
Abstract
Introduction
Psoriasis involvement in special areas (e.g., scalp or nails) is associated with a great disease burden yet it is often inadequately treated with topical treatments. The efficacy and tolerability of apremilast plus existing topical therapy in Japanese patients with mild to moderate plaque psoriasis were demonstrated in PROMINENT, a phase 3b, multicenter, open-label, single-arm study. We evaluated the efficacy of apremilast across disease severities and special areas involved in these patients.
Methods
In PROMINENT, patients received apremilast 30 mg twice daily for 16 weeks in addition to their existing topical therapy, with the option of topical therapy reduction at the discretion of their physician while continuing apremilast treatment from Weeks 16 to 32. We performed a post hoc analysis, assessing apremilast efficacy and safety in Japanese patients stratified by baseline static Physician Global Assessment (sPGA) score (2 [mild] or 3 [moderate]) and special area involvement.
Results
Of patients with baseline sPGA = 2 and sPGA = 3, 62.7% and 30.7%, respectively, achieved sPGA score 0 or 1 at Week 32. At Week 32, improvements in skin, nail, scalp, and quality of life assessments were observed regardless of baseline sPGA score. Improvements in these endpoints at Week 32 were also observed in patients with special area (scalp or nail) involvement (n = 134). Incidence of adverse events was similar between patients with baseline sPGA = 2 and sPGA = 3.
Conclusions
Apremilast in combination with topical therapy may be a beneficial treatment for Japanese patients, who have limited systemic treatment options for mild to moderate psoriasis or psoriasis in special areas.
Trial Registration
NCT03930186.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
It is important to assess whether disease severity or location of psoriasis skin involvement affects the efficacy of psoriasis treatments. |
This analysis assessed the efficacy of apremilast in Japanese patients with mild to moderate psoriasis stratified by psoriasis disease severity and special area involvement. |
Improvements in overall disease severity and signs and symptoms of psoriasis (erythema, pustules, vesicles, and desquamation/scales) were seen in patients regardless of disease severity or special area involvement (scalp or nails) with apremilast treatment over 32 weeks. |
Apremilast in combination with topical therapy may be a beneficial treatment option for Japanese patients with mild to moderate psoriasis and patients with psoriasis in special areas. |
Introduction
In the UPLIFT survey of patients with psoriasis and/or psoriatic arthritis in Japan, approximately 43% of patients with limited skin involvement (body surface area [BSA] ≤ 3%) self-reported moderate or severe disease [1]. Most patients with BSA ≤ 3% also had psoriasis in special areas (including the scalp, face, palms or soles, nails, and genitals), and special area involvement was associated with a greater impact on quality of life. Despite this burden, many patients in UPLIFT reported receiving no treatment or topical treatment only.
Apremilast is an oral phosphodiesterase 4 inhibitor that has demonstrated efficacy and safety in treating moderate to severe psoriasis and mild to moderate psoriasis in global, randomized, placebo-controlled phase 3 clinical trials [2,3,4,5]. Quality of life and clinical outcomes have also been shown to improve with apremilast treatment in patients with psoriasis in special areas [6].
Apremilast is indicated in Japan for the treatment of patients with psoriasis vulgaris with an inadequate response to topical therapy [7]. PROMINENT was a phase 3b multicenter, open-label, single-arm study evaluating apremilast in Japanese patients with mild to moderate plaque psoriasis not adequately controlled by topical therapy alone. Apremilast in combination with existing topical therapy showed efficacy and tolerability over 32 weeks in PROMINENT [8]. Because psoriasis presents across severities and in different areas from one patient to another, there is an opportunity within the PROMINENT dataset to evaluate the efficacy of apremilast across disease severities and special areas involved in Japanese patients. This analysis assessed apremilast efficacy and safety over 32 weeks in patients stratified by baseline static Physician Global Assessment (sPGA) score (2 [mild] or 3 [moderate]) and special area involvement in PROMINENT.
Methods
Study Design
The study design and patient population have been described in detail [8]. Briefly, PROMINENT (NCT03930186) was a phase 3b, open-label, single-arm study conducted at 28 sites in Japan in patients with mild to moderate plaque psoriasis not adequately controlled by topical therapy alone. After a 5-day dose titration, patients received apremilast 30 mg orally twice daily (BID) in addition to existing stable topical therapy for 16 weeks. From Weeks 16 to 32, patients could reduce topical therapy at the physician’s discretion while maintaining apremilast 30 mg BID throughout.
