
Abstract
It has been reported that transforming growth factor-β1 (TGF-β1) signaling plays an important role in the development of diabetic nephropathy (DN). The nuclear transcription co-repressor Ski-related novel protein N (SnoN) is a critical negative regulator of TGF-β1/Smad signal pathway, involving in tubule epithelial–mesenchymal transition (EMT), extracellular matrix (ECM) accumulation, and tubulointerstitial fibrosis. In this study, we focused on miR-23a as a regulator of SnoN. Our purpose is to study the effects of miR-23a on high glucose (HG)-induced EMT process and ECM deposition in HK2 cells. We found that miR-23a was up-regulated in renal tissues of diabetic patients and HG-induced HK2 cells. Besides, the high level of miR-23a was closely associated with decreased SnoN expression. Knockdown of miR-23a increased SnoN expression and in turn suppressed HG-induced EMT and renal fibrogenesis. Introduction of miR-23a decreased SnoN expression and enhanced the profibrogenic effects of HG on HK2 cells. Next, bioinformatics analysis predicted that the SnoN was a potential target gene of miR-23a. Luciferase reporter assay demonstrated that miR-23a could directly target SnoN. We demonstrated that overexpression of SnoN was sufficient to inhibit HG-induced EMT and renal fibrogenesis in HK2 cells. Furthermore, down-regulation of SnoN partially reversed the protective effect of miR-23a knockdown on HG-induced EMT and renal fibrogenesis in HK2 cells. Collectively, miR-23a and SnoN significantly impact on the progression of HG-induced EMT and renal fibrogenesis in vitro, and they may represent novel targets for the prevention strategies of renal fibrosis in the context of DN.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Diabetic nephropathy (DN) is one of the most common complications of diabetes mellitus (DM) [1]. The basement membrane thickening, renal tubal epithelial–mesenchymal transition (EMT), extracellular matrix (ECM) accumulation, glomerulosclerosis, and tubulointerstitial fibrosis (TIF) are pathological characteristics of DN, finally contributing to irreversible renal damage [2,3,4,5]. The pathogenesis of DN is still unclear, and there is limited effectiveness for the current treatments [6]. Therefore, it is necessary to further study its precise molecular mechanisms, developing new effective therapy for DN.
Recent studies have demonstrated that hyperglycemia stimulates EMT of tubular cells, which is a critical mechanism of renal tubulointerstitial fibrosis in DN [7,8,9]. EMT is characterized by the decreased expression of the epithelial surface marker E-cadherin, the increased expressions of the mesenchymal markers, and the disruption of the tubular basement membrane in tubular epithelia cells [10]. In consideration of the important role of EMT in initiating and promoting renal fibrosis in DN, a deeper understanding of the underlying mechanisms of EMT is important to clarify the pathogenesis of DN, and modulation of EMT may provide a novel therapeutic target to potentially inhibit renal fibrogenesis in the diabetic kidney.
miRNAs have been confirmed to play critical roles in cell proliferation, apoptosis, and differentiation, contributing to the pathogenesis of many human diseases such as cancers, diabetes, and diabetic complications including DN [11,12,13]. Deregulated miRNAs and their roles in the progression of diabetic nephropathy have recently attracted substantial attention. However, the role of miRNAs in diabetic nephropathy is not clear, and a few studies have linked miRNAs with diabetes mellitus. In this study, we found that the level of miR-23a is evidently up-regulated in renal tissues from patients with type 2 diabetes mellitus (T2DM) associated renal fibrosis compared with control subjects. Besides, miR-23a were identified to directly target the SnoN gene. The functional analysis confirmed that inhibition of miR-23a increased SnoN expression and suppressed the profibrogenic effects of HG on HK2 cells. Overexpression of miR-23a down-regulated the expression of SnoN and in turn promoted HG-induced EMT and renal fibrogenesis. According to above results, miR-23a may represent a novel target for the prevention strategies of renal fibrosis in the context of DN.
