
Abstract
Elevated serum uric acid (SUA)—hyperuricemia—is caused by overproduction of urate or by its decreased renal and/or intestinal excretion. This disease, which is increasing in prevalence worldwide, is associated with both gout and metabolic diseases. Several studies have reported relationships between apolipoprotein E (APOE) haplotypes and SUA levels in humans; however, their results remain inconsistent. This prompted us to investigate the relationship between APOE polymorphisms and SUA levels. Our subjects were 5,272 Japanese men, premenopausal women, and postmenopausal women. Multiple linear regression analyses revealed the ε2 haplotype of APOE to be independently associated with higher SUA in men (N = 1,726) and postmenopausal women (N = 1,753), but not in premenopausal women (N = 1,793). In contrast, the ε4 haplotype was little related to SUA levels in each group. Moreover, to examine the effect of Apoe deficiency on SUA levels, we conducted animal experiments using Apoe knockout mice, which mimics ε2/ε2 carriers. We found that SUA levels in Apoe knockout mice were significantly higher than those in wild-type mice, which is consistent with the SUA-raising effect of the ε2 haplotype observed in our clinico-genetic analyses. Further analyses suggested that renal rather than intestinal underexcretion of urate could be involved in Apoe deficiency-related SUA increase. In conclusion, we successfully demonstrated that the ε2 haplotype, but not the ε4 haplotype, increases SUA levels. These findings will improve our understanding of genetic factors affecting SUA levels.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Hyperuricemia is caused by the overproduction of urate or by decreased renal [1, 2] and intestinal [3, 4] urate excretion. This common disease is not only associated with gout but also with other common conditions including hypertension [5, 6] and atherosclerotic cardiovascular diseases [7] as well as kidney diseases [8]. Although recent studies have revealed the pathophysiological importance of urate transporters in urate handling in humans [9, 10], other (non-transporter) genetic factors associated with serum uric acid (SUA) levels have also been reported [11,12,13]. Moreover, based on the classically known association between hyperuricemia and hyperlipidemia [14, 15], the influence on SUA levels of genetic factors affecting lipid levels in the blood is likely to be involved. One of these is variation in apolipoprotein E (APOE) polymorphisms; however, as described below, their effects on SUA levels have not been conclusive.
The human APOE gene, which is located on chromosome 19q13.2, has two common non-synonymous single-nucleotide polymorphisms (SNPs)—rs429358 (c.334T>C; p.Cys112Arg) and rs7412 (c.472C>T; Arg158Cys) [16]. Given the lack of simultaneous presence of their minor alleles in one haplotype, three haplotypes are defined, named as ε2, ε3, and ε4. Six diplotypes have been observed as combinations of these three haplotypes (Table 1). Among the three haplotypes, ε3 is the commonest and recognized as the parent form, corresponding to the wild type (WT).
APOE, a glycoprotein constituted of 299 amino acids, is chiefly distributed in very low-density lipoproteins (VLDLs), chylomicrons, and some high-density lipoproteins (HDLs) [17]. It plays multiple roles in the regulation of lipid and lipoprotein levels in the blood [18]. APOE polymorphisms are also reportedly associated not only with lipoprotein metabolism but with atherosclerotic cardiovascular diseases [19], kidney diseases [20], and neurodegenerative diseases [21,22,23,24]. Accordingly, given the observed associations between SUA levels and these disease phenotypes, it is possible that APOE polymorphisms and SUA levels are confounding factors for these disorders. Investigation of the latent relationship between APOE polymorphisms and SUA levels in humans should therefore provide new insights into the pathogenesis of hyperuricemia as well as its associated diseases. Several studies have investigated the relationship between APOE polymorphisms and SUA levels, but their results remain inconsistent.