The study was approved by the institutional review board/ethics committee before commencement and conducted in compliance with Good Clinical Practice, the International Council for Harmonisation Guideline E6, the Declaration of Helsinki, and applicable regulatory requirements. Patients provided written informed consent before study-related procedures.
Patients
Patients enrolled in PROMINENT were biologic-naive adults aged ≥ 20 years with chronic plaque psoriasis for ≥ 6 months before baseline and current treatment with topicals for ≥ 4 weeks before baseline. Patients had mild to moderate psoriasis (sPGA score of 2 [mild] or 3 [moderate]) that was inadequately controlled by topical therapy. Patients were excluded if they had prior exposure to biologics for treatment of psoriasis or had used conventional systemic therapy for psoriasis (e.g., cyclosporine, oral retinoids) within the past 8 weeks.
Assessments
The efficacy and safety results for the overall population have been published [8]. The primary endpoint in PROMINENT was achievement of sPGA score 0 (clear) or 1 (almost clear) at Week 16. A prespecified subgroup analysis of the primary endpoint was conducted at Week 16 to assess response rates by baseline disease severity (sPGA 2 or sPGA 3).
In a post hoc analysis, primary and secondary endpoints were evaluated at Weeks 16 and 32 in subgroups according to baseline disease severity (sPGA 2 or sPGA 3) and special area involvement (Scalp Physician’s Global Assessment [ScPGA] score ≥ 2 and Nail Psoriasis Severity Index [NAPSI] ≥ 1) at baseline. Assessments included the proportions of patients achieving ≥ 50% or ≥ 75% reduction from baseline in Psoriasis Area and Severity Index (PASI) score (PASI-50 and PASI-75), change from baseline in psoriasis-involved BSA, proportions of patients achieving ≥ 75% reduction from baseline in BSA (BSA-75), patients achieving ScPGA score of 0 or 1 (in patients with ScPGA score ≥ 2 at baseline), patients achieving a ≥ 50% reduction from baseline in NAPSI score (NAPSI-50; in patients with NAPSI ≥ 1 at baseline), mean percent change from baseline in pruritus visual analog scale (VAS), mean change from baseline in Shiratori’s Pruritus Severity Score (in daytime and at nighttime), mean change from baseline in Dermatology Life Quality Index (DLQI), the proportion of patients achieving a DLQI score of 0 or 1 (in patients with DLQI ≥ 2 at baseline), and patients achieving a Patient Benefit Index (PBI) ≥ 1.
The type, frequency, and severity of treatment-emergent adverse events (TEAEs) were assessed in subgroups according to baseline disease severity through Week 32.
Statistical Analysis
Descriptive statistics were used to summarize all analyses. No formal statistical testing was performed. Efficacy analyses were performed on the enrolled population, defined as all patients enrolled in the study. Safety analyses were performed on the safety population, defined as all patients who received ≥ 1 dose of the study drug. Nonresponder imputation was used for all categorical variables with missing data. For continuous variables, data are presented as observed.
Results
Baseline Characteristics
A total of 152 patients enrolled in the study, of which 140 (92.1%) completed 16 weeks of treatment. Of those, 136 patients (89.5%) completed 32 weeks of treatment. Of the 152 enrolled patients, 51 (33.6%) had a baseline sPGA score of 2 and 101 (66.4%) had a baseline sPGA score of 3 (Table 1). Mean age was similar between the subgroups (Table 1). Patients in the sPGA 3 group were more likely to be men and had a slightly longer duration of psoriasis compared with patients in the sPGA 2 group. Patients in the sPGA 3 group had more chronic, severe, and symptomatic psoriasis at baseline, with greater moderate to severe involvement of the scalp and a lower reported quality of life compared with patients in the sPGA 2 group. At baseline, approximately 84% of the overall population had scalp involvement (defined as ScPGA ≥ 2) and 50% had nail involvement (defined as NAPSI ≥ 1) [8]; these patients were included in the analysis of the subgroup of patients with special area involvement.
