
Abstract
This paper provides an update and comprehensive review of the analytical methods used for quantifying isoflavones and their metabolites in cow’s milk. Isoflavones are secondary plant metabolites that are similar to 17 β-estradiol in chemical structure. They form one of the most common categories of phytoestrogens. Numerous health benefits have been attributed to isoflavones, but many of these compounds are also considered to be endocrine disruptors, with adverse effects on health. These contradictory trends offer an attractive prospect for future research, and therefore, sensitive and reliable analytical methods are required to clarify various issues about isoflavones. For this review, a structured methodology was used to select 26 relevant articles published between 2005 and 2015 from the Scopus and CAB Abstract databases. The review discusses individual steps of the analytical procedures described in these articles, including sample preparation, instrumental analysis and validation. The most commonly used analytical procedure is sample preparation involving liquid-liquid extraction and an enzymatic hydrolysis step followed by liquid chromatography with mass spectrometry analysis. Currently, however, there is no standardized procedure for the sample preparation and analysis of isoflavones in milk.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
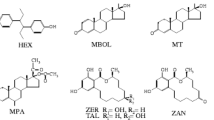
Isoflavones are classified as phytoestrogens, which are widely distributed in the plant kingdom. These plant secondary metabolites are structurally similar to 17 β-estradiol and bind selectively, but weakly, to mammalian estrogen receptors (ER), with a preference for ERβ (Vitale et al. 2013; Baber 2013; Mostrom and Evans 2012). These diphenolic compounds appear to have antioxidant activity and could offer alternative therapies for a range of hormone-dependent conditions, including cancer, menopausal symptoms, cardiovascular disease, and osteoporosis (Ko 2014; Vitale et al. 2013; Baber 2013; Mostrom and Evans 2012; Mortensen et al. 2009). They also can be considered as endocrine disruptors, however, with the potential to have adverse health effects (Wielogorska et al. 2015; Sirotkin and Harrath 2014; Maggioni et al. 2013; Wocławek-Potocka et al. 2013; Patisaul and Jefferson 2010; Afssa 2005). The highest concentrations are found in plants of the Fabaceae family, such as soybean (Glycine max L.) and clover (Trifolium pratense L.) (Ko 2014; Mostrom and Evans 2012). In plants, isoflavones are usually found in conjugated forms as glucosides, acetylglucosides, and malonylglucosides (Kalač 2013; Mostrom and Evans 2012). When ingested by cows, isoflavones appear to be metabolized mainly in the rumen, and the main route of excretion is through feces and urine, with only a small proportion being excreted in milk (Njåstad et al. 2014). A diagram of isoflavone metabolization in cows is shown in Fig. 1. Biochanin A (BA) is demethylated mainly into genistein (GE) and via a ring cleavage into para-ethyl phenol (a compound that apparently has no estrogenic activity) and organic acids (Njåstad et al. 2014; Kalač 2013; Mostrom and Evans 2012). Formononetin (FO) is demethylated into daidzein (DA) and then reduced via hydrogenation and ring scission to equol (EQ) (a microbial metabolite of isoflavone with high estrogenic activity). FO can also be metabolized into other metabolites, such as O-desmethylangolensin (O-DMA) (compounds with low estrogenic activity) (Njåstad et al. 2014; Kalač 2013; Mostrom and Evans 2012; Setchell and Clerici 2010). The aglycones seem to be the most biologically active forms and are quickly absorbed by the rumen and gut mucosa (Vitale et al. 2013; Kalač 2013; Mostrom and Evans 2012; Lundh 1990). Most of them are conjugated by glucuronic acid (a limited amount is conjugated with sulfate) during this absorption process. A small proportion of aglycones reaches the blood circulatory system, but the aglycones are rapidly conjugated in the liver and other tissues (Mostrom and Evans 2012; King et al. 1998; Lundh 1990). These isoflavone metabolites then circulate throughout the body and are excreted in feces and urine or transferred to the milk.

In recent decades, due to the growing interest in isoflavones, a diverse range of analytical methods has been developed for isoflavone determination and quantification in plant material, soil, water, food, and food supplements, as well as in biological matrices, generating a huge amount of scattered information (Fig. 2). The overall analytical method used for isoflavones is highly dependent upon the matrix characteristics, the availability of the techniques, the desired selectivity, and the need to obtain information about the chemical structure of these compounds or simply to unambiguously identify previously targeted compounds. Initially, samples are usually freeze-dried or simply frozen. Depending on the matrix analyzed, isoflavones are extracted by classical maceration or liquid-liquid extraction (LLE), but a promising approach that is increasingly being used is to combine extraction/clean-up techniques such as ultrasound-assisted extraction (UAE), solid-phase extraction (SPE), microwave-assisted extraction (MAE) or matrix solid-phase dispersion (MSPD) (Rostagno et al. 2009). Given that isoflavones are often in conjugated forms, a hydrolysis step is sometimes used when the aim is to find aglycones. The number of analytical separation and detection techniques proposed is also numerous. Depending on the matrix and the information required, many of these techniques have been developed using variants of immunoassays (enzyme-linked immunosorbent assay, ELISA; radioimmunoassay, RIA; and time-resolved fluorescence immunoassay, TR-FIA), gas chromatography (GC), liquid chromatography (LC), and capillary electrophoresis (CE) coupled with various detection modes (mass spectrometry, MS; diode array detection, DAD; fluorescence detection, FLD; and electrochemical detection, ED) (Ko 2014; Rostagno et al. 2009; Valls et al. 2009; Vacek et al. 2008; Stalikas 2007; De Rijke et al. 2006; Grynkiewicz et al. 2005; Wu et al. 2004). Immunoassays are non-chromatographic methods with high sensitivity and specificity for single-component detection, high throughput screening and relatively low cost per sample. For this approach, however, preparing the antibodies for corresponding analytes is a challenging task and it suffers from the likelihood of cross-reactivity between the similar compounds, leading to a possible overestimation of the targeted analyte (Ko 2014; Wu et al. 2004). CE is also a promising technique with a high separation resolution and a low amount of sample required, but it is restricted to laboratories that have the necessary technical capacity (Wu et al. 2004; Wang et al. 2002). The most common analytical method used for quantifying isoflavones remains LC coupled with MS or ultra-violet detection (LC-MS, LC-UV). Thanks to its high sensitivity, MS detection is used preferentially when low amounts of isoflavones are thought to be in the matrix. UV detection is used when isoflavones are thought to be present in larger amounts (Ko 2014).