Hitherto, several human studies have reported that the ε2 and ε4 haplotypes may be associated with higher [25,26,27,28] and lower [29] SUA levels, respectively, than seen with the ε3 haplotype. In contrast, other studies have reported an association between the ε4 haplotype and higher SUA levels [30, 31]. To enhance our understanding of this unresolved question, we herein aimed to investigate the relationships between APOE polymorphisms and SUA levels in a larger population. We also performed animal experiments using Apoe knockout (KO) mice to examine the effect of total Apoe deficiency on SUA levels in terms of urate excretion from the body.
Methods
Study participants
All the procedures used in human studies were approved by the institutional ethical committees (National Defense Medical College and Nagoya University), and were performed in accordance with the Declaration of Helsinki. All of the Japanese individuals in this study were recruited from participants in the Shizuoka area and Daiko area in the Japan Multi-Institutional Collaborative Cohort Study (J-MICC Study) [32, 33]. Written informed consent was obtained from all the subjects.
Among the participants, those who were under treatment for or had past histories of gout/hyperuricemia or dyslipidemia, and female participants for whom there was no information about menopause were excluded. Multiple regression analysis was performed on the resulting 5,272 subjects to evaluate the relationships among SUA levels, APOE gene polymorphisms, and other risk factors. Non-HDL cholesterol level was calculated using the following equation: [Non-HDL cholesterol (non-HDL-C) (mg/dL)] = [Total cholesterol (TC) (mg/dL)] − [HDL cholesterol (HDL-C) (mg/dL)].
Genetic analysis
Genomic DNA was extracted from whole peripheral blood cells. Genotyping of the two SNPs (rs429358 and rs7412) in the APOE gene was performed using the TaqMan method (Thermo Fisher Scientific, Waltham, MA, USA) with a LightCycler 480 (Roche Diagnostics, Mannheim, Germany) [34], with minor modifications. To confirm the genotypes, direct sequencing was also performed on more than 200 samples with the following primers: for forward, 5′-CCTACAAATCGGAACTGGAG-3′, and for reverse, 5′-CCCGGCCTGGTACACTG-3′. DNA sequencing analysis was performed with a 3130xl Genetic Analyzer (Thermo Fisher Scientific) [34].
Experimental assessment of urate excretion pathways
Animals were handled humanely in accordance with the National Cerebral and Cardiovascular Center’s Guidelines for the Care and Use of Laboratory Animals. The experimental protocol and animal use procedures were approved by the Committee of the National Cerebral and Cardiovascular Center. Male Apoe KO mice [35] (Jackson Laboratories, Bar Harbor, ME, USA) and control WT mice (C57BL6/J; Japan SLC, Shizuoka, Japan) were fed a normal rodent laboratory diet (CE-2; CLEA Japan, Tokyo, Japan).
Concentrations of urate and creatinine in collected serum and urine samples were determined by QuantiChrom Uric Acid Assay Kit (BioAssay Systems, Hayward, CA, USA) and Creatinine Assay Kit (Cayman Chemical, Ann Arbor, MI, USA), respectively. To analyze intestinal urate excretion, mice that had fasted overnight were anaesthetized by intraperitoneal injection of urethane and cannulated with polyethylene tubing (Hibiki Size 8) (Sansyo, Tokyo, Japan) at the upper duodenum and the middle jejunum to make an intestinal loop at the upper half of the intestine, in the same way as in our previous study [4]. After the intestinal contents had been removed by the slow infusion of saline and air, efflux buffer (saline containing 0.3 mM potassium oxonate) was introduced into the intestinal loop and both ends of the loop were closed with syringes. After the indicated periods, the efflux buffer in the loop was collected using syringes and the urate concentrations were quantified. Intestinal urate excretion was calculated using the following equation: [Intestinal urate excretion] = [Urate concentration in the intestinal loop] × [Volume of efflux buffer in the intestinal loop] × [Length of the whole small intestine]/[Length of the intestinal loop] as previously described [4].