Materials and methods
Patients and tissue samples
The DN tissues were obtained from DN patients (n = 10) with type 2 diabetes who underwent diagnostic procedures. The healthy control kidney tissues were obtained from patients (n = 10) who underwent nephrectomy as part of their tumor treatment. All these samples were collected from the Urology Department, Cangzhou Central Hospital. Informed consent was obtained from all patients and the study was approved by the Ethics Committee of Cangzhou Central Hospital.
Cell culture and transient transfection
Human kidney 2 (HK-2) is a cortex/proximal tubular cell (PTC) line derived from normal kidney, and purchased from American Type Culture Collection (ATCC, Manassas, VA, USA). Immortalized HK2 cells were cultured in DMEM/F12 containing 10% FBS, 1.20 g/l sodium bicarbonate, 100 U/ml penicillin, and 100 mg/ml streptomycin at 37 °C in 5% CO2.
HK2 cells were transfected with miR-23a mimic or inhibitor (RiboBio, Guangzhou, China) at 50 or 100 nM concentration using Lipofectamine RNAiMax reagent (Invitrogen, USA) according to the manufacturer’s protocols. Following transfection, cells were stimulated with high glucose (HG, 30 mM) for 24 h or were not stimulated, cell were harvested, and SnoN expression was quantified by western blot. Total RNA extracted from HK2 cells was used to determine the levels of miR-23a, E-cadherin, α-SMA, Vimentin, Fibronectin (FN), and Collagen IV (Col IV) by qRT-PCR.
RNA extraction and quantitative real-time-polymerase chain reaction (qRT-PCR)
Total RNA of tissues and HK2 cells was extracted for analyzing miRNA (Qiagen, USA) and mRNA (Axygen, USA) levels according to the manufacturer’s protocols. For quantification of miR-23a, the TaqMan MicroRNA Reverse Transcription Kit and TaqMan miRNA assay (Qiagen, USA) were used to perform reverse transcription and PCR according to the manufacturer’s instructions. U6 was used as the internal control. The gene expressions of E-cadherin, α-SMA, Vimentin and FN, and Col IV were detected using the SYBR Green PCR kits (TAKARA, Japan). Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was amplified as an internal reference. Each sample was assessed in triplicate. Primers used were listed in Table 1.
Western blot analysis
Proteins were extracted using RIPA buffer (Millipore, USA) with protease inhibitor cocktail (Millipore, USA). The protein concentration was quantified by BCA assay kit. Lysates containing equal amounts of proteins were subjected to 10% SDS-PAGE, and then transferred to PVDF membrane (Millipore, USA). Membranes were blocked with 5% nonfat milk diluted with TBST and probed at 4 °C overnight with primary antibody, as follows: anti-SnoN antibody, anti-E-cadherin antibody, anti-α-SMA antibody, and anti-Vimentin antibody (1:1000 dilution, Abcam, USA). Next, membranes were washed by TBST and incubated with a goat anti-rabbit IgG conjugated to horseradish peroxidase (1:1000 dilution, CST, USA) for 2 h at room temperature. Incubation with monoclonal mouse α-tubulin antibody (1:2000 dilution; Sigma, USA) was performed as the loading control. The proteins were visualized using ECL western blotting detection reagents (Millipore, USA). The densitometry of the bands was quantified using the Image J 1.38X software (USA).
Enzyme-linked immunosorbent assay (ELISA)
After treatment, culture medium was centrifuged at 2000×g for 30 min, and the supernatants were determined for FN and Col IV using the commercial ELISA kits (R&D, USA). The absorbance of supernatants was read at 450 nm using a microplate reader.
Dual-luciferase reporter assay
HK2 cells were seeded in 6-well plates (2 × 105/well) and incubated overnight before transfection. Then, pGL3-SnoN-3′UTR wild-type or mutant reporter plasmid, miR-23a inhibitor and inhibitor-NC, or miR-23a mimic and miR-NC, and pRL-TK Renilla luciferase reporter (Promega, USA) were cotransfected into cells using Lipofectamine 3000 (Invitrogen, USA). After that, luciferase activities were quantified using the Dual-Luciferase reporter system (Promega, USA) according to the manufacturer’s protocols. And firefly luciferase activities were normalized to renilla luciferase activities.