Main sample preparation and instrumental analysis methods for the determination of isoflavones and their related compounds. Abbreviations: UAE ultrasound-assisted extraction, SFE supercritical fluid extraction, LLE liquid-liquid extraction, SLE solid-liquid extraction, SPME solid-phase micro-extraction, SPE solid-phase extraction (incorporating the Quechers approach), MAE microwave-assisted extraction, MSPD matrix solid-phase dispersion, CCC counter-current chromatography, LC liquid chromatography (HPLC high-performance liquid chromatography, UPLC ultra-performance liquid chromatography, and UHPLC ultra high-performance liquid chromatography), GC gas chromatography, MS mass spectrometry, UV ultra-violet, DAD diode array detector (also called photodiode array detector, PDA), FLD fluorescence detector, NMR nuclear magnetic resonance, ED electrochemical detection, ECD electron capture detection, FID flame ionization detection, TLC thin layer chromatography, CE capillary electrophoresis, ELISA enzyme-linked immunosorbent assay, RIA radioimmunoassay, and TR-FIA time-resolved fluorescence immunoassay
Milk is one of the most widely consumed foods in the world and several studies have been conducted in recent years to estimate its isoflavone content and to assess whether or not milk can be considered as a useful or dangerous source of isoflavones for humans. An initial answer was presented in two articles, especially with regard to the impact of cattle feed on the EQ content of milk (Kalač 2011, 2013). As far as we know, there has not yet been a specific review of the analytical methods used for quantifying isoflavones in cow’s milk. This paper, therefore, reviews the current situation with regard to sample preparation and analysis for the quantification of isoflavones in cow’s milk, including the most recent advances.
The literature overview was conducted online using the Scopus and CAB Abstracts bibliographic databases. Four keywords were identified: “phytoestrogens”, “isoflavones”, “milk”, and “quantitative analysis”. These keywords and their descriptors were used to create a Boolean equation in the two databases. The most recent studies (2005–2015) were selected. Key data of the selected articles are given in Tables 1 and 2. Where the authors of a selected article refer to a method published before 2005, the analytical protocol from the reference is summarized and presented in these tables. All the analytical procedures described in these articles were analyzed chronologically: initially, sample preparation procedures were compared and discussed, and then the instrumental analysis and validation parameters were examined.
2 Sample preparation
Broadly, an analytical method can be split into two distinct parts: sample preparation and sample analysis. Sample preparation is undertaken before instrumental analysis and should ensure that: (1) the target compounds can be extracted from the matrix; (2) interference that could disrupt the detection of the target analyte or damage the chromatographic column can be eliminated or reduced; (3) to allow analysis of samples with low concentrations of the target compound; and (4) the analyte of interest can be transformed into a more suitable form that could be easily separated, detected and quantified. In Table 1, all the information about sample preparation found in the 26 selected articles is detailed and split into four chronological steps. The table also presents the origin of the milk and the isoflavones quantified. Studies using the same sample preparation procedure were grouped together. Articles where the sample preparation procedure was based on a previous study, but with some modifications, were not grouped (Daems et al. 2015; Flachowsky et al. 2011; Antignac et al. 2009). Thirteen sample preparation procedures were identified and are discussed in the next section.
2.1 Sample conditioning
As shown in Table 1, most milk samples in the experiments reviewed were simply frozen between the sampling and analysis days. In other articles, sample condition was not mentioned. Often, information about the handling of milk samples after collection and before actual analysis was limited. An exception was Wielogórska et al. (2015), who reported that isoflavones (DA and GE) were stable in the matrix for only 2 weeks, indicating that all samples were analyzed immediately after collection or stored at −20 °C for no longer than 2 weeks before being thawed for analytical purposes. Poor storage can lead to errors that cannot be corrected later and that will affect the outcome of the final analysis. How should samples be stored, and for how long, without affecting their original isoflavone profile? For matrices such as soybean, some isoflavones (mainly the conjugated forms) have a fairly unstable character (Rostagno et al. 2009). In ruminants, glucuronide-conjugated compounds are the major form found in biological fluids (Mostrom and Evans 2012; Nielsen et al. 2012). The degradation problem could, therefore, also arise during milk storage and the information about the conjugated form content could be lost. More attention should be given to the storage and handling of milk samples between sampling and analysis in future studies.
If freeze-drying is an easy method for milk-sample storage, it would be interesting to check isoflavone stability under a range of temperature and storage-time conditions. The freezing method could be also compared with other techniques, such as freeze-drying. Isoflavones can be found in powdered milks (Maggioni et al. 2013; Antignac et al. 2009). Therefore, freeze-drying, which is often used for plant matrices because it does not seem to affect these compounds (Hoerger et al. 2011), could be an interesting alternative to the traditional freezing process. These findings need to be taken into account in future studies.
2.2 Hydrolysis
Among the 13 sample preparation procedures listed in Table 1, only four did not include a hydrolysis step. Wielogórska et al. (2015) proposed a method without a hydrolysis step for the quantification of 19 endocrine disruptors in milk, including DA and GE. Rostagi et al. (2013) did not use a hydrolysis step because they sought to quantify puerarin (PU, the 8-C-glucoside of daidzein) from pasteurized toned milk enriched with extracts from Pueraria tuberosa Linn (commonly called “kudzu”). According to these authors, this glycoside form of isoflavone that is abundant in kudzu might be beneficial in the treatment and prevention of some diseases and cancers. The two other procedures were used by Maggioni et al. (2013) and Rastogi et al. (2013) to quantify aglycones in infant powdered milk formulas and glycosides in soy-fortified milk. Integrating a hydrolysis step into the analytical protocol depends on the objectives sought. Hydrolysis is often used when the exact nature and composition of isoflavone glycosides in the matrix analyzed are unknown or when target aglycone compounds are sought. It allows the time required for instrumental analysis to be considerably reduced and facilitates the separation of target compounds. It also makes it unnecessary to use standard references that are difficult to obtain, not available, or often expensive. Introducing an additional step, however, can increase analytical variability.
As shown in Table 1, most of the studies reviewed used a hydrolysis step. Unlike other matrices where three procedures are reported (enzymatic, acidic and, least common, basic hydrolysis) (Rostagno et al. 2009; Schwartz et al. 2009), for milk only enzymatic hydrolysis is carried out to hydrolyze isoflavones. The authors focused on aglycones because these forms appear to be the most biologically active and can be absorbed by the intestinal tract of humans and animals (Ko 2014; Mostrom and Evans 2012). In addition, some isoflavones, such as EQ, seem to be sensitive to acidic conditions (Setchel and Clerici 2010). Enzymatic hydrolysis involves incubating the sample with enzymes at a fixed temperature (within a range of 37 to 52 °C, depending on the study) and over different incubation times (between 1 and ≥16 h). A solution buffer (pH of about 5) is sometimes used to optimize hydrolysis conditions. Unlike plant matrices (Rostagno et al. 2009), only β-glucuronidase/sulfatase from Helix pomatia was involved in the hydrolysis of isoflavones in milk. This enzymatic juice contains glucuronidase and sulfatase activities. The former catalyzes the breakdown of complex carbohydrates (glucuronides) and the latter catalyzes the hydrolysis of sulfate esters, the two main forms of isoflavones found in biological fluids (Ko 2014; Nielsen et al. 2012; Rostagno et al. 2009). Only Kuhnle et al. (2008) reported using a mixed solution of three enzymes: β-glucuronidase from H. pomatia; cellulase from Trichoderma reesi; and β-glucosidase from almonds.
The hydrolysis temperature varied between 37 and 40 °C, but it did reach 52 °C in some studies (Flachowsky et al. 2011; Antignac et al. 2009). The temperature generally used by suppliers to evaluate the glucuronidase and sulfatase activities from these enzymes was 37 °C. Where a buffer solution was used, the pH of the solution varied between 4.6 and 5.2. The optimal pH values given by suppliers were 5.0 and 6.2 for glucuronidase and sulfatase activities, respectively. Hydrolysis time varied, ranging from 1 h (Andersen et al. 2009b) to overnight (Antignac et al. 2009; Kuhnle et al. 2008). During an evaluation of the robustness of their hydrolysis protocol based on a one-variable-at-a-time technique, Daems et al. (2015) demonstrated that an incubation time of 1 h was sufficient for hydrolyzing conjugated forms of EQ. Another important point, but one on which there is little information in the studies, is the minimum enzyme activity needed to hydrolyze all the conjugated forms of isoflavones present in milk. When enzyme activity values were provided by authors or when estimating them was possible, it was found that they varied greatly across the studies. For example, for their analysis of isoflavones (containing EQ) in commercial milk, Hoikkala et al. (2007) used 100 units of glucuronidase activity per ml of sample, whereas Daems et al. (2015) used 2250 units. In the product information data sheets for other biological fluids, Sigma-Aldrich recommends using between 1000 and 20,000 units per mL of β-glucuronidase type H2 from H. pomatia (EC 3.2.1.31), but no specific information is given on the hydrolysis of milk samples. One disadvantage of using H. pomatia juice is that it usually contains appreciable levels of some isoflavones (according to Taylor et al. (2005): 13.38 ng GE.mL−1 enzyme, 0.25 ng DA.mL−1, and 0.58 ng EQ.mL−1) which can affect the quantification. Daems et al. (2015) and Grace and Teale (2006) did not detect any trace of EQ, and Grace and Teale (2006) found that GE was the major isoflavone in H. pomatia juice. These observations show that isoflavone concentrations appear to fluctuate from one enzyme batch to another. The hydrolysis with this enzymatic solution also showed interfering peaks with isoflavone signals in the chromatogram of human milk extracts (Franke and Cluster 1996). There are two ways of addressing these problems. One is a pre-cleanup of the enzymatic preparation by SPE (Kuhnle et al. 2007; Grace and Teale 2006). The second way, probably the easier of the two, involves working with a blank sample where the milk has been replaced by a buffer solution in order to estimate the isoflavone content in the enzymatic mixture (Taylor et al. 2005).
2.3 Isolation of target compounds
Solvent extraction is the main step used for the recovery and isolation of bioactive compounds from a matrix before instrumental analysis and it can be performed before or after the hydrolysis step. Additional steps are sometimes necessary to remove lipids or proteins, which can cause interferences during instrumental analysis. Fat is one of the major causes of the matrix effect in LC-MS (Jiang et al. 2012; Trontelj 2012) and most of the protocols in Table 1 therefore indicate defatting and deproteinizing steps. Lipids can be removed by LLE with an apolar solvent (e.g., n-heptane or n-hexane) and centrifugation. Proteins are removed by precipitation in acidic conditions and centrifugation. Deproteinization is often used at the same time that the defatting or extraction of target compounds is done because organic solvents can also precipitate the proteins (Trontelj 2012).
Liquid-liquid extraction is the most widely used technique for extracting isoflavones from milk. The extractions are repeated two to three times and the extracts are pooled. Among the analytical protocols listed in Table 1, only three used SLE to recover isoflavones in milk samples (Maggioni et al. 2013; Rastogi et al. 2013; Kuhnle et al. 2008). Only Rastogi et al. (2013) deliberately used the freeze-drying process, whereas two other studies used this extraction technique because they were analyzing milk samples in powder form. Optimizing extraction is a challenging task given the wide polarity range of isoflavones. As an example, compared with DA, GE has an additional hydroxyl group and therefore DA and GE are expected to have different solubilities (Harjo et al. 2007). In general, conjugated forms are more polar than aglycones and are extracted with polar solvents such as alcohols (MeOH, EtOH) or acetonitrile, often mixed with varying proportions of water. For isoflavone quantification in milk, a hydrolysis step is almost always used in order to remove the polar groups from conjugated forms, leading to the formation of isoflavone aglycones, which are less polar and almost insoluble in water (Vitale et al. 2013; Ishii et al. 2013; Harjo et al. 2007). This is why less polar solvents (diethyl ether or ethyl acetate), with no water added, are used for extraction in milk. The choice of solvent composition should be determined empirically according to the target molecules that the researchers wish to extract.
Looking more closely at the order in which the hydrolysis and isolation steps are done, some authors conducted the hydrolysis step first and then the extraction step, whereas others used the reverse order. This raises the question: is one protocol better than the other? If the less polar solvents are more efficient for extracting aglycone forms, when the hydrolysis is performed after the extraction step the conjugated isoflavones might not be completely extracted, leading to an underestimation of the overall isoflavone content. Some authors (Daems et al. 2015; Křížová et al. 2011a) reported that isoflavones are distributed more widely in the aqueous fraction than in the lipid fraction. In contrast, Tsen et al. (2014) reported that isoflavones are distributed mostly in the lipid fraction and that the defatting process, therefore, reduces the overall amount of isoflavones in a sample. These are all matters that need to be addressed in future studies.
2.4 Purification and concentration extracts
After the extraction of isoflavones in milk, the extract is subjected to a series of post-treatment steps before instrumental analysis. In almost all protocols, centrifugation is performed. As well as eliminating the cream (fat) and the precipitated proteins, it also isolates other small solid impurities. A filtration step is often performed just before the sample extract injection (Wielogórska et al. 2015; Daems et al. 2015; Tsen et al. 2014; Rastogi et al. 2013; Flachowsky et al. 2011; Andersen et al. 2009b; Nielsen et al. 2009; Hoikkala et al. 2007; Uzzan et al. 2007) to prevent damage to the analytical columns.
In milk, isoflavones are usually present in low concentrations (a few ppb or, sometimes, higher for microbial metabolites) (Kalač 2013) and, therefore, it is necessary to concentrate the target analyte before instrumental analysis. SPE and evaporation were the most commonly used enrichment techniques. SPE using C18 bonded silica (Flachowsky et al. 2011; Antignac et al. 2009; Kuhnle et al. 2008) or a universal polymeric reversed-phase sorbent called OASIS-HLB from Waters (Wielogórska et al. 2015; Maggioni et al. 2013; Mustonen et al. 2009; Hoikkala et al. 2007) were reported to provide a clean concentrated isoflavone extract. Probably due to its typical chemistry, OASIS-HLB has the advantage of greater retention and good recoveries even if the sorbent runs dry (Rostagno et al. 2009). The final extracts are then evaporated until dry and re-suspended with the polar solvent used in the instrumental analysis.
3 Instrumental analysis
After these preparation steps, the sample extract can be injected into instruments that will allow the target compounds to be separated, detected and quantified. The data in the 26 selected articles are presented in Table 2. When different authors used the same analytical methods, the data presented are derived from all the information in the articles. Thirteen analytical methods were identified and are discussed below.
3.1 Analyte separation
Unlike in other matrices (plants, foods or other biological fluids), there are few separation methods reported in the literature for the quantification of isoflavones in milk. Over the past 10 years, the only analytical separation technique used has been reverse-phase LC with a binary solvent system that contains acidified water and an acidified polar organic solvent, such as methanol or acetonitrile (Table 2).
With the LC technique, a guard column is often placed before the analytical column in order to retain all impurities remaining in the sample extract (Rastogi et al. 2013; Křížová et al. 2011a, b; Antignac et al. 2009; Třináctý et al. 2009; Hoikkala et al. 2007; Uzzan et al. 2007). This also enables the compounds from the extract to start being separated. These small columns are regularly changed and allow the lifetime of the main analytical column to be increased. They often have a stationary phase similar to that of the main column. Columns chosen for isoflavone separation in milk are exclusively packed with reversed-phases, mainly C18 with an internal diameter of 2.0–4.6 mm, a length of 50–250 mm, and a particle size of 3–5 μm. Two other stationary phase chemistries are sometimes also mentioned in the articles: C8 (Rastogi et al. 2013; Maggioni et al. 2013) and diphenyl (Kuhnle et al. 2008). Two recent studies report on the use of ultra high-performance LC (UHPLC) columns with a particle size of 1.8 μm, an internal diameter of 2.1 mm, and a length of 100 mm (Wielogórska et al. 2015; Daems et al. 2015). This technology allows separations that have positive effects on both resolution and accuracy. It also increases sample-throughput due to reduced analysis times (Gumustas et al. 2013; Valls et al. 2009). The reduction of analysis time is illustrated in Table 2. For classical LC analysis, the average analysis time is about 35 min, ranging from a minimum of 14 min (Steinshamn et al. 2008) to a maximum of 60 min (Flachowsky et al. 2011). In the two studies using UHPLC it was between 5 and 10.5 min. These times could be further reduced by manipulating the column temperature, flow rate, mobile phase composition, and solvent gradients.
The mobile phase flow rate depends upon its composition, column type and temperature. In the selected articles, the flow rates varied between 0.2 and 1 mL.min−1, the temperature from 25 to 45 °C, and the injection volume from 10 to 50 μL. The mobile phase is usually a mixture of water and an organic solvent (methanol or acetonitrile) with a small proportion of formic or acetic acids (up to a max. of 1%). The differing hydrophobicities of isoflavones indicate that the gradient elution mode should be used (Rostagno et al. 2009). Since isoflavones exhibit a weak acidic nature, the use of acids in the mobile phase can enhance chromatographic resolution and peak shape (Rostagno et al. 2009; Wu et al. 2004). Acidification of the mobile phase also significantly increases the isoflavone limit of detection (LOD) by promoting positive ion formation in the MS source (Wu et al. 2004).
3.2 Analyte detection
As shown in Table 2, the most commonly used detection methods combined with LC are MS and UV detectors. They can be used alone, in combination (Andersen et al. 2009b; Nielsen et al. 2009) or with other types of detectors (Mustonen et al. 2009; Hoikkala et al. 2007). Hoikkala et al. (2007) used UV-DAD at 262 nm for GE, O-DMA and BA and they used FLD for DA and FO (λex at 254 nm and λem at 465 nm), as well as for EQ (λex at 254 nm and λem at 310 nm). Although the LC-FLD method is often more sensitive than the UV absorption methods, the number of isoflavones that are naturally fluorescent is fairly limited (Ko 2014). In addition, the acidification of the mobile phase has a quenching effect that can reduce FLD sensitivity (Vacek et al. 2008).
Overall, LC-UV and LC-MS are the two main detection modes used for quantifying isoflavones. In milk, however, MS is preferred to UV detection. UV detection uses the isoflavone characteristic of having at least one aromatic ring that strongly absorbs UV light, with a maximum wavelength ranging from 230 to 280 nm (Ko 2014; Rostagno et al. 2009; Stalikas 2007; Wu et al. 2004). A range of 300 to 550 nm was sometimes also used as a second maximum wavelength and attributed to the substitution pattern and conjugation of the C-ring (Stalikas 2007). This is not the case with all isoflavones. For example, the microbial metabolites EQ and O-DMA exhibit poor UV absorption characteristics, making UV detection unsuitable for their measurement (Ko 2014; Setchel and Clerici 2010). For isoflavones that exhibit this UV absorption characteristic, the UV–visible spectra of many of them are very similar (Vacek et al. 2008). Another disadvantage of this method is its low sensitivity, which restricts its use to quantification in matrices likely to have a large amount of isoflavones, such as plants, plant-derived samples and supplemented foods (Ko 2014; Wu et al. 2004). Among the studies presented in Table 2, only two used UV as the sole mode of detection (Rastogi et al. 2013; Uzzan et al. 2007) and both focused on analyzing fortified milk.
In order to overcome the problems of non-specificity and lack of sensitivity, researchers have focused on MS detection. Usually, this method is highly sensitive, with low limits of quantification (LOQ) under or around the ppb level (see Table 2). Electrospray ionization (ESI) and atmospheric pressure chemical ionization (APCI) are the two sources most commonly used for quantifying isoflavones in milk. In the selected articles, these sources were used in positive and/or negative mode, except for APCI, which was used exclusively in positive mode with select reaction monitoring (SRM). In the ESI, multiple reaction monitoring (MRM) was used. ESI and APCI interfaces appeared to be suitable for the analysis of isoflavones, but ESI is better suited for the conjugated forms. ESI is the softest and most suitable method for ionization without the in-source fragmentation of the relatively weak glucuronide ether or ester bonds. Thermally labile conjugates form molecular ions in ESI, but with APCI these conjugates decompose because they are not stable enough (Trontelj 2012; Hoikkala et al. 2003). In their comparison of ESI and APCI sources for the quantitative determination of phytoestrogens in urine, Rybak et al. (2008) showed that, for EQ, ESI was better in terms of measurement precision, sensitivity and LOD.
The benefits of LC-MS include high precision and sensitivity, suitability for determining isoflavones and their metabolites or conjugated forms, less samples manipulation than for GC, and applicability to non-volatile analytes with direct injection of the liquid samples. The higher price of this technology remains its major disadvantage compared with other separation and detection technologies. LC-MS, however, is now a fast-moving field, with many new detection and chromatographic techniques being developed. With their shorter analysis time, lower solvent consumption, better resolution properties, and better sensitivity than classical LC, UPLC and UHPLC with MS appear to be the fastest and easiest methods to implement and to produce reliable results.
3.3 Validation parameters
As far as we could ascertain, method validations were seldom discussed in the previous review articles on isoflavones and related compounds. Where they were mentioned, the validation parameters are shown in Table 2 (when several works used the same analytical procedure, the various data in each study were collected and summarized in this table).
The validation parameters taken into account in this table come from the EMA (2015) guidelines, which describe the suitable criteria for the validation of analytical methods used in veterinary drug residue depletion studies. The guidelines could apply to the quantification of isoflavones in milk because these estrogenic compounds can be active in very low amounts. As shown in Table 2, most of the studies used external calibration as the quantification mode for determining the isoflavone content of milk, whereas Steinshamn et al. (2008) and related studies used a standard addition protocol. Among these studies, Andersen et al. (2009a) reported that the LOD for FO and EQ were 0.047 and 0.170 ng.mL−1, respectively. In the other selected articles, the validation parameters most often omitted were sample or extract stability and the robustness of the analytical method proposed. The most comprehensive validation procedures were reported by Wielogórska et al. (2015) (for DA, GE and other endocrine disruptors) and Daems et al. (2015) (for EQ).
4 Conclusion
This paper summarizes the current analytical methods used for quantifying isoflavones and their metabolites in cow’s milk. Careful examination of methodological aspects showed that most of the studies used LLE to extract isoflavones from milk. Given that aglycones are the most biologically active forms of isoflavones, most of the analytical protocols used a hydrolysis step with β-glucuronidase/sulfatase from H. pomatia.
Although non-chromatographic immunoassays and CE with MS or ED detectors are seen as promising techniques for quantifying isoflavones, reverse-phase LC-MS is the only analytical method used in the past 10 years. LC-MS is now a fast-moving field, with many new detection and chromatographic techniques being developed over the past decade that achieve high sensitivity and reduce analysis time. The low isoflavone content (about ppb value) naturally present in milk, except in the case of some isoflavone metabolites, justifies the use of MS as a detection method. The new chromatographic technologies, UPLC and UHPLC, reduce the time needed and achieve good resolution.
Although some analytical protocols have been used frequently by researchers, there is still no standardized procedure available for the sample preparation and determination of these phytoestrogenic compounds in milk. A rigorous validation referring to a guideline would be enough to ensure the reliability of the chromatographic results. For sample preparation, however, a standardized procedure is interesting to compare the results from the different studies.
References
Adler SA, Purup S, Hansen-Møller J, Thuen E, Gustavson AM, Steinshamn H (2014) Phyto-oestrogens and their metabolites in milk produced on two pastures with different botanical compositions. Livest Sci 163:62–68
Adler AS, Purup S, Hansen-Møller J, Thuen E, Steinshamn H (2015) Phytoestrogens and their metabolites in bulk-tank milk: effects of farm management and season. PLoS One 10(5):e0127187
Afssa (2005) Sécurité et bénéfices des phyto-estrogènes apportés par l’alimentation—Recommandations, France. https://www.anses.fr/sites/default/files/documents/NUT-Ra-Phytoestrogenes.pdf (accessed March 2015)
Andersen C, Weisbjerg MR, Hansen-Møller J, Sejrsen K (2009a) Effect of forage on the content of phyto-oestrogens in bovine milk. Animal 3(4):617–622
Andersen C, Nielsen TS, Purup S, Kristensen T, Eriksen J, Søegaard K, Sørensen J, Fretté XC (2009b) Phyto-oestrogens in herbage and milk from cows grazing white clover, red clover, lucerne or chicory-rich pastures. Animal 3(8):1189–1195
Antignac J-P, Gaudin-Hirret I, Naegeli H, Cariou R, Elliott C, Le Bizec B (2009) Multi-functional sample preparation procedure for measuring phytoestrogens in milk, cereals, and baby-food by liquid-chromatography tandem mass spectrometry with subsequent determination of their estrogenic activity using transcriptomic assay. Anal Chim Acta 637:55–63
Baber RJ (2013) Phytoestrogens in health: the role of isoflavones. In: VR Preedy (ed.) Isoflavones: Chemistry, Analysis, Function and Effects (No. 5), UK, Chapter 1, 1-13
Daems F, Jasselette C, Romnee J-M, Planchon V, Lognay G, Froidmont É (2015) Validating the use of an ultra-performance liquid chromatography with tandem mass spectrometry method to quantify equol in cow’s milk. Dairy Sci Technol 95(3):303–319
De Rijke E, Out P, Niessen WMA, Ariese F, Gooijer C, Brinkman UAT (2006) Analytical separation and detection methods for flavonoids. J Chromatogr A 1112:31–63
EMA (2015) VICH GL49: studies to evaluate the metabolism and residue kinetics of veterinary drugs in food-producing animals—validation of analytical methods used in residue depletion studies, UK. http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2011/04/WC500105053.pdf (accessed February 2015)
Flachowsky G, Hünerberg M, Meyer U, Kammerer DR, Carle R, Goerke M, Eklund M (2011) Isoflavone concentration of soybean meal from various origins and transfer of isoflavones into milk of dairy cows. J Verbr Lebensm 6:449–456
Franke AA, Custer LJ (1996) Daidzein and genistein concentrations in human milk after soy consumption. Clin Chem 42(6):955–964
Grace PB, Teale P (2006) Purification of the crude solution from Helix pomatia for use as β-glucuronidase and aryl sulfatase in phytoestrogen assays. J Chromatogr B 832:158–161
Grynkiewicz G, Hsycińska H, Ramza J, Zagrodzka J (2005) Chromatographic quantification of isoflavones (why and how). Acta Chromatogr 15:31–65
Gumustas M, Kurbanoglu S, Uslu B, Ozkan SA (2013) UPLC versus HPLC on drug analysis: advantageous, applications and their validation parameters. Chromatographia 76:1365–1427
Harjo B, Wibowo C, Zhang EJN, Luo KQ, Ng KM (2007) Development of process alternatives for separation and purification of isoflavones. Ind Eng Chem Res 46:181–189
Hoerger CC, Praplan AP, Becker L, Wettstein FE, Hungerbühler K, Bucheli TD (2011) Quantification of five isoflavones and coumestrol in various solid agroenvironmental matrices using 13C3-labeled internal standards. J Agric Food Chem 59:847–856
Hoikkala AA, Schiavoni E, Wähälä K (2003) Analysis of phyto-oestrogens in biological matrices. Br J Nutr 89:S5–S18
Hoikkala A, Mustonen E, Saastamoinen I, Jokela T, Taponen J, Saloniemi H, Wähälä K (2007) High levels of equol in organic skimmed Finnish cow milk. Mol Nutr Food Res 51:782–786
Höjer A, Adler S, Purup S, Hansen-Møller J, Martinsson K, Steinshamn H, Gustavsson AM (2012) Effects of feeding dairy cows different legume-grass silages on milk phytoestrogen concentration. J Dairy Sci 95(5):4526–4540
Ishii K, Hosoda K, Furuta T (2013) Isoflavones: high-performance liquid chromatographic analysis of glucuronic acid- and sulfuric acid-conjugated metabolites of daidzein and genistein in human plasma and urine. In: VR Preedy (ed) Isoflavones: chemistry, analysis, function and effects (No. 5), UK, Chapter 13, 196-217
Jiang H, Cao H, Zhang Y, Fast DM (2012) Systematic evaluation of supported liquid extraction in reducing matrix effect and improving extraction efficiency in LC–MS/MS based bioanalysis for 10 model pharmaceutical compounds. J Chromatogr B 891–892:71–80
Kalač P (2011) The effects of silage feeding on some sensory and health attributes of cow’s milk: a review. Food Chem 125:307–317
Kalač P (2013) Fresh and ensiled forages as a source of estrogenic equol in bovine milk: a review. Czech J Anim Sci 58(7):296–303
King RA, Mano MM, Head RJ (1998) Assessment of isoflavonoid concentrations in Australian bovine milk samples. J Dairy Res 65:479–489
Ko K-P (2014) Isoflavones: chemistry, analysis, functions and effects on health and cancer. Asian Pac J Cancer Prev 15(17):7001–7010
Krajčová A, Schulzová V, Lojza J, Křížova L, Hajšlová J (2010) Phytoestrogens in bovine plasma and milk–LC-MS/MS analysis. Czech J Food Sci 28(4):264–274
Křížová L, Třináctý J, Hajšlová J, Havlíková Š (2011a) The effect of technological processing on the content of isoflavones in bovine milk and dairy products. In: Tzi-Bun Ng (ed.) Soybean – Applications and Technology, Croatia, Chapter 6, 95-110
Křížová L, Veselý A, Třináctý J, Schulzová V, Hurajová A, Hajšlová J, Kvasničková E, Havlíková Š (2011b) Changes in isoflavones concentrations in cheese during processing and ripening. Acta Univ Agric et Silvic Mendel Brun 59:153–162
Kuhnle GGC, Dell’Aquila C, Low Y-L, Kussmaul M, Bingham SA (2007) Extraction and quantification of phytoestrogens in foods using automated solid-phase extraction and LC/MS/MS. Anal Chem 79:9234–9239
Kuhnle GG, Dell’Aquila C, Aspinall SM, Runswick SA, Mulligan AA, Bingham SA (2008) Phytoestrogen content of animal origin: dairy products, eggs, meat, fish, and seafood. J Agric Food Chem 56(21):10099–10104
Lundh TJ-O (1990) Conjugation of the plant estrogens formononetin and daidzein and their metabolite equol by gastrointestinal epithelium from cattle and sheep. J Agric Food Chem 38:1012–1016
Maggioni S, Bagnati R, Pandelova M, Schramm K-W, Benfenati E (2013) Genistein and dicarboximide fungicides in infant formulae from the EU market. Food Chem 136:116–119
Mortensen A, Kulling SE, Schwartz H, Rowland I, Ruefer CE, Rimbach G, Cassidy A, Magee P, Millar J, Hall WL, Birkved FK, Sorensen IK, Sontag G (2009) Analytical and compositional aspects of isoflavones in food and their biological effects. Mol Nutr Food Res 53:S266–S309
Mostrom M, Evans TJ (2012) Phytoestrogens. In: Gupta RC (ed) Veterinary toxicology—basic and clinical principles (2nd edn), Netherlands, Chapter 76, 1012-1028
Mustonen EA, Tuori M, Saastamoinen I, Taponen J, Wähälä K, Saloniemi H, Vanhatalo A (2009) Equol in milk of dairy cows is derived from forage legumes such as red clover. Brit J Nutr 102:1552–1556
Nielsen TS, Nørgaard JV, Purup S, Fretté XC, Bonefeld-Jørgensen EC (2009) Estrogenic activity of bovine milk high or low in equol using immature mouse uterotrophic responses and an estrogen receptor transactivation assay. Cancer Epidemiol 33:61–68
Nielsen TS, Purup S, Wärri A, Godschalk RW, Hilakivi-Clarke L (2011) Effects of maternal exposure to cow’s milk high or low in isoflavones on carcinogen-induced mammary tumorigenesis among rat offspring. Cancer Prev Res 4(5):694–701
Nielsen TS, Höjer A, Gustavsson A-M, Hansen-Møller J, Purup S (2012) Proliferative effect of whey from cow’s milk varying in phyto-oestrogens in human breast and prostate cancer cells. J Dairy Res 79:143–149
Njåstad KM, Adler SA, Hanse-Møller J, Thuen E, Gustavsson A-M, Steinshamn H (2014) Gastrointestinal metabolism of phytoestrogens in lactating dairy cows fed silages with different botanical composition. J Dairy Sci 97:7735–7750
Patisaul HB, Jefferson W (2010) The pros and cons of phytoestrogens. Front Neuroendocrin 31:400–419
Rastogi S, Katara A, Pandey MM, Arora S, Singh RRB, Rawat AKS (2013) Physical stability and HPLC analysis of Indian kudzu (Pueraria tuberosa Linn.) fortified milk. Evid Based Complement Alternat Med, vol. 2013, Article ID 368248, 6 pages
Rostagno MA, Villares A, Guillamón A, García-Lafuente A, Martínez JA (2009) Sample preparation for the analysis of isoflavones from soybeans and soy foods. J Chromatogr A 1216:2–29
Rybak ME, Parker DL, Pfeiffer CM (2008) Determination of urinary phytoestrogens by HPLC–MS/MS: a comparison of atmospheric pressure chemical ionization (APCI) and electrospray ionization (ESI). J Chromatogr B 861:145–150
Saviranta NMM, Julkunen-Tiitto R, Oksanen E, Karjalainen RO (2010) Leaf phenolic compounds in red clover (Trifolium pratense L.) induced by exposure to moderately elevated ozone. Environ Pollut 158:440–446
Schwartz H, Sontag G, Plumb J (2009) Inventory of phytoestrogen databases. Food Chem 113:736–747
Setchell KDR, Clerici C (2010) Equol: history, chemistry and formation. J Nutr 140:1355S–1362S
Sirotkin AV, Harrath AH (2014) Phytoestrogens and their effects. Eur J Pharmacol 741:230–236
Skaanild MT, Nielsen TS (2010) The influence of bovine milk high or low in isoflavones on hepatic gene expression in mice. J Toxicol, vol. 2010, Article ID 423179, 5 pages
Stalikas CD (2007) Extraction, separation, and detection methods for phenolic acids and flavonoids. J Sep Sci 30:3268–3295
Steinshamn H, Purup S, Thuen E, Hansen-Møller J (2008) Effects of clover-grass silages and concentrate supplementation on the content of phytoestrogens in dairy cow milk. J Dairy Sci 91:2715–2725
Taylor JI, Grace PB, Bingham SA (2005) Optimization of conditions for the enzymatic hydrolysis of phytoestrogen conjugates in urine and plasma. Anal Biochem 341:220–229
Třináctý J, Křížová L, Schulzová V, Hajšlová J, Hanuš O (2009) The effect of feeding soybean-derived phytoestogens on their concentration in plasma and milk of lactating dairy cows. Arch Anim Nutr 63(3):219–229
Trontelj J (2012) Quantification of glucuronide metabolites in biological matrices by LC-MS/MS. In: Prasain JK (ed) Tandem mass spectrometry–applications and principles USA, Chapter 23, 531-558
Tsen SY, Siew J, Lau EKL, Roslee FAB, Chan HM, Loke WM (2014) Cow’s milk as a dietary source of equol and phenolic antioxidants: differential distribution in the milk aqueous and lipid fractions. Dairy Sci Technol 94:625–632
Uzzan M, Nechrebeki J, Labuza TP (2007) Thermal and storage stability of nutraceuticals in a milk beverage dietary supplement. J Food Sci 72(3):E109–E114
Vacek J, Klejdus B, Lojková L, Kubán V (2008) Current trends in isolation, separation, determination and identification of isoflavones: a review. J Sep Sci 31:2054–2067
Valls J, Millán S, Martí MP, Borràs E, Arola L (2009) Advanced separation methods of food anthocyanins, isoflavones and flavanols. J Chromatogr A 1246:7143–7172
Vitale DC, Piazza C, Melilli B, Drago F, Salomone S (2013) Isoflavones: estrogenic activity, biological effect and bioavailability. Eur J Drug Metab Pharmacokinet 38:15–25
Wang C-C, Prasain JK, Barnes S (2002) Review of the methods used in the determination of phytoestrogens. J Chromatogr B 777:3–28
Wielogórska E, Elliot CT, Danaher M, Chevallier O, Connolly L (2015) Validation of an ultra high performance liquid chromatography-tandem mass spectrometry method for detection and quantification of 19 endocrine disruptors in milk. Food Control 48:48–55
Wocławek-Potocka I, Mannelli C, Boruszewska D, Kowalczyk-Zieba I, Waśniewski T, Skarżyński DJ (2013) Diverse effects of phytoestrogens on the reproductive performance: cow as a model. Int J Endocrinol, vol. 2013, Article ID 650984, 15 pp
Wu Q, Wang M, Simon JE (2004) Analytical methods to determine phytoestrogenic compounds. J Chromatogr B 812:325–355
Acknowledgments
The authors wish to thank the Public Service of Wallonia (PhytoHealth Project, Moerman funded) for providing financial assistance during the course of this research.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

Cite this article
Daems, F., Romnee, JM., Heuskin, S. et al. Analytical methods used to quantify isoflavones in cow’s milk: a review. Dairy Sci. & Technol. 96, 261–283 (2016). https://doi.org/10.1007/s13594-015-0276-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13594-015-0276-8