
Abstract
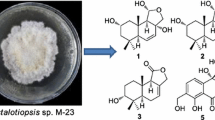
Seven new drimane-type sesquiterpennoids, phellinuins A–G (1–7), together with one known compound 3β,11,12-trihydroxydrimene (8) were isolated from the cultures of mushroom Phellinus tuberculosus. Their structures were elucidated on the basis of NMR and MS spectroscopic data and by comparison with data reported in the literature.
Graphical Abstract

Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
Phellinus is a genus of fungi in the family Hymenochaetaceae. Many species cause white rot. Its fruiting bodies, often growing on wood, are resupinate, sessile, and perennial. The flesh is tough and woody or cork-like, and brown in color [1]. The fungus Phellinus tuberculosus has a wide distribution in Yunnan province of China [2]. The crude extract of mushroom P. tuberculosus was reported to possess antioxidant activity, which exhibited potent radical scavenging activity [3]. However, the chemical constituents of P. tuberculosis has not reported yet. As our continuous search for natural products from higher fungi [4–7], we carried out the chemical investigation on cultures of P. tuberculosus, which resulted in the isolation of seven new drimane-tpye sesquiterpennoids named phellinuins A–G (1–7) and one known compound (8) (Fig. 1). The structures of new compounds were determined on the basis of extensive spectroscopic analysis including NMR, MS, IR data, while the known compound was identified as 3β,11,12-trihydroxydrimene (8) by comparison with data reported in literature [8]. This paper describes their isolation and structural elucidation.

Structures of compounds 1–8
2 Results and Discussion
Compound 1 was obtained as a colorless oil. Its molecular formula C15H26O4 was revealed on the basis of the HREIMS at m/z 270.1827 (calcd for C15H26O4, 270.1831 [M]+), suggesting three degrees of unsaturation. The 1H NMR (Table 1) and 13C NMR data (Table 2) shows the presence of two methyls, six methylenes, four methines, and three quaternary carbons. In addition, the IR spectrum showed the presence of hydroxy group (3405 cm−1). Apart from one double bond, the remaining two degrees of unsaturation in 1 were assumed to be a bicyclic sesquiterpenoid. Detailed analysis of NMR data suggested that compound 1 should be a drimane-type sesquiterpenoid with a similar planar structure to that of 3α,11,15-trihydroxydrimene [9]. Analysis of 2D NMR data suggested that only Me-12 was oxygenated into an oxymethylene in 1, which was suggested by the HMBC correlations from δH 4.23 (1H, d, J = 11.8 Hz, H-12a) and 3.95 (1H, d, J = 11.8 Hz, H-12b) to δC 138.7 (s, C-8). The HMBC data further supported that the other parts of the planar structure of 1 were the same to those of 3α,11,15-trihydroxydrimene (Fig. 2) [9]. In the ROESY spectrum (Fig. 2), the correlation of H-5/Me-15 suggested that C-14 was β oriented, while the correlation of Me-15/H-3, as well as the constant coupling of H-3 (dd, J = 11.8, 3.6 Hz), indicated OH-3 to be β oriented. On the basis of these data, the ROESY correlations of H-5/H-9 and Me-13/H-11 indicated that H-9 was α oriented, while Me-13 was β oriented. Therefore, compound 1 was established as 3β,11,12,14-tetrahydroxydrimene and named as phellinuin A.

The main HMBC, COSY, and ROESY correlations of 1–7
Compound 2 was isolated as amorphous powder. The molecular formula was established to be C15H26O4 on the basis of the HREIMS at m/z 270.1828 (calcd for C15H26O4, 270.1831 [M]+). The 1D NMR data (Tables 1 and 2) were very similar to those of compound 1, which indicated that both compounds had the same structure. However, the ROESY correlations of H-5/H-15 and H-15/H-3 indicated that Me-15 was oxygenated into an oxymethylene in 2, while C-14 should be a methyl (Fig. 2). Detailed analyses of other 2D NMR data suggested that the other parts of 2 were the same to those of 1. Therefore, the structure of compound 2 was established as 3β,11,12,15-tetrahydroxydrimene and named phellinuin B.
Compound 3, a colorless oil, possessed a molecular formula C17H28O5, as deduced from HREIMS at m/z 312.1935 (calcd for C17H28O5, 312.1937 [M]+). The IR spectrum displayed the absorption bands for C=O (1722 cm−1), OH (3418 cm−1), and C=C (1642 cm−1). All the NMR data suggested that compound 3 was closely related to 1 except one more O-acetyl group in 3. The O-acetyl group was substituted at C-11—as revealed by HMBC correlations from δH 4.30 (1H, dd, J = 12.0, 4.2 Hz, H-11a) and 4.20 (1H, overlap, H-11b) to δC 173.0 (s, OAc) and 52.2 (d, C-9). The other 2D NMR data suggested that the other parts of 3 were the same to those of 1 (Fig. 2). Thus, the structure of compound 3 was elucidated as phellinuin C as shown in Fig. 1.
Compound 4 was also obtained as a colorless oil. HREIMS gave one pseudomolecular ion at m/z 312.1947 (calcd for C17H28O5, 312.1937). The 1D NMR data (Tables 1 and 2) were very similar with those of compound 3. However, the HMBC correlations from δH 4.70 (1H, d, J = 12.6 Hz, H-12a) and 4.53 (1H, d, J = 12.6 Hz, H-12b) to δC 134.4 (s, C-8) and 173.0 (s, OAc) suggested that the O-acetyl group was substituted at C-12 in 4 rather than at C-11 in 3. The other parts of structure 4 were established to be the same with those of 3 by 2D NMR correlations (Fig. 2). Therefore, compound 4 was identified as phellinuin D.
Compound 5 was established as an O-acetyl derivative of 2, which was supported by the HMBC correlations from δH 4.32 (1H, dd, J = 11.4 and 4.2 Hz, H-11a) and 4.21 (1H, dd, J = 11.4 and 6.0 Hz, H-11b) to δC 137.2 (s, C-8) and 173.0 (s, OAc). The other 2D NMR data suggested that the others parts of 5 were the same to those of 2. Therefore, the structure of compound 5 was established and named as phellinuin E.
Compounds 6 and 7 were identified as O-acetyl analogues of the known compound 3β,11,12-trihydroxydrimene (8) [8]. The HMBC data suggested that the O-acetyl group was substituted at C-11 in 6 and C-12 in 7, respectively (Fig. 2). Analyses of other 2D NMR data suggested that the other parts were the same to those of 8 (Fig. 2). Therefore, the structures of compounds 6 and 7 were established and named as phellinuin F (6) and phellinuin G (7), respectively.
3 Experimental Section
3.1 General Experimental Procedures
Optical rotations were measured on a Jasco-P-1020 polarimeter. IR spectra were obtained by using a Bruker Tensor 27 FT-IR spectrometer with KBr pellets. NMR spectra were acquired with instruments of Avance Ш 600 and Bruker DRX-500. HREIMS were measured on a waters autoSpec Primier P776 instruments. Silica gel (200–300 mesh, Qingdao Marine Chemical Inc., China) and Sephadex LH-20 (Amersham Biosciences, Sweden) were used for column chromatography (CC). Fractions were monitored by TLC and spots were visualized by heating silica gel plates immersed in vanillin-H2SO4 in EtOH, in combination with Agilent 1200 series HPLC system (Eclipse XDB-C18 column, 5 μm, 4.6 × 150 mm).
3.2 Material and Cultural Conditions
Fruiting bodies of Phellinus tuberculosus were collected at Jingdong, Yunnan Province, China in 2003 and identified by Prof. Zhu-Liang Yang of Kunming Institute of Botany, CAS. The voucher specimen (NO.CGBWSHF00118) was deposited at herbarium of Kunming Institute of Botany. Culture medium: glucose (5 %), pork peptone (0.15 %), yeast (0.5 %), KH2PO4 (0.05 %), MgSO4 (0.05 %), The initial pH was adjusted to 6.0, the fermentation was first carried out on an Erlenmeyer flask for 6 days till the mycelium biomass reached to the maximum. Later it was transferred to a fermentation tank (20 L) at 24 °C and 250 rpm for 20 days, ventilation was set to 1.0 vvm (vvm: air volume/culture volume/min).
3.3 Extraction and Isolation
The culture broth (20 L) was concentrated under vacuum, extracted three times with EtOAc. The organic layer was evaporated in vacuum to give a crude extract (3.1 g), which was separated by Sephadex LH-20 (MeOH) CC to afford fractions A–C. Fraction B (2.5 g) was separated by reversed-phased C18 column (MeOH-H2O, 30–100 %) to give sub-fractions B1 and B5. The sub-fraction B1 (110 mg) was further purified by silica gel (CHCl3-MeOH, 20/1) to yield 1 (5.8 mg), Fraction B2 was separated by Sephadex LH-20 (MeOH) to obtain B2-1 (50 mg), which was further isolated and purified by silica gel CC (CHCl3-MeOH, 20/1) to obtain 2 (31.6 mg). Fraction B3 was purified by Sephadex LH-20 (MeOH) and silica gel (CHCl3-MeOH, 30/1) to get 5 (2.2 mg) and 8 (8.6 mg). The sub-fraction B4 was purified by preparative HPLC (MeOH-H2O, 40 %, 10 mL/min) to obtain 3 (7.9 mg) and 4 (2.5 mg). Fraction B5 was separated by Sephadex LH-20 (MeOH) column chromatography, then further separated on silica gel (CHCl3-MeOH, 30/1) to give 6 (17.4 mg) and 7 (1.0 mg).
