
Abstract
Five new guanacastane-type diterpenes, named guanacastepenes P–T (1–5), were isolated from cultures of the fungus Psathyrella candolleana. Their structures were elucidated on the basis of extensive spectroscopic methods. All of the compounds were tested for their 11β-hydroxysteroid dehydrogenase (11β-HSD1) inhibitory activity. Compound 3 exhibited inhibitory activity against both human and mouse isozymes of 11β-HSD1 with IC50 values of 6.2 and 13.9 μM, respectively.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
1 Introduction
Guanacastepene A, was the first member of the guanacastane family isolated from an unidentified endophytic fungus (CR 115) [1]. This was followed by isolation of guanacastepenes B–O from the same fungus by the same group [2]. Heptemerones A–G were later isolated from cultures of Coprinus heptemerus [3, 4], as well as radianspenes A–M from Coprinus radians [5], which attracted interest of the guanacastanes in the synthetic organic community [6–18]. The total synthesis of guanacastepenes with a novel 5/7/6 ring system was considered a challenging synthetic target [19], whereas synthesis of analogues offered the best prospects for capitalizing on the promising antibiotic activity, as well as avoiding the side effect of hemolytic activity against human red blood cells [10, 20]. However, no other bioactivities, except for antibiotic effects were reported for this type of diterpenes. As a part of a search for naturally occurring secondary metabolites with diverse structures from higher fungi in China, investigations of chemical components from Psathyrella candolleana cultures were carried out, which led to the isolation of a series of new guanacastane-type diterpenes, guanacastepenes P–T (1–5, Fig. 1). Their structures were elucidated by means of spectroscopic methods. These compounds shared a larger conjugated system including an α,β-unsaturated ketone moiety in the highly oxygenated five-member ring system comparing to the known guanacastane-type diterpenes. All of these compounds were evaluated for their cytotoxic and anti-herpes simplex viruses (HSV) activities, while compounds 1, 3, and 5 were evaluated for inhibitory activities against one isozyme of 11β-hydroxysteroid dehydrogenases (11β-HSD1).

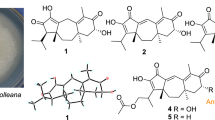
Structures of compounds (1–5)
2 Results and Discussion
Guanacastepene P (1) was obtained as a light yellow oil. Its molecular formula C20H28O3 was established from the positive ion HRESIMS ([M+Na]+, 339.1930), indicating 7 degrees of unsaturation. The 13C NMR (DEPT) spectrum suggested 20 carbon resonances (Table 2), classified as five methyl groups, four aliphatic methylenes, three methines (one olefinic and one oxygenated), and eight quaternary carbons (one carbonyl and five olefinic). Thus, four degrees of unsaturation were accounted for by the three double bonds and the one carbonyl group, while the remaining three degrees of unsaturation suggested that compound 1 should possess a three-ring system. Further inspection of the 1H NMR spectrum and 1H–1H COSY correlations (Fig. 2) resulted in the deduction of an isopropyl group (Me-19–H-18–Me-20), two connected methylene groups (H2-9–H2-10), a chain of CH2–CH2–CH–OH (H2-7–H2-6–H-5–OH), an isolated olefinic hydrogen (H-2), and three isolated methyl groups (H-15, H-16 and H-17).

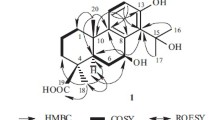
1H–1H COSY and HMBC correlations for 1 and 5
Connectivity among the above mentioned fragments and the contribution of the three-ring system were established by analysing 1H, 13C long-range correlations extracted from HMBC experiments (Fig. 2). The correlations of H-18 to C-11, C-12 and C-13 and HO-13 to C-12 and C-14 established the connection of C-11–C-12–C-13–C-14, while the correlations of H-17 to C-11, C-12 and C-1, and H-2 to C-1 and C-11 indicated the presence of a five-membered ring A. Subsequently, the correlations of H-9 to C-8 and H-10 to C-11 supported the connection of C-8–C-9–C-10–C-11. Besides, correlations of H-16 to C-8 and C-3, as well as H-2 to C-8 were evident for a seven-membered ring B. The last ring C and the links of remaining blocks were clearly shown by HMBC correlations of H-15 to C-3, C-4 and C-5, H-5 to C-4 and C-6, H-6 to C-4 and C-5, and H-7 to C-8 and C-3. Finally, compound 1 possessed a backbone of a 5/7/6 ring system related to that of guanacastepene A [1]. Biogenetically, the stereoconfigurations of methyls of Me-16 and Me-17 were α and β oriented, respectively [4]. In the ROESY spectrum (Fig. 3), correlations between H-17, H-9β and H-10β were observed, suggesting both were β oriented, while correlations of H-5/H-6α, and H-6α/H-16 indicated that H-5 was α oriented.