Statistical analysis
For statistical analysis calculations in the human studies, R software (version 3.1.1) (http://www.r-project.org/) was used. Student’s t test was employed for comparison of SUA levels in humans. We also carried out a multiple regression analysis to evaluate the independent effect of APOE polymorphisms on SUA levels, adjusting for confounding factors such as serum creatinine levels and non-HDL cholesterol levels. In animal experiments, analyses were performed using JMP software version 12.0 (SAS Institute, Cary, NC, USA). Student’s t test was used for comparison of urate concentrations in mice. All P values were two-tailed, and a P value of < 0.05 was considered to be statistically significant.
Results
Effects of APOE haplotypes on SUA levels in humans
To investigate the effects of APOE haplotypes on SUA levels, we examined the associations between APOE haplotypes and SUA levels in 5,272 Japanese individuals. SUA levels in carriers and non-carriers of APOE ε2 and ε4, respectively, are summarized in Tables 2 and 3. The call rates for two SNPs (rs429358 and rs7412) that determine the APOE haplotypes were 100%; these SNPs in the control group were in Hardy–Weinberg equilibrium (P > 0.05). Due to sex differences in SUA levels, which are also affected by menopause, we divided the study participants into three groups: men (N = 1,726), premenopausal women (N = 1,793), and postmenopausal women (N = 1,753). As shown in Table 2, SUA levels were significantly higher in postmenopausal than in premenopausal women (P = 3.1 × 10−52). The ε2 haplotype was associated with higher SUA levels in men (P = 0.033) and in postmenopausal women (P = 0.048); however, interestingly, the ε2 haplotype did not affect SUA levels among premenopausal women (P = 0.61) (Table 2). The ε4 haplotype was not significantly related to SUA levels in men, premenopausal women, or postmenopausal women (Table 3). We therefore conducted further analyses focusing on the ε2 haplotype.
To examine the quantitative effect on SUA levels of harboring the ε2 haplotype, we next performed a multiple linear regression analysis that included variables associated with increased SUA levels. A previous study showed that in healthy adults, the correlation coefficient for an association of non-HDL cholesterol with SUA was higher than those for associations of triglycerides and other lipid parameters including TC, HDL-C, and LDL-C [36]. Based on this information, we chose non-HDL-C levels as a covariate among available lipid parameters. In the multiple linear regression analysis adjusted for age, body mass index (BMI), serum creatinine levels, and non-HDL-C levels, the ε2 haplotype was independently associated with higher SUA levels in men (P = 0.015) and postmenopausal women (P = 0.005), while this was not in the case in premenopausal women (P = 0.55) (Table 4). Although these associative analyses could not uncover the molecular mechanisms lying behind the APOE ε2-associated increase of SUA levels, the fact that the APOE E2 protein is defective in its binding ability to the APOE receptors, unlike APOE E4 protein [37] and APOE E3 protein [38], suggests that APOE dysfunction might lead to increased SUA. To address this hypothesis, we further conducted in vivo analyses using male Apoe KO mice as described below.
Effects of Apoe knockout on SUA levels and urate secretion in mice
To examine whether deficiency in Apoe function can affect SUA levels, we performed animal experiments using male Apoe KO mice. Given that like APOE deficiency, ε2 homozygosity is also associated with an increased risk of type III hyperlipoproteinemia in humans [36], together with the fact that knock-in mice carrying the human APOE E2 allele in place of the mouse Apoe gene cause type III hyperlipoproteinemia and spontaneous atherosclerosis in mice [39], Apoe KO mice can be a model mimicking ε2/ε2 carriers.
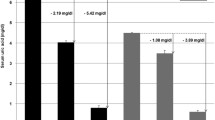
As expected, SUA levels of Apoe KO mice were significantly higher than those of WT mice (P = 0.021) (Fig. 1A). We then investigated the latent mechanisms in terms of urate excretion from the body. As shown in Fig. 1B, there was no significant difference in urate excretion from the intestine between Apoe KO mice and WT mice; however, the urinary urate/creatinine ratios were significantly lower in Apoe KO mice than those in WT mice (P = 0.022) (Fig. 1C). There was no difference in body weight between Apoe KO mice and WT mice (28.1 ± 1.0 g vs. 28.0 ± 3.2 g; P = 0.937). These results suggest that higher SUA levels in Apoe KO mice could be caused by a decrease not in intestinal but in renal urate excretion.

Serum uric acid levels and urate excretion activities in Apoe knockout mice. A Serum uric acid levels. B Intestinal urate excretion. There were no significant differences in intestinal urate excretion between WT and KO mice at each time point. C Renal urate excretion. WT wild type mice, KO Apoe knockout mice. Data are expressed as mean ± standard error (SE). n = 12 (A), 9 (B), and 12 (C). *P < 0.05 (t test)
Discussion
The present study demonstrates the ε2 haplotype, not ε4 haplotype, to be associated with higher SUA levels in a Japanese population (Tables 2 and 3). This association was observed in male subjects and in postmenopausal female subjects, but not in premenopausal female subjects. To the best of our knowledge, this is the first report showing the effect of the ε2 haplotype on SUA levels to be different in women between before and after menopause. In other words, in contrast to the ε4 haplotype, the ε2 haplotype can have different effects on human SUA levels in difference populations, possibly in response to their levels of sex hormones. Although this study cannot support a previously reported association between the ε4 haplotype and lower SUA levels in southern Iranian subjects [29], our results are consistent with several previous studies showing that the ε2 haplotype was associated with higher SUA levels in Caucasian [20, 28] and Chinese [25, 26] populations.
Our findings suggest that the presence of a menstrual cycle, absent in men and postmenopausal women, might attenuate the effect of the ε2 haplotype causing higher SUA levels in premenopausal women. In women, the mean values of SUA levels reportedly increase after menopause [40], suggesting that female hormone-mediated regulatory mechanisms are involved in lowering SUA. This notion is supported by a mechanistic insight [41] showing that in the kidney of mice, estradiol and progesterone suppressed the protein levels of physiologically important renal urate re-absorbers [i.e., urate transporter 1 (Urat1/Slc22a12) [42] and glucose transporter 9 (Glut9/Slc2a9) [43]] and sodium-coupled monocarboxylate transporter 1 (Smct1/Slc5a8, a functional co-operator of Urat1 [44]), respectively, which could theoretically result in an increase in renal urate excretion and therefore decreased SUA levels.
In this study, we examined Apoe KO mice as a suitable model of ε2 homozygous subjects. We showed, for the first time, that Apoe KO mice had higher SUA levels than did WT mice (Fig. 1). As a plausible explanation of this phenotype, we found that Apoe KO mice exhibited lower renal urate excretion than WT mice, which was accompanied by little difference in intestinal urate excretion. Given that spontaneously developed hypercholesterolemia in Apoe-deficient mice promoted early renal dysfunction [45], the higher SUA levels and lower renal urate excretion in Apoe KO mice observed in this study might be associated with hypercholesterolemia-related renal dysfunction. This should be addressed in more detail in future. Also, while hyperuricemia is commonly associated with metabolic syndrome, dyslipidemia, and chronic kidney disease in humans, further studies are needed to elucidate any latent mechanisms linking the ε2 haplotype and higher SUA levels in humans.
Additionally, our multiple linear regression analyses adjusted for age, BMI, serum creatinine levels, and non-HDL-C levels revealed the ε2 haplotype to be independently associated with increased SUA levels (Table 4). Hence, despite the limited data currently available, the human phenotype in SUA levels associated with ε2 haplotype may not be explained by renal dysfunction alone. To address this point, further human studies will be required in addition to biochemical and histological investigations of the kidney using Apoe KO mice and/or such mice with the human APOE ε2.
In conclusion, we have demonstrated that the ε2 haplotype, not the ε4 haplotype, of APOE is associated with higher SUA levels in humans. Results of in vivo experiments suggest that renal underexcretion of urate might be involved in the observed Apoe deficiency-related SUA increase; however, further studies are required to uncover the details of the mechanisms in question. Our findings will enhance our understanding of the genetic factors affecting SUA levels in humans.