Statistical analysis
All the experiments were repeated at least three times. Data are expressed as mean ± standard error of the mean (SEM). Analysis of the pairwise comparisons of independent groups is used by two-tailed Student’s t test. One-way analysis of variance (ANOVA) was used to compare three or more independent groups. All statistical calculations were performed using GraphPad Prism 5.0 (GraphPad Software, Inc., USA), with P < 0.05 considered to indicate a statistically significant difference.
Results
The level of miR-23a was increased and SnoN expression was decreased in human kidneys with DN and HG-induced HK2 cells
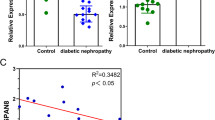
Male were 7 patients and female were 3 patients, and the mean age in diabetic nephropathy patients was 51 ± 12 (years). The mean systolic blood pressure was 145 ± 10 mmHg, and the diastolic blood pressure was 92 ± 11 mmHg. The serum Alb was 35.5 ± 5.1 g/l; HbA1C was 6.9 ± 0.6%; and serum creatinine (Scr) was 168 ± 90 μmol/l. To confirm the potential role of miR-23a in the processes of renal fibrosis, we determined the level of miR-23a by qRT-PCR in renal tissues from patients with T2DM associated renal fibrosis and control subjects. Our data demonstrated that the level of miR-23a was dramatically up-regulated in renal fibrosis tissues compared to that in control groups (Fig. 1a). Next, we studied the level of miR-23a in HG-induced HK2 cells. As expected, HK2 cells were treated with normal glucose (NG, 5.5 mM) or high glucose (HG, 30 mM) for 6, 12, 24, or 48 h, after which the level of miR-23a was examined. Compared with control group, our results indicated that HG could significantly increase the level of miR-23a in a time-dependent manner (Fig. 1b). Subsequently, the online database (TargetScan 6.2) showed that SnoN was predicted to be a direct target of miR-23a. Then, we determined the protein level of SnoN in renal fibrosis tissues and HG-induced HK2 cells, respectively. The results indicated that the expression of SnoN was significantly down-regulated in renal fibrosis tissues and HG-induced HK2 cells at protein level (Fig. 1c, d). From above outcomes, we predicted that miR-23a might negatively regulate SnoN.

Levels of miR-23a and SnoN in renal fibrosis tissues and HG-induced HK2 cells. a Level of miR-23a in normal (n = 10) and renal fibrosis tissues (n = 10) were detected by qRT-PCR. b HK2 cells were treated with normal glucose (NG, 5.5 mM) or high glucose (HG, 30 mM) for 6, 12, 24, or 48 h. Level of miR-23a was analyzed by qRT-PCR and normalized to U6. c Protein expression of SnoN in normal (n = 10) and renal fibrosis tissues (n = 10) were detected by western blot. d HK2 cells were treated with NG (5.5 mM) or HG (30 mM) for 6, 12, 24, or 48 h. Level of SnoN was analyzed by western blot. The data shown are mean ± SEM, n = 4. **P < 0.01, ***P < 0.001 vs. normal or NG
Effects of miR-23a on HG-induced EMT of HK2 cells
To confirm whether the level of miR-23a was closely related to HG-stimulated EMT of HK2 cells, we transfected HK2 cells with miR-23a inhibitor or mimic. First, we evaluated the efficiency of miRNA transfection via the qRT-PCR assay. After transfection for 48 h, the level of miR-23a was significantly up-regulated after transfection with miR-23a mimic, and the miR-23a level was dramatically down-regulated in HK2 cells transfected with miR-23a inhibitor (Fig. 2a). Notably, knockdown of miR-23a significantly attenuated HG-induced down-regulation of the epithelial marker E-cadherin, and up-regulation of mesenchymal markers such as Vimentin, α-SMA when compared to the group treated with HG alone (Fig. 2b), which suggested that miR-23a down-regulation could reverse the effects of HG on EMT of HK2 cells. However, overexpression of miR-23a aggravated HG-stimulated EMT of HK2 cells (Fig. 2b). Taken together, these results indicated that decreased miR-23a inhibited HG-induced EMT in HK2 cells.

Effect of miR-23a on HG-induced EMT of HK2 cells. HK2 cells were transfected with miR-23a mimic or inhibitor. a Level of miR-23a in HK2 cells were determined by qRT-PCR. HK2 cells were treated with HG (30 mM) for 24 h after transfection with miR-23a mimic or inhibitor. b The mRNA and protein expressions of α-SMA, Vimentin and E-cadherin were determined by qRT-PCR and western blot, respectively. The data shown are mean ± SEM, n = 4. **P < 0.01, ***P < 0.001 vs. NG; # P < 0.05, ## P < 0.01, ### P < 0.001 vs. miR-NC, miR-inhibitor, or vehicle + HG
Effects of miR-23a on HG-stimulated fibrogenesis of HK2 cells
To study the effect of miR-23a on the HG-induced fibrogenesis in HK2 cells, we transfected either miR-23a mimic or inhibitor into the HK2 cells and then stimulated them with HG. Next, we detect the expressions of FN and Col IV. Our data indicated that knockdown of miR-23a dramatically inhibited HG-induced expressions of FN and Col IV in HK2 cells, whereas introduction of miR-23a significantly aggravated HG-induced expressions of FN and Col IV (Fig. 3a). Similar to our qRT-PCR results, transfection of cultured HK2 cells with miR-23a inhibitor reduced the secretion of FN and Col IV into the culture medium, whereas transfection with miR-23a mimic increased their secretion in comparison with miR-NC transfection (Fig. 3b). These results suggested that miR-23a inhibited HG-induced fibrogenesis in HK2 cells.

Effects of miR-23a on HG-induced renal fibrogenesis in HK2 cells. HK2 cells were treated with HG (30 mM) for 24 h after transfection with miR-23a mimic or inhibitor. a The mRNA expressions of FN and Col IV were determined by qRT-PCR, respectively. b Secreted levels of FN and Col IV in the culture medium were detected by ELISA assay. All data are presented as mean ± SEM, n = 4. **P < 0.01, ***P < 0.001 vs. NG; # P < 0.05, ## P < 0.01 vs. vehicle + HG
MiR-23a directly targets SnoN in HK2 cells
Using TargetScan 6.2, an online database, our purpose was to predict the target gene and found that SnoN was a binding target of miR-23a, we performed western blot to determine the protein level of SnoN in HK2 cells after transfection with miR-23a mimic or inhibitor. We found that the protein expression of SnoN was dramatically reduced after miR-23a overexpression (Fig. 4a), but was significantly enhanced after miR-23a knockdown (Fig. 4a). To further confirm whether miR-23a directly target SnoN, we cloned human SnoN-WT 3′-UTR into the pGL3-luciferase reporter vector, and the putative miR-23a binding site in the SnoN 3′-UTR was mutated (SnoN-MUT 3′-UTR) (Fig. 4b). Our data showed that miR-23a mimic significantly inhibited the luciferase activity, whereas miR-23a inhibitor promoted the luciferase activity in HK2 cells transfected with WT 3′-UTR of SnoN (Fig. 4c). The SnoN-MUT 3′-UTR blocked the effects of miR-23a. These results showed that miR-23a could directly and negatively regulate SnoN.

SnoN was a direct target of miR-23a. HK2 cells were transfected with miR-23a mimic or inhibitor. a Protein level of SnoN was determined by western blot. SnoN expression was normalized to α-tubulin. b Schematic representation of SnoN 3′UTRs showing putative miRNA target site. c Analysis of the relative luciferase activities of SnoN-WT and SnoN-MUT. All data are presented as mean ± SEM, n = 4. **P < 0.01 vs. miR-NC or miR-inhibitor
Introduction of SnoN had the similar effects with knockdown of miR-23a
To further study the function of SnoN in HK2 cells, HK2 cells were overexpressed SnoN expression by transfection with pcDNA-SnoN. The western blot analysis showed that the protein expression of SnoN was significantly increased after 48 h in HK2 cells transfected with pcDNA-SnoN (Fig. 5a). Furthermore, overexpression of SnoN could significantly attenuate HG-induced down-regulation of E-cadherin and up-regulation of α-SMA, Vimentin (Fig. 5b). Next, introduction of SnoN dramatically down-regulated HG-induced expressions of FN and Col IV in HK2 cells, which indicated that increased SnoN expression inhibited HG-induced fibrogenesis (Fig. 5c, d). Our these findings demonstrated that SnoN up-regulation had very similar effects with miR-23a knockdown in HK2 cells.

SnoN overexpression also inhibits HG-induced EMT and renal fibrogenesis in HK2 cells. HK2 cells were treated with HG (30 mM) for 24 h after transfection with pcDNA-SnoN or pcDNA3.1. a Protein expression of SnoN was determined by western blot. b Expressions of α-SMA, Vimentin and E-cadherin were determined by qRT-PCR, respectively. c mRNA expressions of FN and Col IV were determined by qRT-PCR, respectively. d Secreted levels of FN and Col IV in the culture medium were detected by ELISA assay. All data are presented as mean ± SEM, n = 4. **P < 0.01, ***P < 0.001 vs. NG; # P < 0.05, ## P < 0.01 vs. vehicle + HG
Down-regulation of SnoN partially blocked the effects of miR-23a knockdown on HG-induced EMT and renal fibrogenesis of HK2 cells
To demonstrate that miR-23a directly targeted SnoN to inhibit HG-induced EMT and renal fibrogenesis of HK2 cells, we cotransfected HK2 cells with miR-23a inhibitor and si-SnoN. We found that the protein level of SnoN was dramatically decreased after transfection with miR-23a inhibitor and si-SnoN compared with miR-23a inhibitor and si-NC in HK2 cells (Fig. 6a). Moreover, down-regulation of SnoN promoted HG-induced EMT in HK2 cells transfected with miR-23a inhibitor (Fig. 6b). Next, Our results showed that silencing of SnoN could reverse the inhibitory effect of miR-23a knockdown on HG-induced expressions of FN and Col IV in HK2 cells (Fig. 6c, d). Therefore, our data clearly confirmed that knockdown of miR-23a inhibited HG-induced EMT and renal fibrogenesis in HK2 cells by directly targeting SnoN, and overexpression of SnoN was essential for the miR-23a inhibitor-induced suppression of EMT and renal fibrogenesis in HK2 cells.

SnoN was involved in the effects of miR-23a on HG-induced EMT and renal fibrogenesis of HK2 cells. HK2 cells were transfected with either miR-23a inhibitor with si-SnoN, and then treated with HG (30 mM) for 24 h. a Protein expression of SnoN was determined by western blot. SnoN expression was normalized to α-tubulin. b Expressions of α-SMA, Vimentin, and E-cadherin were determined by qRT-PCR, respectively. c mRNA expressions of FN and Col IV were determined by qRT-PCR, respectively. d Secreted levels of FN and Col IV in the culture medium were detected by ELISA assay. All data are presented as mean ± SEM, n = 4. **P < 0.01, ***P < 0.001 vs. NG; # P < 0.05, ## P < 0.01 vs. vehicle + HG; & P < 0.05, && P < 0.01 vs. HG + miR-23a inhibitor
Discussion
Increasing studies have already confirmed a link between altered levels of miRNAs and many diseases [14]. Consequently, it is not surprising that miRNAs may also be involved in the development and progression of diabetic nephropathy. The role of miR-23a has been studied in some biological processes. miR-23a inhibits PTEN to promote activation of AKT/ERK pathways and EMT in osteosarcoma cells [15]. Introduction of miR-23a suppresses E-cadherin expression and stimulates EMT in A549 lung adenocarcinoma cells [16]. Conversely, overexpression of miR-23a in HEC-1-A cells increased E-cadherin expression and decreased the expression of vimentin and α-SMA, leading to suppression of EMT [17]. However, the roles of miR-23a in DN remain unclear. In this study, miR-23a was considered as a deregulated miRNA in HG-induced EMT and renal fibrogenesis. This miRNA may act as a promoter of HG-induced EMT and renal fibrogenesis, and it was found to be involved in the development and progression of DN by regulation of SnoN.
Diabetic nephropathy, one of the most common diabetic complications, is a chronic progressive kidney disease [18]. It is characterized by glomerular basement membrane thickening, mesangial expansion (hypertrophy), ECM accumulation, and renal fibrosis [19]. EMT is an essential process during the development and progress of DN. There is increasing interest in the role of miRNAs in all kinds of chronic diseases including diabetic complications. In this study, we investigated the potential role of miR-23a as regulator of SnoN during EMT and renal fibrogenesis in DN. We have demonstrated that knockdown of miR-23a was sufficient to suppress HG-induced EMT and fibrogenesis in HK2 cells, indicating that it is an important regulatory event in EMT changes and renal fibrogenesis. Moreover, since SnoN was a direct target of miR-23a and significantly decreased by treatment with HG, we found that SnoN overexpression could prevent HG-induced EMT and fibrogenesis in HK2 cells, suggesting that it was sufficient to regulate the progression of EMT and renal fibrogenesis and it might play an important role in preventing renal fibrosis in DN.
More and more studies demonstrated that EMT of the tubular epithelial cells plays a functional role in the occurrence and progression of DN and organ fibrosis including renal fibrosis [20,21,22]. In addition, the previous findings have demonstrated that inhibition of specific program of EMT in tubular epithelial cells markedly ameliorates renal fibrosis of different models including diabetes, emphasizing the crucial role of EMT in DN [23, 24]. Increasing reports demonstrated that specific miRNAs have critical roles in the progression of DN. For example, knockdown of miR-21 suppressed HG-induced production of fibrotic and inflammatory markers in kidney cells [25]. Xu et al. reported that HG down-regulated miR-181a-5p to increase pro-fibrotic gene expression by targeting the early growth response factor 1 in HK-2 cells [26]. It has been reported that miR-23a have different roles in the proliferation, invasion, and EMT of different cancer cells [27,28,29]. A previous study had demonstrated that miR-23a suppressed paclitaxel-induced apoptosis and promoted the cell proliferation and colony formation ability of gastric adenocarcinoma cells [27]. MiR-23a could function as a tumor suppressor in osteosarcoma, and suppressed proliferation of osteosarcoma cells by targeting SATB1 [28, 29]. In this study, since miR-23a was markedly increased in renal fibrosis tissues, we focused on it and found that its level was also significantly up-regulated in HK2 cells treated with HG. Furthermore, our results showed that miR-23a knockdown could suppress HG-induced EMT in HK2 cells. The most likely reason for this inconsistence is that there may be different molecular implications on miR-23a-regulated EMT in different cell types or diseases. Thus, the specific signaling pathways regulated by miR-23a in the EMT process of HK2 cells deserve to be further investigated. Furthermore, our data revealed that miR-23a could inhibit HG-induced SnoN expression, and down-regulation of miR-23a caused increased SnoN expression at protein level, demonstrating an inverse regulatory relationship between miR-23a and SnoN in HK2 cells. More importantly, our study has demonstrated for the first time that knockdown of miR-23a was sufficient to attenuate the effects of HG on EMT and renal fibrogenesis through the up-regulation of its downstream target SnoN in HK2 cells. SnoN has been also identified as a potential therapeutic target against renal fibrosis [30]. These results support our study which highlights the beneficial effect of miR-23a on regulation of renal fibrosis in DN.
The previous reports demonstrated that SnoN interacts with Smad to block TGF-β1/Smad signaling, leading to suppressing the transcriptional activation of TGF-β1 target genes [31]. Therefore, SnoN negatively regulates the physiological effects of TGF-β1 [32,33,34]. Besides, it has been reported that HG can increase the expression of TGF-β1 in kidney cells [35, 36]. In this study, we also confirmed that introduction of SnoN could attenuate HG-induced EMT and renal fibrogenesis in HK2 cells. Our results are consistent with the previous studies [30]. For further study, we demonstrated that down-regulation of SnoN significantly reversed the effects of miR-23a inhibitor on HG-induced EMT and renal fibrogenesis of HK2 cells. Altogether, we confirmed an inverse regulatory relationship between miR-23a and SnoN in kidney cells, and knockdown of miR-23a may provide an attractive approach for preventing renal fibrosis of DN.
In conclusion, we confirmed that miR-23a knockdown prevents HG-induced EMT and renal fibrogenesis in HK2 cells by regulation of SnoN expression. Further understanding of the functional significance of these antifibrotic miRNAs and their specific targets will provide viable therapeutic avenues for preventing renal fibrosis in DN.
References
Satirapoj B. Nephropathy in diabetes. Adv Exp Med Biol. 2012;771:107–22.
Sun YM, Su Y, Li J, et al. Recent advances in understanding the biochemical and molecular mechanism of diabetic nephropathy. Biochem Biophys Res Commun. 2013;433(4):359–61.
Kolset SO, Reinholt FP, Jenssen T. Diabetic nephropathy and extracellular matrix. J Histochem Cytochem. 2012;60(12):976–86.
Herbach N. Pathogenesis of diabetes mellitus and diabetic complications. Studies on diabetic mouse models. Der Pathol. 2012;2012(Suppl 2):318–24.
Liu Y. New insights into epithelial–mesenchymal transition in kidney fibrosis. J Am Soc Nephrol. 2010;21(2):212–22.
Bonventre JV. Can we target tubular damage to prevent renal function decline in diabetes? Semin Nephrol. 2012;32(5):452–562.
Burns WC, Twigg SM, Forbes JM, et al. Connective tissue growth factor plays an important role in advanced glycation end product-induced tubular epithelial-to-mesenchymal transition: implications for diabetic renal disease. J Am Soc Nephrol. 2006;17(9):2484–94.
Lv ZM, Wang Q, Wan Q, et al. The role of the p38 MAPK signaling pathway in high glucose-induced epithelial–mesenchymal transition of cultured human renal tubular epithelial cells. PLoS One. 2011;6(7):e22806.
Lee YJ, Han HJ. Troglitazone ameliorates high glucose-induced EMT and dysfunction of SGLTs through PI3K/Akt, GSK-3beta, Snail1, and beta-catenin in renal proximal tubule cells. Am J Physiol Renal Physiol. 2009;298(5):F1263–75.
Liu Y. Epithelial to mesenchymal transition in renal fibrogenesis: pathologic significance, molecular mechanism, and therapeutic intervention. J Am Soc Nephrol. 2004;15(1):1–12.
Esquela-Kerscher A, Slack FJ. Oncomirs—microRNAs with a role in cancer. Nat Rev Cancer. 2006;6(4):259–69.
Lagos-Quintana M, Rauhut R, Lendeckel W, et al. Identification of novel genes coding for small expressed RNAs. Science. 2001;294(5543):853–8.
Xiao C, Rajewsky K. MicroRNA control in the immune system: basic principles. Cell. 2009;136(1):26–36.
Mo YY. MicroRNA regulatory networks and human disease. Cell Mol Life Sci. 2012;69(21):3529–31.
Tian K, Di R, Wang L. MicroRNA-23a enhances migration and invasion through PTEN in osteosarcoma. Cancer Gene Ther. 2015;22(7):351–9.
Cao M, Seike M, Soeno C, et al. MiR-23a regulates TGF-β-induced epithelial–mesenchymal transition by targeting E-cadherin in lung cancer cells. Int J Oncol. 2012;41(3):869–75.
Liu P, Wang C, Ma C, et al. MicroRNA-23a regulates epithelial-to-mesenchymal transition in endometrial endometrioid adenocarcinoma by targeting SMAD3. Cancer Cell Int. 2016;16(1):67.
Ziyadeh FN. The extracellular matrix in diabetic nephropathy. Am J Kidney Dis. 1993;22(5):736–44.
Arai H. Diabetes mellitus related common medical disorders: recent progress in diagnosis and treatment topics: I. Pathophysiology, diagnosis and treatment: 9. Dyslipidemia. Nihon Naika Gakkai Zasshi. 2013;102(4):890–4.
Wei J, Shi Y, Hou Y, et al. Knockdown of thioredoxin-interacting protein ameliorates high glucose-induced epithelial to mesenchymal transition in renal tubular epithelial cells. Cell Signal. 2013;25(12):2788–96.
Kanlaya R, Khamchun S, Kapincharanon C, et al. Protective effect of epigallocatechin-3-gallate (EGCG) via Nrf2 pathway against oxalate-induced epithelial mesenchymal transition (EMT) of renal tubular cells. Sci Rep. 2016;6:30233.
Kalluri R, Weinberg RA. The basics of epithelial–mesenchymal transition. J Clin Investig. 2009;119(6):1420–8.
Hills CE, Squires PE. The role of TGF-β and epithelial-to mesenchymal transition in diabetic nephropathy. Cytokine Growth Factor Rev. 2011;22(3):131–9.
Wang B, Koh P, Winbanks C, et al. miR-200a prevents renal fibrogenesis through repression of TGF-β2 expression. Diabetes. 2011;60(1):280–7.
Zhong X, Chung AC, Chen HY, et al. miR-21 is a key therapeutic target for renal injury in a mouse model of type 2 diabetes. Diabetologia. 2013;56(3):663–74.
Xu P, Guan MP, Bi JG, et al. High glucose down-regulates microRNA-181a-5p to increase pro-fibrotic gene expression by targeting early growth response factor 1 in HK-2 cells. Cell Signal. 2017;31:96–104.
Liu X, Ru J, Zhang J, et al. miR-23a targets interferon regulatory factor 1 and modulates cellular proliferation and paclitaxel-induced apoptosis in gastric adenocarcinoma cells. PLoS One. 2013;8(6):e64707.
He Y, Meng C, Shao Z, et al. MiR-23a functions as a tumor suppressor in osteosarcoma. Cell Physiol Biochem. 2014;34(5):1485–96.
Wang G, Li B, Fu Y, et al. miR-23a suppresses proliferation of osteosarcoma cells by targeting SATB1. Tumour Biol. 2015;36(6):4715–21.
Liu L, Shi M, Wang Y, et al. SnoN upregulation ameliorates renal fibrosis in diabetic nephropathy. PLoS One. 2017;12(3):e0174471.
Zeglinski MR, Hnatowich M, Jassal DS, et al. SnoN as a novel negative regulator of TGF-β/Smad signaling: a target for tailoring organ fibrosis. Am J Physiol Heart Circ Physiol. 2015;308(2):H75–82.
Deheuninck J, Luo K. Ski and SnoN, potent negative regulators of TGF-beta signaling. Cell Res. 2009;19(1):47–57.
Luo K. Ski and SnoN: negative regulators of TGF-beta signaling. Curr Opin Genet Dev. 2004;14(1):65–70.
Tan R, Zhang J, Tan X, et al. Downregulation of SnoN expression in obstructive nephropathy is mediated by an enhanced ubiquitin-dependent degradation. J Am Soc Nephrol. 2006;17(10):2781–91.
Liu L, Wang Y, Yan R, et al. Oxymatrine inhibits renal tubular EMT induced by high glucose via upregulation of SnoN and inhibition of TGF-β1/Smad signaling pathway. PLoS One. 2016;11(3):e0151986.
Wang D, Guan MP, Zheng ZJ, et al. Transcription factor Egr1 is involved in high glucose-induced proliferation and fibrosis in rat glomerular mesangial cells. Cell Physiol Biochem. 2015;36(6):2093–107.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no competing interests.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Xu, H., Sun, F., Li, X. et al. Down-regulation of miR-23a inhibits high glucose-induced EMT and renal fibrogenesis by up-regulation of SnoN. Human Cell 31, 22–32 (2018). https://doi.org/10.1007/s13577-017-0180-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13577-017-0180-z