Guanacastepene Q (2), purified as a light yellow oil, had a molecular formula of C20H26O3 according to its HRESIMS at m/z 315.1962 ([M+H]+). The 13C NMR (Table 2) spectroscopic data were similar to those of compound 1. The main difference was that an oxygenated methine at δC 70.3 (C-5) in 1 was replaced by a keto carbonyl group at δC 197.5 in 2, constructing a 3,4-unsaturated-5-keto moiety, which led to the downfield shift of C-3 (∆δ 20.8 ppm) and C-6 (∆δ 4.4 ppm), and the upfield shift of C-4 (∆δ 6.4 ppm) in compound 2, consistent with the HMBC correlations of H-6, H-7 and H-15 to C-5. Detailed analysis of 1D and 2D NMR data (HSQC, HMBC, 1H–1H COSY, ROESY) suggested that the other parts of 2 were the same as those of 1. Therefore, compound 2 was established as shown.
Guanacastepene R (3), a light yellow oil, had a molecular formula of C20H26O4 on the basis of its HRESIMS at m/z 353.1737 ([M+Na]+), 16 mass units higher than that of 2. The 1D NMR spectroscopic data (Tables 1 and 2) were quite similar to those of 2, except that the signals for a methyl group in 2 were replaced by the signals for an oxygenated methylene group, which was confirmed by the HMBC correlations of δH 3.62 (1H, d, J = 7.0 Hz, OH-15) to δC 56.4 (t, C-15) and δC 136.3 (s, C-4). Careful analysis of the spectroscopic data finally established the structure of 3 as guanacastepene R, as shown.
Guanacastepene S (4) had the same molecular formula of C20H28O3 (HRESIMS ([M+Na]+ at m/z 339.1927) as 1 and was obtained as a colorless oil. Its 13C NMR spectrum (Table 2) displayed 20 carbon signals corresponding to five methyls (three singlets and two doublets), five methylenes, two methines and eight quaternary carbons (four olefinic and two carbonyl carbons), consistent with a similar structure of compound 2. Significant differences were one less double bond and absence of an olefinic hydrogen in 4. The H-18 correlation with a methine proton at δH 1.97 (1H, d, J = 2.7 Hz, H-12) in the 1H–1H COSY spectrum, and the key HMBC correlations of the OH signal at δH 7.84 (1H, br. s, OH-14) to a keto carbonyl carbon δC 202.9 (s, C-13) and the double bond signal at δC 148.1 (s, C-14) indicated a different enolization of the 13,14-diketone system (to a 13-keto-14-enol) in 4. Further analysis of 2D NMR spectroscopic data suggested that the other parts were the same to those of 2, except the olefinic carbon at C-2 in 2 being replaced by a methylene moiety in 4, as established by the HMBC correlations of δH 3.50 and 3.60 (each 1H, d, J = 19.0 Hz, H-2) to C-1. On the basis of the ROESY experiment, H-12 was elucidated to be α oriented by correlations of H-17 with H-18 and H-19. Thus compound 4 was assigned as guanacastepene S.
Guanacastepene T (5) possessed the molecular formula C20H30O3, as determined by its HRESIMS at m/z 341.2091 ([M+Na]+), indicating six degrees of unsaturation. The 13C NMR spectroscopic data (Table 2) together with analysis of its HSQC spectrum indicated 20 carbons, including four methyls, five methylenes (one olefinic), six methines (two oxygenated and one olefinic), and five quaternary carbons (two olefinic and one carbonyl carbons). Comprehensive analysis of the 1H–1H COSY and HMBC spectra (Fig. 2), suggested that 5 shared the same guanacastepene skeleton with 1, except for differences in the number and location of functional groups. The 1H–1H COSY correlations of H-19/H-18/H-12/H-13, combined with HMBC correlations of H-13 with C-14, C-12 and C-1, and H-2 with C-1 and C-14 suggested the presence of a single 2(1)-en-14-one system in 5. Besides, the trisubstituted double bond between C-3 and C-4 in 1 was changed into a terminal double bond between C-4 and C-15 in 5, consistent with the 1H NMR chemical shifts of δH 5.11 and 5.22 (each 1H, br. s, H-15), which was also supported by the HMBC correlations of H-15 to C-3 and C-4, and H-3 to C-2 and C-4. The 1H–1H COSY data, as well as HMBC correlations of H-5 to C-3 and C-15 also gave the information that C-5 was a methine substituted by a hydroxy group. Likewise, C-9 was confirmed as a hydroxy substituted methine based on the 1H–1H COSY and HMBC correlations as shown in Fig. 2. Based on the proposed biogenetic orientation of Me-16 and Me-17, the configurations of H-3 and HO-9 were determined to be β and α oriented, respectively, owing to the strong correlation signals of H-3 with H-17 and H-9, and H-9 with H-17 in the ROESY spectrum. In addition, the stereoconfiguration of H-5 was determined to be α oriented, as indicated by the observation of the ROESY correlations the same to those of compound 1. Consequently, compound 5 was elucidated as guanacastepene T.

Key ROESY correlations of 1
Compounds 1–5 were evaluated for their cytotoxicity against five human cancer cell lines. None was found to possess significant activity with IC50 values less than 40 μM. In addition, they were evaluated for anti-HSV (herpes simplex viruses) activity. However, none exhibited activity. Furthermore, compounds 1, 3 and 5 were tested for inhibitory activities against one isozyme of 11β-HSD1. Of these, only compound 3 exhibited inhibitory activity against both human and mouse isozymes of 11β-HSD1 with IC50 values of 6.2 and 13.9 μM, while glycyrrhizinic acid (positive control) had IC50 values of 4.2 and 6.5 nM, respectively.
Five new guanacastane-type diterpenes, guanacastepenes P–T, each containing a 5/7/6 ring system, were isolated from cultures of fungus P. candolleana. Until now, totally 33 guanacastane-type diterpenes have been obtained from kinds of fungi, and some of them showed antibiotic and antitumor activities. Our present research enriched the structure diversity of the guanacastane family and this is also the first report to show that guanacastane-type diterpenes possessed inhibitory activity against human 11β-HSD1.
3 Experimental
3.1 General Experimental Procedures
Optical rotations (OR) were recorded on a JASCO P-1020 digital polarimeter, while the UV and IR spectra were obtained on a Shimadzu UV2401PC and a Bruker Tensor 27 FT-IR (KBr pellets) spectrometers. NMR spectra were acquired on Bruker AM-400, DRX-500 and Avance III 600 MHz spectrometers with tetramethylsilane (TMS) used as an internal standard at room temperature. High-resolution (HR) ESI-MS were recorded on an API QSTAR Pulsar spectrometer. Silica gel (200–300 mesh, Qingdao Marine Chemical Ltd., People’s Republic of China) and Sephadex LH-20 (Amersham Biosciences, Sweden) were used for open column chromatography (CC). Preparative HPLC was performed on an Agilent 1100 liquid chromatography system equipped with a Zorbax SB-C18 column (21.2 mm × 150 mm). Fractions were monitored by TLC. Spots were visualized by heating silica gel plates immersed in vanillin–H2SO4 in EtOH.
3.2 Fungal Material and Cultivation Conditions
The fungus P. candolleana (Pers.) Fr. was collected at Kunming Institute of Botany in Yunnan Province, People’s Republic of China, in 2003. The fungus was identified by Prof. Zhu-Liang Yang at the Kunming Institute of Botany. A specimen (No. KIB20030828) was deposited at Kunming Institute of Botany, Chinese Academy of Sciences.