Primary Endpoint: sPGA Reduction
At Week 16, 62.7% of patients with a baseline sPGA 2 and 30.7% of patients with baseline sPGA 3 achieved sPGA 0 or 1 (Fig. 1). These results were maintained through Week 32 (sPGA 2: 62.7%; sPGA 3: 29.7%).

Proportions of patients achieving sPGA 0/1 over time. Enrolled population. Error bars represent 95% CI. Nonresponder imputation used for missing data. CI confidence interval sPGA static Physician Global Assessment
Skin Involvement
At Week 16, 90.2% of patients with baseline sPGA 2 and 74.3% of patients with baseline sPGA 3 achieved PASI-50 (Fig. 2a). These results were maintained through Week 32 (sPGA 2: 84.3%; sPGA 3: 70.3%). At Week 16, 52.9% of patients with baseline sPGA 2 and 38.6% with baseline sPGA 3 achieved PASI-75 (Fig. 2b). These results were maintained through Week 32 (sPGA 2: 58.8%; sPGA 3: 40.6%).

Improvements in skin involvement over time. a Achievement of PASI-50. b Achievement of PASI-75. c Change from baseline in BSA. d Achievement of BSA-75. Enrolled population. For figures a, b, and d, error bars represent 95% CI. For PASI-50, PASI-75, and BSA-75, nonresponder imputation used for missing data. For change in BSA, data as observed. BSA, body surface area; BSA-75, a ≥ 75% reduction from baseline in BSA; CI confidence interval; PASI psoriasis area and severity index; PASI-50/PASI-75, a ≥ 50%/ ≥ 75% reduction from baseline in PASI; SE standard error; sPGA static Physician Global Assessment
Mean BSA decreased by 70.3% in patients with baseline sPGA 2 and by 53.9% in patients with baseline sPGA 3 at Week 16 (Fig. 2c). Similar decreases for patients with sPGA 2 (68.0%) and patients with sPGA 3 (55.7%) were observed at Week 32. At Week 16, 56.9% of patients with baseline sPGA 2 and 30.7% with baseline sPGA 3 achieved BSA-75 (Fig. 2d). These results were maintained through Week 32 (sPGA 2: 51.0%; sPGA 3: 35.6%).
Itch
Mean pruritus VAS decreased by 13.2 mm in patients with baseline sPGA 2 and by 19.4 mm in patients with baseline sPGA 3 at Week 16 (Fig. 3a). Similar decreases for patients with baseline sPGA 2 (15.9 mm) and patients with baseline sPGA 3 (19.7 mm) were observed at Week 32. Mean change from baseline in Shiratori’s Pruritus Severity Score in daytime was − 0.6 in patients with baseline sPGA 2 and − 0.8 in patients with baseline sPGA 3 at Week 16 (Fig. 3b). Similar changes were seen for patients with baseline sPGA 2 (− 0.7) and patients with baseline sPGA 3 (− 0.7) at Week 32. Mean Shiratori’s Pruritus Severity Score at nighttime decreased by 0.4 in patients with baseline sPGA 2 and by 0.8 in patients with baseline sPGA 3 at Week 16 (Fig. 3c). Similar decreases were seen at Week 32 for patients with baseline sPGA 2 (0.6) and patients with baseline sPGA 3 (0.8).

Improvements in pruritus over time. a Change in pruritus VAS. b Change in Shiratori’s Pruritus Severity Score in daytime. c Change in Shiratori’s Pruritus Severity Score at nighttime. Enrolled population. Data as observed. VAS visual analog scale; SE standard error; sPGA static Physician Global Assessment
Psoriasis in Special Areas
At baseline, 78.4% (40/51) of patients in the sPGA 2 group and 87.1% (88/101) in the sPGA 3 group had an ScPGA score ≥ 2. Of these, 67.5% of patients in the sPGA 2 group and 45.5% of patients in the sPGA 3 group achieved ScPGA 0 or 1 at Week 16. These improvements in psoriasis were maintained at Week 32 (sPGA 2: 72.5%; sPGA 3: 40.9%) (Fig. 4).