References
Neogi T. Clinical practice. Gout. N Engl J Med. 2011;364:443–52.
Matsuo H, Nakayama A, Sakiyama M, et al. ABCG2 dysfunction causes hyperuricemia due to both renal urate underexcretion and renal urate overload. Sci Rep. 2014;4:3755.
Matsuo H, Tsunoda T, Ooyama K, et al. Hyperuricemia in acute gastroenteritis is caused by decreased urate excretion via ABCG2. Sci Rep. 2016;6:31003.
Ichida K, Matsuo H, Takada T, et al. Decreased extra-renal urate excretion is a common cause of hyperuricemia. Nat Commun. 2012;3:764.
Forman JP, Choi H, Curhan GC. Uric acid and insulin sensitivity and risk of incident hypertension. Arch Intern Med. 2009;169:155–62.
Cannon PJ, Stason WB, Demartini FE, Sommers SC, Laragh JH. Hyperuricemia in primary and renal hypertension. N Engl J Med. 1966;275:457–64.
Feig DI, Kang DH, Johnson RJ. Uric acid and cardiovascular risk. N Engl J Med. 2008;359:1811–21.
Talaat KM, el-Sheikh AR. The effect of mild hyperuricemia on urinary transforming growth factor beta and the progression of chronic kidney disease. Am J Nephrol. 2007;27:435–40.
Dalbeth N, Choi HK, Joosten LAB, et al. Gout. Nat Rev Dis Primers. 2019;5:69.
Major TJ, Dalbeth N, Stahl EA, Merriman TR. An update on the genetics of hyperuricaemia and gout. Nat Rev Rheumatol. 2018;14:341–53.
Nakatochi M, Kanai M, Nakayama A, et al. Genome-wide meta-analysis identifies multiple novel loci associated with serum uric acid levels in Japanese individuals. Commun Biol. 2019;2:115.
Tin A, Marten J, Halperin Kuhns VL, et al. Target genes, variants, tissues and transcriptional pathways influencing human serum urate levels. Nat Genet. 2019;51:1459–74.
Boocock J, Leask M, Okada Y, et al. Genomic dissection of 43 serum urate-associated loci provides multiple insights into molecular mechanisms of urate control. Hum Mol Genet. 2020;29:923–43.
Darlington LG, Scott JT. Plasma lipid levels in gout. Ann Rheum Dis. 1972;31:487–9.
Berkowitz D. Gout, hyperlipidemia, and diabetes interrelationships. JAMA. 1966;197:77–80.
Rall SC Jr., Weisgraber KH, Mahley RW. Human apolipoprotein E. The complete amino acid sequence. J Biol Chem. 1982;257:4171–8.
Hatters DM, Peters-Libeu CA, Weisgraber KH. Apolipoprotein E structure: insights into function. Trends Biochem Sci. 2006;31:445–54.
Utermann G, Hees M, Steinmetz A. Polymorphism of apolipoprotein E and occurrence of dysbetalipoproteinaemia in man. Nature. 1977;269:604–7.
Song Y, Stampfer MJ, Liu S. Meta-analysis: apolipoprotein E genotypes and risk for coronary heart disease. Ann Intern Med. 2004;141:137–47.
Liberopoulos EN, Miltiadous GA, Cariolou M, Kalaitzidis R, Siamopoulos KC, Elisaf MS. Influence of apolipoprotein E polymorphisms on serum creatinine levels and predicted glomerular filtration rate in healthy subjects. Nephrol Dial Transplant. 2004;19:2006–12.
Peng H, Wang C, Chen Z, et al. APOE epsilon2 allele may decrease the age at onset in patients with spinocerebellar ataxia type 3 or Machado–Joseph disease from the Chinese Han population. Neurobiol Aging. 2014;35(2179):e15–8.