Phellinuin A (1): colorless oils, [α] 17.3D − 10.3 (c 0.1 MeOH); IR (KBr) vmax 3405, 2963, 2930, 2871, 1665, 1443 cm−1; 1H NMR data (see Table 1); 13C NMR data (see Table 2); ESIMS (neg.) m/z 539 [2M − H]−; HREIMS m/z 270.1827 [M]+ (calcd for C15H26O4, 270.1831).
Phellinuin B (2): amorphous powder, [α] 20.6D 0 (c 0.3 MeOH); IR (KBr) vmax 3345, 2930, 2878, 1666, 1442, 1057, 1983 cm−1; 1H NMR data (see Table 1); 13C NMR data (see Table 2); ESIMS (pos.) m/z 539 [2M − H]−; HREIMS m/z 270.1828 [M]+ (calcd for C15H26O4, 270.1831).
Phellinuin C (3): colorless oil, [α] 20.6D − 4.2 (c 0.2 MeOH); IR (KBr) vmax 3418, 2966, 2933, 2859, 1722, 1642, 1454, 1385, 1256 cm−1; 1H NMR data (see Table 1); 13C NMR data (see Table 2); ESIMS (pos.) m/z 335 [M + Na]+; HREIMS m/z 312.1935 [M]+ (calcd for C17H28O5, 312.1937).
Phellinuin D (4): colorless oil, [α] 20.8D − 5.4 (c 0.3 MeOH); IR (KBr) vmax 3421, 2961, 2932, 2858, 1736, 1631, 1443, 1384, 1254 cm−1; 1H NMR data (see Table 1); 13C NMR data (see Table 2); ESIMS (pos.) m/z 335 [M + Na]+; HREIMS m/z 312.1947 [M]+ (calcd for C17H28O5, 312.1937).
Phellinuin E (5): colorless oil, [α] 21.1D + 5.6 (c 0.002 MeOH); IR (KBr) vmax 3424, 2932, 2891, 1736, 1634, 1456 cm−1; 1H NMR data (see Table 1); 13C NMR data (see Table 2); ESIMS (pos.) m/z 335 [M + Na]+; HREIMS m/z 312.1930 [M]+ (calcd for C17H28O5, 312.1937).
Phellinuin F (6): colorless oil, [α] 20.4D − 2.8 (c 0.4 MeOH); IR (KBr) vmax 3441, 2965,2931, 2857, 1737, 1639, 1442 cm−1; 1H NMR data (see Table 1); 13C NMR data (see Table 2); ESIMS (pos.) m/z 319 [M + Na]+; HREIMS m/z 296.1986 [M]+ (calcd for C17H28O4, 296.1988).
Phellinuin G (7): colorless oil, [α] 20.4D − 6.5 (c 0.1 MeOH); IR (KBr) vmax 3442, 2933, 2869, 1721, 1665, 1460, 1254 cm−1; 1H NMR data (see Table 1); 13C NMR data (see Table 2); ESIMS (pos.) m/z 4319 [M + Na]+; HREIMS m/z 296.1982 [M]+ (calcd for C17H28O4, 296.1988).
References
J.B. Ellis, M.B. Ellis, Fungi Without Gills (Hymenomycetes and Gasteromycetes): An Identification Handbook (Chapman and Hall, London, 1990)
X.X. Zhang, Y.C. Dai, China Flora 29, 111 (2005)
P. Seephonkai, S. Samchai, A. Thongsom, S. Sunaart, B. Kiemsanmuang, K. Chakuton, Zhongguo Tianran Yaowu 9, 441–445 (2011)
J.H. Ding, T. Feng, B.K. Cui, K. Wei, Z.H. Li, J.K. Liu, Tetrahedron Lett. 21, 2651–2654 (2013)
X.Y. Yang, T. Feng, Z.H. Li, Y. Sheng, X. Yin, J.K. Liu, Org. Lett. 14, 5382–5384 (2012)
S. Wang, L. Zhang, L.Y. Liu, Z.J. Dong, Z.H. Li, J.K. Liu. Nat. Prod. Bioprospect. 2, 240–244 (2012)
S. Wang, Z.H. Li, Z.J. Dong, J.K. Liu, T. Feng, Fitoterapia 91, 194–198 (2013)
J.R. Sierra, J.T. Lopez, M.J. Cortes, Phytochemistry 25, 253–254 (1986)
J.Y. Zhao, J.H. Ding, Z.H. Li, Z.J. Dong, T. Feng, H.B. Zhang, J.K. Liu, J. Asian Nat. Prod. Res. 15, 305–309 (2013)
Acknowledgments
This work was financially supported by National Natural Sciences Foundation of China (U1132607) and Youth Innovation Promotion Association, CAS (2011312D11019).
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
This article is published under license to BioMed Central Ltd. Open Access This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article
Cite this article
He, JB., Feng, T., Zhang, S. et al. Seven New Drimane-Type Sesquiterpenoids from Cultures of Fungus Phellinus tuberculosus. Nat. Prod. Bioprospect. 4, 21–25 (2014). https://doi.org/10.1007/s13659-014-0002-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13659-014-0002-x