Achievement of ScPGA 0/1 over time in patients with baseline ScPGA score ≥ 2. Enrolled population. Error bars represent 95% CI. Nonresponder imputation used for missing data. CI confidence interval; ScPGA scalp physician global assessment; sPGA static Physician Global Assessment
Among the population at baseline, 41.2% (21/51) of patients with sPGA 2 and 54.5% with sPGA 3 had NAPSI ≥ 1 (Table 1). At Week 16, 52.4% and 41.8% of patients in the sPGA 2 and sPGA 3 groups, respectively, achieved NAPSI-50. These response rates continued to increase through Week 32 for both groups (sPGA 2: 66.7%; sPGA 3: 54.5%) (Table 2).
Outcomes in Patients with Special Area Involvement at Baseline
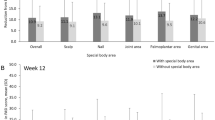
Most patients had involvement in at least one special area at baseline, regardless of psoriasis severity (sPGA 2: 42/51 [82.4%]; sPGA 3: 92/101 [91.1%]) (Supplementary Table S1). Improvements in sPGA, skin involvement (PASI-50, PASI-75), pruritus, scalp and nail involvement, and patient-reported outcomes were seen in patients with special area involvement, regardless of psoriasis severity, at Weeks 16 and 32 (Supplementary Table S2).
Patient-Reported Outcomes
Among patients with DLQI ≥ 2 at baseline, similar proportions of patients in the sPGA 2 and sPGA 3 groups achieved DLQI response at Week 16 (sPGA 2: 44.1%; sPGA 3: 40.0%) (Fig. 5a), and response was maintained at Week 32 (sPGA 2: 58.8%; sPGA 3: 36.5%). Mean DLQI decreased by 51.9% and 58.8% at Weeks 16 and 32, respectively, in the sPGA 2 group and by 44.8% and 41.5% in the sPGA 3 group (Fig. 5b). Similar proportions of patients achieved PBI ≥ 1 at Week 16 in the sPGA 2 (94.1%) and sPGA 3 (90.1%) groups, which was maintained through Week 32 (Fig. 5c).

Patient-reported outcomes over 32 weeks of apremilast treatment. a Achievement of DLQI 0/1 in patients with DLQI ≥ 2 at baseline. b Mean change from baseline in DLQI in the enrolled population. c Achievement of PBI ≥ 1 in the enrolled population. For DLQI 0/1 and PBI ≥ 1, nonresponder imputation was used for missing data. For change in DLQI, data are as observed. For figures a and c, error bars represent 95% CI. CI confidence interval; DLQI Dermatology Life Quality Index; PBI Patient Benefit Index; SE standard error; sPGA static Physician Global Assessment
Safety
Incidences of TEAEs through Week 32 were similar between patients with baseline sPGA 2 (72.5%) and baseline sPGA 3 (77.2%) (Table 3). Incidences of serious TEAEs and TEAEs leading to withdrawal were low in both groups. Serious TEAEs included gastroenteritis (n = 1), depression (n = 1), unilateral deafness (n = 1), increased blood creatine phosphokinase (n = 1), and thoracic vertebral fracture (n = 1). The most common (≥ 5%) TEAEs were nausea, diarrhea, nasopharyngitis, soft feces, and headache. No new safety findings relative to the known safety profile were observed.
Discussion
In this study of apremilast treatment in Japanese patients with mild to moderate psoriasis, improvements were seen regardless of baseline disease severity, with approximately 90% of all patients reporting treatment benefit as measured by PBI ≥ 1 throughout the study. Improvements in sPGA response, skin involvement, special area involvement, and patient-reported outcome measures at Week 16 were maintained through Week 32.
A trend toward greater responses in the population with mild disease was observed in the achievement of sPGA 0 or 1, ScPGA 0 or 1, and DLQI 0 or 1; sPGA 0 or 1 response rates at Weeks 16 and 32 in the mild (sPGA 2) group were twice those in the moderate (sPGA 3) group. The lower response rates in the sPGA 3 group are likely due to the higher baseline scores and the greater decreases needed to achieve response relative to patients with sPGA 2. For other efficacy measures such as PASI-75, PASI-50, and BSA-75, comparable improvements across sPGA 2 and sPGA 3 subgroups were observed.
Efficacy was consistent when evaluated in patients with special area involvement, with improvements in efficacy measures seen regardless of psoriasis severity at Weeks 16 and 32. Most patients in this study had special area involvement, which, per the International Psoriasis Council reclassification criteria, represent a population warranting systemic therapy given the inadequate response to topical therapy and impact of special areas on quality of life [9].