Mata IF, Leverenz JB, Weintraub D, et al. APOE, MAPT, and SNCA genes and cognitive performance in Parkinson disease. JAMA Neurol. 2014;71:1405–12.
Farrer LA, Cupples LA, Haines JL, et al. Effects of age, sex, and ethnicity on the association between apolipoprotein E genotype and Alzheimer disease. A meta-analysis. APOE and Alzheimer Disease Meta Analysis Consortium. JAMA. 1997;278:1349–56.
Saunders AM, Strittmatter WJ, Schmechel D, et al. Association of apolipoprotein E allele epsilon 4 with late-onset familial and sporadic Alzheimer’s disease. Neurology. 1993;43:1467–72.
Wang C, Yan W, Wang H, Zhu J, Chen H. APOE polymorphism is associated with blood lipid and serum uric acid metabolism in hypertension or coronary heart disease in a Chinese population. Pharmacogenomics. 2019;20:1021–31.
Wu J, Qiu L, Guo XZ, et al. Apolipoprotein E gene polymorphisms are associated with primary hyperuricemia in a Chinese population. PLoS ONE. 2014;9:e110864.
Liberopoulos EN, Miltiadous GA, Athyros VG, et al. Effect of apolipoprotein E polymorphism on serum uric acid levels in healthy subjects. J Investig Med. 2005;53:116–22.
Cardona F, Tinahones FJ, Collantes E, Escudero A, Garcia-Fuentes E, Soriguer FJ. The elevated prevalence of apolipoprotein E2 in patients with gout is associated with reduced renal excretion of urates. Rheumatology (Oxford). 2003;42:468–72.
Bazrgar M, Karimi M. Is the apolipoprotein E4 allele always hazardous? Serum uric acid level as a conflict. Genet Test Mol Biomark. 2012;16:920–3.
Sun YP, Zhang B, Miao L, et al. Association of apolipoprotein E (ApoE) polymorphisms with risk of primary hyperuricemia in Uygur men, Xinjiang, China. Lipids Health Dis. 2015;14:25.
Alvim RO, Freitas SR, Ferreira NE, et al. APOE polymorphism is associated with lipid profile, but not with arterial stiffness in the general population. Lipids Health Dis. 2010;9:128.
Asai Y, Naito M, Suzuki M, et al. Baseline data of Shizuoka area in the Japan Multi-Institutional Collaborative Cohort Study (J-MICC Study). Nagoya J Med Sci. 2009;71:137–44.
Hamajima N, J-MICC Study Group. The Japan Multi-Institutional Collaborative Cohort Study (J-MICC Study) to detect gene-environment interactions for cancer. Asian Pac J Cancer Prev. 2007;8:317–23.
Sakiyama M, Matsuo H, Shimizu S, et al. Common variant of leucine-rich repeat-containing 16A (LRRC16A) gene is associated with gout susceptibility. Hum Cell. 2014;27:1–4.
Shibata MA, Harada-Shiba M, Shibata E, et al. Crude alpha-mangostin suppresses the development of atherosclerotic lesions in Apoe-deficient mice by a possible M2 macrophage-mediated mechanism. Int J Mol Sci. 2019;20:1722.
Ghiselli G, Schaefer EJ, Gascon P, Breser HB Jr. Type III hyperlipoproteinemia associated with apolipoprotein E deficiency. Science. 1981;214:1239–41.
Utermann G. Apolipoprotein E polymorphism in health and disease. Am Heart J. 1987;113:433–40.
Weisgraber KH, Innerarity TL, Mahley RW. Abnormal lipoprotein receptor-binding activity of the human E apoprotein due to cysteine-arginine interchange at a single site. J Biol Chem. 1982;257:2518–21.
Sullivan PM, Mezdour H, Quarfordt SH, Maeda N. Type III hyperlipoproteinemia and spontaneous atherosclerosis in mice resulting from gene replacement of mouse Apoe with human Apoe*2. J Clin Invest. 1998;102:130–5.
Akizuki S. Serum uric acid levels among thirty-four thousand people in Japan. Ann Rheum Dis. 1982;41:272–4.
Takiue Y, Hosoyamada M, Kimura M, Saito H. The effect of female hormones upon urate transport systems in the mouse kidney. Nucleosides Nucleotides Nucleic Acids. 2011;30:113–9.
Enomoto A, Kimura H, Chairoungdua A, et al. Molecular identification of a renal urate anion exchanger that regulates blood urate levels. Nature. 2002;417:447–52.
Matsuo H, Chiba T, Nagamori S, et al. Mutations in glucose transporter 9 gene SLC2A9 cause renal hypouricemia. Am J Hum Genet. 2008;83:744–51.
So A, Thorens B. Uric acid transport and disease. J Clin Invest. 2010;120:1791–9.
Balarini CM, Oliveira MZ, Pereira TM, et al. Hypercholesterolemia promotes early renal dysfunction in apolipoprotein E-deficient mice. Lipids Health Dis. 2011;10:220.
Acknowledgements
We express our sincere thanks to all the participants in this study. Our heartfelt gratitude goes to the members of the Shizuoka area and Daiko area in the Japan Multi-Institutional Collaborative Cohort Study (J-MICC Study) for their support. Especially, the authors also acknowledge N. Hamajima, H. Tanaka, K. Takeuchi, and K. Wakai for sample collection and their continuous encouragement of our study. We are grateful to M. Horiuchi (National Cerebral and Cardiovascular Center Research Institute) for skilled technical assistance in the animal experiments. We are indebted to J. Abe, M. Kawaguchi, M. Miyazawa, and K. Morichika (National Defense Medical College) for genetic analyses and helpful discussions. YT is an Excellent Young Researcher in MEXT Leading Initiative for Excellent Young Researchers.
Funding
This work was supported by the National Cerebral and Cardiovascular Center’s Intramural Research Fund (No. 25-2-5) for Cardiovascular Diseases. The research conducted by the National Defense Medical College was also supported by JSPS KAKENHI (Nos. 25293145, 15K15227, 16H06279 (PAGS), 221S0002, 17H04128, 20H00566, 20H00568, 20H04100, 20K23152, and 21H03350); the Ministry of Defense of Japan; the Kawano Masanori Memorial Foundation for Promotion of Pediatrics; and the Gout and Uric Acid Foundation of Japan. The research conducted by Nagoya University Graduate School of Medicine was supported by a JSPS KAKENHI Grant [nos. 16H06277 (CoBiA)] and Grants-in-Aid for Scientific Research on Priority Areas (no. 17015018) and Innovative Areas (no. 221S0001) from the Ministry of Education, Culture, Sports, Science and Technology of Japan.
Author information
Authors and Affiliations
Contributions
MO, MS, and HM conceived and designed this study; MH-S, and NS assisted in the research design; MN, AH, SK, RO, and HM collected and analyzed participants’ clinical data; YT, MS, YK, AN, SS, TH, MN, and HM performed genetic analyses; MO, YT, MS, YK, AN, and HM performed statistical analyses; MO, YT, YY, TT, and HS conducted or supported the animal experiments; MO and HM organized this collaborative study; MS, MA, KT, KI, MH-S, and NS provided intellectual input and assisted with preparation of the manuscript; MO, YT, MS, and HM wrote and revised the manuscript. MO, YT, and MS contributed equally to this work.
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethics approval
All procedures used in the human study were approved by the institutional ethical committees at the National Defense Medical College (No. 2914) and Nagoya University (No. 2010-0939-7), and were performed in accordance with the Declaration of Helsinki.
Informed consent
Written informed consent was obtained from each subject participating in this study.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Ogura, M., Toyoda, Y., Sakiyama, M. et al. Increase of serum uric acid levels associated with APOE ε2 haplotype: a clinico-genetic investigation and in vivo approach. Human Cell 34, 1727–1733 (2021). https://doi.org/10.1007/s13577-021-00609-w
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13577-021-00609-w