Basidiomata small to medium-sized. Pileus 3–7 cm in diam, campaniform then flattened, with moist umbonate center; surface glabrous, light honey-yellow to brown, yellow-brown at the apex, fading to grayish when dry; white universal veil gradually fall off with age. Context white, relatively thin. Lamellae adnate, narrow and length unequal, crowded, dirty white, grayish to pallid purple-brown; edge dirty white, coarse. Stipe slender and fistulous, cylindrical, 3–8 × 0.2–0.7 cm, white, weak and fragile, surface densely floccose-fibrillose or reticulate. Spores print dark purple-brown; spores smooth, ellipsoid, 6.5–9.0 × 3.5–5.0 μm, germ pore was visible. Pleurocystidia smooth and thin-walled, hyaline in KOH, more or less lageniform and apex rounded, 34–50 × 8–16 μm.
The culture medium consisted of glucose (5 %), peptone from porcine meat (0.15 %), yeast powder (0.5 %), KH2PO4 (0.5 %) and MgSO4. Fermentation was carried out on a shaker at 160 RPM for 25 days.
3.3 Extraction and Isolation
The culture broth (21 L) was filtered, and the filtrate was extracted with ethyl acetate (20 L × 3), while the mycelium was extracted three times with CHCl3–MeOH (1:1). The EtOAc layer together with the mycelium extract was concentrated under reduced pressure to give a crude extract (10 g), and the latter was applied to a silica gel column eluted with a gradient of CHCl3/MeOH (1:0→0:1) to obtain five fractions (1–5). Fraction 3 was subjected to Sephadex LH-20 (CHCl3/MeOH 1:1) to give two subfractions (3a–3b). Fraction 3a was separated by semipreparative HPLC (MeCN/H2O, 1:9→2:8, 30 min) to give three mixtures, then purified separately by Sephadex LH-20 CC (acetone) to give 2 (2.1 mg), 4 (2.0 mg) and 5 (4.8 mg), respectively. In the same way, subfraction 3b was purified by semipreparative HPLC (MeCN/H2O, 1:9→2:8, 40 min) to yield 1 (3.1 mg) and 3 (3.3 mg).
3.4 Guanacastepene P (1)
Light yellow oil; \( [\alpha ]_{\text{D}}^{ 1 6} \) − 507.0 (c 0.70, MeOH); IR (KBr) νmax 3444, 2925, 1632 cm−1; UV (MeOH) λmax (log ε) 307 (4.16), 206 (4.16); For 1H (400 MHz) and 13C NMR (150 MHz) spectroscopic data (Me2CO-d6), see Tables 1 and 2; positive ion HRESIMS m/z 339.1930 (calcd for C20H28O3Na [M+Na]+, 339.1936).
3.5 Guanacastepene Q (2)
Light yellow oil; \( [\alpha ]_{\text{D}}^{ 1 6} \) − 318.3 (c 1.6, MeOH); IR (KBr) νmax 3433, 2963, 2925, 1669, 1627, 1409, 1152 cm−1; UV (MeOH) λmax (log ε) 289 (4.03), 236 (3.87), 203 (3.91); For 1H (400 MHz) and 13C NMR (100 MHz) spectroscopic data (Me2CO-d6), see Tables 1 and 2; positive ion HRESIMS m/z 315.1962 (calcd for C20H27O3 [M+H]+, 315.1960).
3.6 Guanacastepene R (3)
Light yellow oil; \( [\alpha ]_{\text{D}}^{ 1 6} \) − 303.7 (c 1.9, MeOH); IR (KBr) νmax 3433, 2924, 1630, 1104 cm−1; UV (MeOH) λmax (log ε) 285 (3.99), 232 (3.82), 203 (3.93); For 1H (400 MHz) and 13C NMR (150 MHz) spectroscopic data (Me2CO-d6), see Tables 1 and 2; positive ion HRESIMS m/z 353.1737 (calcd for C20H26O4 [M+H]+, 353.1728).
3.7 Guanacastepene S (4)
Colorless oil; \( [\alpha ]_{\text{D}}^{ 2 1} \) − 167.5 (c 1.9, MeOH); IR (KBr) νmax 3431, 2924, 2346, 1630 cm−1; UV (MeOH) λmax (log ε) 285 (3.82), 201 (3.86); For 1H (400 MHz) and 13C NMR (150 MHz) spectroscopic data (Me2CO-d6), see Tables 1 and 2; positive ion HRESIMS m/z 339.1927 (calcd for C20H28O3 [M+Na]+, 339.1927).
3.8 Guanacastepene T (5)
Colorless oil; \( [\alpha ]_{\text{D}}^{ 2 1} \) + 0.75 (c 0.8, MeOH); IR (KBr) νmax 3440, 2923, 1631 cm−1; UV (MeOH) λmax (log ε) 252 (3.71), 201(3.52); For 1H (400 MHz) and 13C NMR (150 MHz) spectroscopic data (CDCl3), see Tables 1 and 2; positive ion HRESIMS m/z 341.2091 (calcd for C20H30O3 [M+Na]+, 341.2092).
3.9 Cytotoxicity Assay
Human myeloid leukemia HL-60, hepatocellular carcinoma SMMC-7721, lung cancer A-549 cells, breast cancer MCF-7 and colon cancer SW480 cell lines were used in the cytotoxic assay. All cell lines were cultured in RPMI-1640 or DMEM medium (Hyclone, USA), supplemented with 10 % fetal bovine serum (Hyclone, USA) in 5 % CO2 at 37 °C. The cytotoxicity assay was performed according to the MTT [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide[ method in 96-well microplates [21]. Briefly, adherent cells (100 µL) were seeded into each well of 96-well cell culture plates and allowed to adhere for 12 h before drug addition, while suspended cells were seeded just before drug addition with initial density of 1 × 105 cells/mL. Each tumor cell line was exposed to the test compound dissolved in DMSO at concentrations of 0.0625, 0.32, 1.6, 8, and 40 μM in triplicates for 48 h, with cisplatin (Sigma, USA) as a positive control. After compound treatment, cell viability was detected and a cell growth curve was graphed. IC50 values were calculated by Reed and Muench’s method [22].
3.10 Antiviral Assay
Confluent Vero cells in a 96-well tissue plate were inoculated in triplicate with virus suspension (50 μL) [HSV/Blue, at multiplicity of infection (MOI) 1] and culture medium (50 μL) containing testing compounds at different concentrations. Cells were lysed with 1 % Nonidet P-40 in DMEM at 24 h postinfection. Lysates from each well were mixed with chlorophenol red-β-d-galactopyranoside [CPRG; Boehringer, Ingelheim, Germany), and β-galactosidase (β-Gal)] activity was measured by taking absorbance readings at 570 nm every 2 min for a total of 25 readings. The slope of the line was used to quantify β-Gal activity as milli-optical density units/min (mOD/min). The 50 % inhibitory concentration (IC50) was defined as the concentration of the antiviral drug that reduced the mOD/min values by 50 % relative to the virus control. Inhibitory concentrations were calculated using the probit regression method [23].
3.11 Inhibitory Activities Against 11β-HSD1 Assay
Inhibitory activities of the compounds on human or mouse 11β-HSD1 were determined using scintillation proximity assay (SPA). Microsomes containing 11β-HSD1 were used according to our previous studies [24, 25]. Full-length cDNAs of human or murine 11β-HSD1 were isolated from cDNA libraries provided by the NIH Mammalian Gene Collection. The cDNAs were cloned into pcDNA3 expression vectors. HEK-293 cells were transfected with the pcDNA3-derived expression plasmid and selected by cultivation in the presence of G418 (700 μg/mL). The microsomal fraction overexpressing 11β-HSD1 was prepared from the HEK-293 cells, which were stable transfected with 11β-HSD1. The fraction was then used as the enzyme source for SPA. Microsomes containing human or mouse 11β-HSD1 were incubated with NADPH and [3H] cortisone. The product, [3H] cortisol, was specifically captured by a monoclonal antibody coupled to protein A-coated SPA beads. All tests were done in twice with glycyrrhizinic acid as a positive control. IC50 (X ± SD, n = 2) values were calculated by using Prism Version 4 (GraphPad Software, San Diego, CA, USA).
References
S.F. Brady, M.P. Singh, J.E. Janso, J. Clardy, J. Am. Chem. Soc. 122, 2116–2117 (2000)
S.F. Brady, S.M. Bondi, J. Clardy, J. Am. Chem. Soc. 123, 9900–9901 (2001)
M. Kettering, C. Valdivia, O. Sterner, H. Anke, E. Thines, J. Antibiot. 58, 390–396 (2005)
C. Valdivia, M. Kettering, H. Anke, E. Thines, O. Sterner, Tetrahedron 61, 9527–9532 (2005)
Y.X. Qu, Y.Y. Li, X.M. Qian, Y.M. Shen, Phytochemistry 78, 190–196 (2012)
M.A. Battiste, P.M. Pelphrey, D.L. Wright, Chemistry 12, 3438–3447 (2006)
K.M. Brummond, D. Gao, Org. Lett. 5, 3491–3494 (2003)
X.H. Du, H.V. Chu, O. Kwon, Org. Lett. 5, 1923–1926 (2003)
G.B. Dudley, D.S. Tan, G. Kim, J.M. Tanski, S.J. Danishefsky, Tetrahedron Lett. 42, 6789–6791 (2001)
G.B. Dudley, S.J. Danishefsky, Org. Lett. 3, 2399–2402 (2001)
C.M. Gampe, E.M. Carreira, Angew. Chem. Int. Ed. 50, 2962–2965 (2011)
C.C. Hughes, J.J. Kennedy-Smith, D. Trauner, Org. Lett. 5, 4113–4115 (2003)
C.C. Hughes, A.K. Miller, D. Trauner, Org. Lett. 7, 3425–3428 (2005)
C.C. Li, C.H. Wang, B. Liang, X.H. Zhang, L.J. Deng, S. Liang, J.H. Chen, Y.D. Wu, Z. Yang, J. Org. Chem. 71, 6892–6897 (2006)
S.N. Lin, G.B. Dudley, D.S. Tan, S.J. Danishefsky, Angew. Chem. Int. Ed. 41, 2188–2191 (2002)
C.A. McGowan, A.K. Schmieder, L. Roberts, M.F. Greaney, Org. Biomol. Chem. 5, 1522–1524 (2007)
G. Mehta, J.D. Umarye, Org. Lett. 4, 1063–1066 (2002)
Y. Oonishi, A. Taniuchi, Y. Sato, Synthesis 28, 84–2892 (2010)
S. Iimura, L.E. Overman, R. Paulini, A. Zakarian, J. Am. Chem. Soc. 128, 13095–13101 (2006)
M.P. Singh, J.E. Janso, S.W. Luckman, S.F. Brady, J. Clardy, M. Greenstein, W.M.J. Maiese, J. Antibiot. 53, 256–261 (2000)
T. Mosmann, J. Immunol. Methods 65, 55–63 (1983)
L.J. Reed, H. Muench, Am. J. Hyg. 27, 493–497 (1938)
Q.C. Zhu, Y. Wang, T. Peng, J. Biomol. Screen. 15, 1016–1020 (2010)
H.Y. Yang, W. Dou, J. Lou, Y. Leng, J.H. Shen, J Bioorg. Med. Chem. Lett. 18, 1340–1345 (2008)
H.Y. Yang, Y. Shen, J.H. Chen, Q.F. Jiang, Y. Leng, J.H. Shen, Eur. J. Med. Chem. 44, 1167–1171 (2009)
Acknowledgments
This project was supported by the National Natural Sciences Foundation of China (U1132607, 81373289).
Conflict of interest
All authors declare no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
This article is published under license to BioMed Central Ltd. Open Access This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article

Cite this article
Yin, X., Feng, T., Li, ZH. et al. Five New Guanacastane-Type Diterpenes from Cultures of the Fungus Psathyrella candolleana. Nat. Prod. Bioprospect. 4, 149–155 (2014). https://doi.org/10.1007/s13659-014-0020-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13659-014-0020-8