Results of this study in Japanese patients are consistent with other global studies of apremilast in patients with mild to moderate psoriasis inadequately controlled by topical therapy. In ADVANCE, a global phase 3 clinical study of apremilast in patients with mild to moderate psoriasis, significant improvements were observed in sPGA, ScPGA, BSA, PASI, and quality of life at 16 weeks compared with placebo and maintained at Week 32 [5, 10]. Similarly, improvements in these outcomes were observed over 32 weeks in PROMINENT. Although sPGA response with apremilast at Week 16 was higher in PROMINENT (62.7%, sPGA 2 group; 30.7%, sPGA 3 group) than in ADVANCE (21.6%), the primary endpoint in ADVANCE was slightly more stringent; an sPGA score of 0 (clear) or 1 (almost clear) and ≥ 2-point reduction from baseline. This may in part explain the higher sPGA response rate in PROMINENT. In addition, it should be noted that ADVANCE was a double-blind, placebo-controlled trial while PROMINENT was open-label. EMBRACE, a global, placebo-controlled, phase 4 study of apremilast in patients with limited skin involvement and plaque psoriasis in special areas, showed significant improvements in quality-of-life outcomes and skin involvement, including BSA, PASI, itch, and skin discomfort/pain, at 16 weeks compared with placebo [6]. Our study shows that these benefits were maintained up to 32 weeks in Japanese patients. Apremilast was well tolerated, and rates of TEAEs and the most common TEAEs were similar between the sPGA 2 and sPGA 3 groups. The adverse events were consistent with the known safety profile of apremilast [5, 6].
Apremilast may be useful in patients with milder disease to control disease activity. Psoriasis is marked by systemic inflammation irrespective of disease severity with a significant burden and may be inadequately controlled by topicals. This highlights the need to introduce systemic therapy early in the disease course. Indeed, we observed comparable improvements in quality of life (DLQI) and patient satisfaction (PBI) at Weeks 16 and 32 between patients with sPGA 2 and sPGA 3, reflecting the value of apremilast treatment across disease severity levels.
Limitations of the study include its open-label design, the lack of a placebo comparator, and the use of a post hoc analysis. In addition, discontinuations, and a focus on those with special area involvement, led to a loss of sample size in some analyses.
This study supports the addition of apremilast in patients not responding to topical therapy. Apremilast may potentially be a beneficial treatment option for Japanese patients who have limited systemic treatment options for mild to moderate psoriasis.
Conclusion
Apremilast was safe and effective in Japanese patients with mild disease and patients with psoriasis in special areas, demonstrating an effective systemic treatment option for patients not adequately controlled by topical therapy.
Data Availability
Qualified researchers may request data from Amgen clinical studies. Complete details are available at http://www.amgen.com/datasharing.
References
Torii H, Kishimoto M, Tanaka M, Noguchi H, Chaudhari S. Patient perceptions of psoriatic disease in Japan: results from the Japanese subgroup of the Understanding Psoriatic Disease Leveraging Insights for Treatment (UPLIFT) survey. J Dermatol. 2022;49:818–28.
Ohtsuki M, Okubo Y, Komine M, Imafuku S, Day RM, Chen P, et al. Apremilast, an oral phosphodiesterase 4 inhibitor, in the treatment of Japanese patients with moderate to severe plaque psoriasis: Efficacy, safety and tolerability results from a phase 2b randomized controlled trial. J Dermatol. 2017;44:873–84.
Papp K, Reich K, Leonardi CL, Kircik L, Chimenti S, Langley RG, et al. Apremilast, an oral phosphodiesterase 4 (PDE4) inhibitor, in patients with moderate to severe plaque psoriasis: results of a phase III, randomized, controlled trial (Efficacy and Safety Trial Evaluating the Effects of Apremilast in Psoriasis [ESTEEM 1]). J Am Acad Dermatol. 2015;73:37–49.
Paul C, Cather J, Gooderham M, Poulin Y, Mrowietz U, Ferrandiz C, et al. Efficacy and safety of apremilast, an oral phosphodiesterase 4 inhibitor, in patients with moderate to severe plaque psoriasis over 52 weeks: a phase III, randomized, controlled trial (ESTEEM 2). Br J Dermatol. 2015;173:1387–99.
Stein Gold L, