
Abstract
Two new neolignans selaginellol (1) and selaginellol 4′-O-β-d-glucopyranoside (2), together with seven known compounds (3–9), were isolated from the whole plant of Selaginella moellendorffii. The structures of the new isolates were determined through spectroscopic data analysis. Compounds 1–9, as well as compounds 10–18 previously isolated from the species, were measured for the activity against platelet aggregation induced by ADP or collagen. Three neoligans (8, 11, and 12), one flavanone (14), and one alkaloid (16) showed inhibitory activity against ADP- or collagen-induced platelet aggregation as compared with tirofiban. The dihydrobenzofuran neolignans (8, 11, and 12) are more potent than the benzofuran neolignan (13) and other types of neolignans (1–7). Glucosidation of the dihydrobenzofuran neolignans (11 and 12) is helpful for the activity.
Graphical Abstract
Two new neolignans selaginellol (1) and selaginellol 4′-O-β-d-glucopyranoside (2) were isolated from the whole plant of Selaginella moellendorffii. Several compounds from this plant showed the activity against platelet aggregation induced by ADP or collagen.

Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
The family Selaginellaceae Willk. includes the single genus Selaginella Beauv. Selaginella is a nearly worldwide genus of about 700 species, with 72 of them in China and more than 20 species used in traditional Chinese medicine [1, 2]. Several Selaginella species including S. delicatula (Desv. ex Poir.) Alston, S. moellendorffii Hieron., S. nipponica Franch. & Sav., S. sanguinolenta (L.) Spring, S. stauntoniana Spring, and S. tamariscina (P. Beauv.) Spring are used in promotion of blood circulation (Huoxue in Chinese) [1]. Traditional Chinese medicines with the functions of “Huoxue” and/or “Huayu” (removing blood stasis) are claimed to be useful in antiplatelet therapies and the treatment of thrombotic diseases [3, 4]. Previously, a pyrrolidinoindoline alkaloid selaginellic acid with antiplatelet activity was found from the whole plant of S. moellendorffii [5, 6]. This result prompted us to further investigate the plant which led to the isolation of nine compounds (1–9, Fig. 1) including two new neolignans (1 and 2). Compounds 1–9, as well as those (10–18) previously isolated from the plant [5, 7, 8], were evaluated for antiplatelet activity. The structural elucidation of the new compounds and the bioassay results are reported.

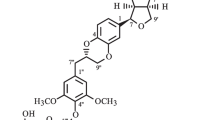
The chemical structures of 1–9 from Selaginella moellendorffii
2 Results and Discussion
The HRESIMS analysis of selaginellol (1) gave an [M+Na]+ ion at m/z 415.1729 appropriate for a molecular formula of C21H28O7 requiring eight sites of unsaturation. The IR absorption signals revealed the presence of hydroxy (3428 cm−1) and aromatic (1614, 1518, 1496, and 1461 cm−1) groups. The 1H NMR data of 1 (Table 1) exhibited three methoxy groups [δ H 3.82 (3H, s) and 3.70 (6H, s)], and two 1,2,3,5-tetrasubstituted benzene rings [δ H 6.30 (2H, s); 6.63 (d, J = 1.6 Hz) and 6.47 (d, J = 1.6 Hz)]. The 13C NMR data of 1 (Table 1) showed the signals for three methoxy groups (δ C 56.5 × 2 and 56.4), two phenyl rings, five methylenes including two oxygenated ones (δ C 65.9, 62.2, 37.9, 35.8, and 32.8), and one methine (δ C 45.4). According to above NMR signal characteristics [8], compound 1 might be a neolignan.
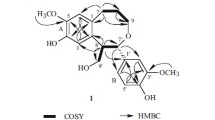
The 1H–1H COSY correlations (Fig. 2) exhibited two partial structures from C-7 to C-9 and C-7′ to C-9′. Based on the HMBC correlations (Fig. 2) from H-2 and H-6 to C-4, H2-7 to C-2 and C-6, H-8 to C-1, H2-8′ to C-1′, H2-7′ to C-2′ and C-6′, H-2′ and H-6′ to C-4′, 3-OMe to C-3, 5-OMe to C-5, and 5′-OMe to C-5′, two phenylpropanoid moieties, namely 4-(3-hydroxypropyl)-2,6-dimethoxyphenol and 4-(3-hydroxypropyl)-2-methoxyphenol, were confirmed. The two fragments were linked through C-8-C-3′ by the HMBC correlations from H2-7 and H2-9 to C-3′ as well as H-8 to C-2′ and H-2′ to C-8. Therefore, the relative configuration of 1 was elucidated as 3,5,5′-trimethoxy-8,3′-neoligna-4,4′,9,9′-tetraol. The absolute configuration of selaginellol (1) was elucidated as (8R)-3,5,5′-trimethoxy-8,3′-neoligna-4,4′,9,9′-tetraol by comparing its electronic circular dichroism (ECD) spectrum [Δε −0.12 (273)] with that of a known analogue secodihydrodehydrodiconiferyl alcohol tetraacetate [9].

Key 2D NMR correlations of 1 and 2
According to the HREIMS ion at m/z 554.2365 [M]+ (calcd for C27H38O12, 554.2363), the molecular formula of compound 2 was determined as C27H38O12 with nine degrees of unsaturation. The IR absorption signals showed the presence of hydroxy (3425 cm−1) and aromatic (1614, 1518, and 1461 cm−1) groups. The NMR data (Table 1) of 2 were very similar to those of 1, except that more signals for a β-glucopyranosyl moiety [δ H 4.57 (d, J = 7.5 Hz); δ C 105.6, 75.9, 77.8, 71.1, 78.0, and 62.4] were observed. As demonstrated in the 1H–1H COSY, HMBC and ROESY correlations (Fig. 2), compound 2 was determined to be the β-glucopyranoside of selaginellol (1). The HMBC correlation from H-1″ to C-4′ indicated that the β-glucopyranosyl part was located at C-4′. According to our previously acidic hydrolysis of rel-(7R,8S)-3,3′,5-trimethoxy-4′,7-epoxy-8,5′-neoligna-4,9,9′-triol 4-O-β-d-glucopyranoside (11) [8], the sugar in the plant is d-glucose. The absolute configuration of the aglycone was elucidated to be the same as that of selaginellol (1) by comparison of its ECD spectrum [Δε −0.37 (273)] with that of 1. Therefore, compound 2 is selaginellol 4′-O-β-d-glucopyranoside.
The known compounds were determined as (−)-syringaresinol (3) [10], (−)-lariciresinol (4) [11], 7S,7′S,8R,8′R-icariol A2 (5) [12], lyoniside (6) [13], (−)-8,8′-bisdihydrosiringenin (7) [14], (7S,8R)-3,3′,5-trimethoxy-4′,7-epoxy-8,5′-neoligna-4,9,9′-triol (8) [8], and dihydrosinapyl alcohol (9) [15], by comparing their NMR data (for all known compounds) and optical rotation values (for the neolignans) with those reported in the literature.
All of these compounds (1–9), along with those previously isolated from the plant, including (7S,8R)-4,9-dihydroxy-3,3′,5-trimethoxy-4′,7-epoxy-8,5′-neolignan-9′-oic acid methyl ester (10) [8], rel-(7R,8S)-3,3′,5-trimethoxy-4′,7-epoxy-8,5′-neoligna-4,9,9′-triol 4-O-β-d-glucopyranoside (11) [8], rel-(7R,8S)-3,3′,5-trimethoxy-4′,7-epoxy-8,5′-neoligna-4,9,9′-triol 9-O-β-d-glucopyranoside (12) [8], 3,3′,5-trimethoxy-4′,7-epoxy-8,5′-neolign-7-ene-4,9,9′-triol 9-O-β-d-glucopyranoside (13) [8], 5-carboxymethyl-7,4′-dihydroxyflavanone 7-O-β-d-glucopyranoside (14) [7], N-selaginelloyl-l-phenylalanine (15) [5], paucine 3′-O-β-d-glucopyranoside (16) [8], paucine (17) [8], and N 1-cis-p-coumaroylagmatine (18) [8], were evaluated for the inhibitory activity against platelet aggregation induced by ADP or collagen. As shown in Table 2, compounds 8, 11, 12, 14, and 16 showed potential inhibitory activity against ADP-induced platelet aggregation with IC50 values of 80.84, 35.76, 42.47, 27.70, and 59.19 µM, respectively, as compared with the positive control tirofiban (IC50 = 25.32 µM). Compounds 8, 11, 12, and 14 also showed the activity against collagen-induced platelet aggregation with IC50 values of 146.70, 31.17, 24.57, and 26.25 µM, respectively, as compared with the positive control tirofiban (IC50 = 148.20 µM). The dihydrobenzofuran neolignans (8, 11, and 12) are more potent than the benzofuran neolignan (13) and other types of neolignans (1–7). Glucosidation of the dihydrobenzofuran neolignans (11 and 12) is helpful for the activity as compared the bioassay result of 11 and 12 with that of 8.
3 Experimental Section
3.1 General Experimental Procedures
Optical rotations were recorded using a JASCO P-1020 Polarimeter (Jasco Corp., Tokyo, Japan). UV spectra were taken on a Shimadzu UV-2401 PC spectrophotometer (Shimadzu, Kyoto, Japan). ECD spectra were recorded on a Chirascan CD spectrometer (Applied Photophysics Ltd., Leatherhead, UK). IR spectra were measured on a Bruker Tensor 27 FTIR Spectrometer (Bruker Corp., Ettlingen, Germany) with KBr disks. 1H and 13C NMR spectra were collected on Bruker Avance 400, DRX-500 or Avance III-600 spectrometers (Bruker Bio-Spin GmbH, Rheinstetten, Germany) with TMS as an internal standard. ESIMS and HRESIMS analyses were carried out on an API QSTAR Pulsar 1 spectrometer (Applied Biosystems/MDS Sciex, Foster City, CA, USA). HREIMS were carried out on a Waters AutoSpec Premier p776 spectrometer (Waters, Millford, MA, USA). Silica gel G (80–100 and 300–400 mesh, Qingdao Meigao Chemical Co., Ltd., Qingdao, China), C18 silica gel (40–75 μm, Fuji Silysia Chemical Ltd., Aichi, Japan), Sephadex LH-20 (GE Healthcare Bio-Sciences AB, Uppsala, Sweden), and D101 macroporous resin (Qingdao Marine Chemical Ltd., Qingdao, China) were used for column chromatography, and silica gel GF254 (Qingdao Meigao Chemical Co., Ltd.) was used for preparative TLC as precoated plates. TLC spots were visualized under UV light at 254 nm and by dipping into 5 % H2SO4 in alcohol followed by heating. Semipreparative HPLC was performed on an Agilent 1200 series pump (Agilent Technologies, Santa Clara, USA) equipped with a diode array detector and an Agilent Zorbax SB-C18 column (5.0 μm, ϕ 9.4 × 250 mm).
3.2 Plant Material
The whole plant of S. moellendorffii was collected from Jingxi County of Guangxi Zhuang Autonomous Region in 2008. A voucher specimen (No. JX0801) was identified by one of the authors (Chun-Lin Long) and deposited at the Key Laboratory of Economic Plants and Biotechnology, Kunming Institute of Botany, Chinese Academy of Sciences.
3.3 Extraction and Isolation
The air-dried, powdered S. moellendorffii plants (15 kg) were exhaustively extracted with MeOH (45 L × 3) at 60 °C. The solvent was removed to give a residue (0.89 kg). The crude extract was subjected to chromatography on a D101 macroporous resin column eluted successively with H2O, 35 % EtOH, and 95 % EtOH to give three portions (I–III), respectively. Portion II (398 g) was subjected to column chromatography (silica gel G; CHCl3/MeOH, 1:0 → 0:1, v/v) to yield six fractions (A–F). Fr. A was subjected to column chromatography (silica gel G; petroleum ether/EtOAc, 15:1 → 0:1, v/v) to yield four fractions (A1–A4). Fr. A1 was purified by column chromatography (silica gel G; CHCl3-acetone, 15:1, v/v) to obtain 9. Fr. A2 was chromatographed on a Sephadex LH–20 column (MeOH) to give subfractions A2–1 and A2–2. Subfraction A2–1 was subjected to chromatography on a silica gel G column (CHCl3-acetone, 20:1, v/v) and then further purified by semi-preparative HPLC (MeCN/H2O, 30:70, v/v) to yield 4 (14.9 mg, t R = 13.099 min). Subfraction A2-2 was purified by preparative TLC (CHCl3/MeOH, 10:1, v/v) to obtain 3 (24.6 mg). Fr. A3 was chromatographed over a C18 silica gel column (MeOH/H2O, 30:70, v/v), a Sephadex LH-20 column (MeOH), and a silica gel G column (CHCl3/MeOH/H2O, 50:1:0.25), and purified by semi-preparative HPLC (MeOH/H2O, 40:60, v/v) to obtain 5 (15.5 mg, t R = 5.864 min). Fr. A4 was chromatographed on a Sephadex LH-20 column (MeOH), a C18 silica gel (MeOH/H2O, 50:50, v/v), and a silica gel G column (CHCl3/MeOH, 60:1, v/v), and purified by semi-preparative HPLC (MeCN/H2O, 30:70, v/v) to yield 7 (2.0 mg, t R = 7.716 min), 8 (2.0 mg, t R = 8.917 min) and 1 (4.0 mg, t R = 13.652 min). Fr. D was chromatographed on a C18 silica gel column (MeOH/H2O, 30:70, v/v), a Sephadex LH-20 column (MeOH), and a silica gel G column (CHCl3/MeOH/H2O, 100:10:0.5, v/v), and purified by semi-preparative HPLC (MeOH/H2O, 40:60, v/v) to yield 2 (7.8 mg, t R = 12.138 min) and 6 (4.4 mg, t R = 18.734 min).
3.3.1 Selaginellol (1)
Pale yellow oil (MeOH); [α] 24D −50.4 (c 0.40, MeOH); UV (CH3OH) λ max (log ε) 280 (3.31), 228 (3.98) nm; ECD Δε (c 0.010, MeOH) −0.12 (273), −3.84 (214), +3.63 (197); IR (KBr) v max 3428, 1614, 1518, 1496, 1461, 1431, 1289, 1217, 1114 cm−1; 1H and 13C NMR data, see Table 1; positive ion ESIMS m/z 415 [M+Na]+; positive ion HRESIMS m/z 415.1729 [M+Na]+ (calcd for C21H28O7Na+, 415.1727).
3.3.2 Selaginellol 4′-O-β-d-glucopyranoside (2)
Pale yellow solid (MeOH); [α] 22D −57.8 (c 0.26,MeOH); UV (CH3OH) λ max (log ε) 274 (3.80) nm; ECD Δε (c 0.011, MeOH) −0.37 (273), −6.17 (210), +3.20 (200); IR (KBr) v max 3425, 1615, 1518, 1461, 1428, 1325, 1216, 1113, 1071 cm−1; 1H and 13C NMR data, see Table 1; positive ion ESIMS m/z 577 [M+Na]+; HREIMS m/z 554.2365 [M]+ (calcd for C27H38O12, 554.2363).
3.4 In Vitro Platelet Aggregation Assay
In vitro platelet aggregation was conducted using the turbidimetric method with a minor modification [16, 17]. Briefly, blood was withdrawn from the carotid artery of New Zealand rabbits,and anticoagulated with 3.8 % sodium citrate (1:9 citrate/blood, v/v) and centrifuged for 15 min at 950 rpm to prepare platelet-rich plasma (PRP) or 10 min at 3000 rpm to obtain platelet-poor plasma (PPP). The platelet concentration was adjusted to 3 × 108 platelets/mL. PRP in 270 μL was preincubated at 37 °C for 5 min in the cuvette with 20 μL of sample or vehicle (saline), and then platelet aggregation was induced by 10 μL ADP (10 μM) or collagen (2.5 μg/mL). The maximum platelet aggregation rate was determined within 5 min with continuous stirring at 37 °C using four-channel aggregometer (Beijing Steellex Science Instrument Company, China).
For each compound, five concentrations were chosen and a percentage inhibition-concentration curve was derived. From this curve the IC50 value was calculated as the concentration of inhibitor causing a 50 % inhibition of the aggregation using SPSS software.
References
Z.Y. Wu, T.Y. Zhou, P.G. Xiao, Xinghua Bencao Gangyao, vol. 3 (Shanghai Scientific and Technological Press, Shanghai, 1990), pp. 626–631
X.C. Zhang, H.P. Nooteboom, M. Kato, Selaginellaceae, in Flora of China, ed. by Z.Y. Wu, P.H. Raven, D.Y. Hong (Science Press, Beijing and Missouri Botanical Garden Press, St. Louis, 2013), pp. 37–66
Y. Liu, H.J. Yin, D.Z. Shi, K.J. Chen, Evid.-Based Compl. Alt. Med. 2012, Article ID 184503 (2012)
C. Chen, F.Q. Yang, Q. Zhang, F.Q. Wang, Y.J. Hu, Z.N. Xia, Evid.-Based Compl. Alt. Med. 2015, Article ID 876426 (2015)
Y.H. Wang, C.L. Long, F.M. Yang, X. Wang, Q.Y. Sun, H.S. Wang, Y.N. Shi, G.H. Tang, J. Nat. Prod. 72, 1151–1154 (2009)
Y. Kong, X.L. Su, Y.H. Wang, H.M. Niu, Chinese Patent CN 104274441A, 27 Oct 2014
H.S. Wang, L. Sun, Y.H. Wang, Y.N. Shi, G.H. Tang, F.W. Zhao, H.M. Niu, C.L. Long, L. Li, Arch. Pharm. Res. 34, 1283–1288 (2011)
Y.H. Wang, Q.Y. Sun, F.M. Yang, C.L. Long, F.W. Zhao, G.H. Tang, H.M. Niu, H. Wang, Q.Q. Huang, J.-J. Xu, L.J. Ma, Helv. Chim. Acta 93, 2467–2477 (2010)
W.C. Su, J.M. Fang, Y.S. Cheng, Phytochemistry 40, 563–566 (1995)
A.K. Chakravarty, S. Mukhopadhyay, S.K. Moitra, B. Das, Indian J. Chem. Sect B 33, 405–408 (1994)
N. Erdemoglu, E. Sahin, B. Sener, S. Ide, J. Mol. Struct. 692, 57–62 (2004)
H. Yamauchi, R. Kakuda, Y. Yaoita, K. Machida, M. Kikuchi, Chem. Pharm. Bull. 55, 346–347 (2007)
H.T. Le, C.T.A. Minh, T.H. Kim, P. Van Kiem, N.D. Thuan, M. Na, Arch. Pharmacal Res. 35, 87–92 (2012)
M.A. Rahman, T. Katayama, T. Suzuki, T. Nakagawa, J. Wood Sci. 53, 161–167 (2007)
H.Q. Huang, M.N. Yan, X.L. Piao, China J Chin. Mat. Med. 36, 2211–2214 (2011)
M. Nishikawa, H. Hidaka, J. Clin. Invest. 69, 1348–1355 (1982)
T.L. Aghaloo, P.K. Moy, E.G. Freymiller, J. Oral Maxil. Surg. 60, 1176–1181 (2002)
Acknowledgments
This work was funded by the Ministry of Science & Technology of China (2012FY110300), the National Natural Science Foundation of China (3116140345), and the Minzu University of China (YLDX01013, 2015MDTD16C).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of Interest
The authors declare no conflict of interest.
Additional information
Jing-Xian Zhuo and Yue-Hu Wang contributed equally to this work.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Zhuo, JX., Wang, YH., Su, XL. et al. Neolignans from Selaginella moellendorffii . Nat. Prod. Bioprospect. 6, 161–166 (2016). https://doi.org/10.1007/s13659-016-0095-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13659-016-0095-5