 1 M NaOH and filtered. The filtrate was extracted with CHCl3 (3 × 2 mL). The aqueous layer was evaporated to dryness. The dried residue was dissolved in 0.5 mL anhydrous pyridine and treated with l-cysteine methyl ester hydrochloride (1.0 mg) stirred at 60 °C for 1 h. Trimethylsilylimidazole (0.5 mL) was added to the reaction mixtures, and these were kept at 60 °C for 30 min. The supernatants (2 μL) were analyzed by GC, respectively, under the following conditions: H2 flame ionization detector. Column: 30QC2/AC-5 quartz capillary column (30 m × 0.32 mm). Column temperature: 180–280 °C with the rate of 3 °C/min, and the carrier gas was N2 (1 mL/min); injector temperature: 250 °C; split ratio: 1/50. The configurations of d-glucose and l-rhamnose for compounds 1–5 were determined by comparison of the retention times of the corresponding derivatives with those of standard d-glucose and l-rhamnose giving a single peak at 19.01 and 15.43 min, respectively. These assignments of absolute configurations are based on the assumption that the corresponding enantiomeric sugar derivatives of d-cysteinyl methyl ester would in fact be separable from the l-cysteinyl derivatives under our GC conditions.
3.9 Platelet Aggregation Assays
Turbidometric measurements of platelet aggregation of the samples were performed in a Chronolog Model 700 Aggregometer (Chronolog Corporation, Havertown, PA, USA) according to Born’s method [15, 16]. The blood from the rabbits by cardiac puncture, was anticoagulated with 3.8 % sodium citrate (9:1, v/v). Platelet-rich plasma (PRP) was prepared shortly after blood collection by spinning the sample at 180 g for 10 min at 22 °C. The PRP was carefully removed and the remaining blood centrifuged at 2400 g for 10 min to obtain platelet-poor plasma (PPP). The centrifuge temperature was maintained at 22 °C. Platelet counts were adjusted by the addition of PPP to the PRP to achieve a count of 500 × 109 L−1. Platelet aggregation studies were completed within 3 h of preparation of PRP. Immediately after preparation of PRP, 250 μL was incubated in each of the test tubes at 37 °C for 5 min and then 2.5 μL of inducers (or compounds) was added. The change of optical density as a result of platelet aggregation was recorded. The extent of aggregation was estimated by the percentage of maximum increase in light transmission, with the buffer representing 100 % transmittance. Arachidonic acid (AA) was used as a positive control.
3.10 Cytotoxic Assay
Compounds 1–6 were evaluated for their cytotoxicities against two human cancer cell lines (HepG2 and HEK293) using the MTT method described in the literature elsewhere [17]. (−)-OddC (Troxacitabine) was used as a positive control with IC50 values of 0.17 ± 0.02 μM and 0.30 ± 0.03 μM to the two cell lines, respectively. The experiments were conducted in three independent replicates, and IC50 > 20 μM was considered to be inactive.
References
The Editorial Board of Flora of China, Flora of China (Science Press, Beijing, 1980), p. 17
B.B. Xie, H.Y. Liu, W. Ni, C.X. Chen, Y. Lu, L. Wu, Q.T. Zheng, Chem. Biodivers. 3, 1211–1218 (2006)
B.B. Xie, H.Y. Liu, W. Ni, C.X. Chen, Steroids 74, 950–955 (2009)
Y. Lu, C.X. Chen, W. Ni, H.Y. Liu, Steroids 75, 982–987 (2010)
Y. Lu, B.B. Xie, C.X. Chen, W. Ni, H. Yan, H.Y. Liu, Helv. Chim. Acta 94, 92–97 (2011)
H.Y. Liu, C.X. Chen, Y. Lu, J.Y. Yang, W. Ni, Nat. Prod. Bioprospect. 2, 11–15 (2012)
X.D. Zhang, C.X. Chen, J.Y. Yang, W. Ni, H.Y. Liu, Helv. Chim. Acta 95, 1087–1093 (2012)
B.B. Xie, C.X. Chen, Y.H. Guo, Y.Y. Li, Y.J. Liu, W. Ni, L.M. Yang, N.B. Gong, Y.T. Zheng, R.R. Wang, Y. Lu, H.Y. Liu, Planta Med. 79, 1063–1067 (2013)
Y.A. Si, H. Yan, W. Ni, Z.H. Liu, T.X. Lu, C.X. Chen, H.Y. Liu, Nat. Prod. Bioprospect. 4, 315–318 (2014)
C.M. Xiao, J. Huang, X.M. Zhong, X.Y. Tang, Helv. Chim. Acta 92, 2587–2595 (2009)
P.K. Agrawal, Steroids 70, 715–724 (2005)
P.K. Agrawal, Phytochemistry 31, 3307–3330 (1992)
R. Kasai, M. Okihara, J. Asakawa, K. Mizutani, O. Tanaka, Tetrahedron 35, 1427–1432 (1979)
Yunnan Food, Drug Administration, The Yunnan Chinese Materia Medica Standards ((Yi Nationality Medicine (III)) (Yunnan Scientific and Technological Press, Kunming, 2010), pp. 5–6
G.V.R. Born, Nature 194, 927–929 (1962)
G.V.R. Born, J. Physiol. 168, 178–195 (1963)
T. Mosmann, J. Immunol. Methods 65, 55–63 (1983)
Acknowledgments
This work was supported by the National Natural Science Funding of China (Grants 31170333 and 31570363).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
All authors do not have any financial/commercial conflicts of interest.
Additional information
Yu Chen and Yong-Ai Si have contributed equally to this work.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Chen, Y., Si, YA., Ni, W. et al. Ypsiyunnosides A–E, Five New Cholestanol Glycosides from Ypsilandra yunnanensis . Nat. Prod. Bioprospect. 6, 173–182 (2016). https://doi.org/10.1007/s13659-016-0098-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13659-016-0098-2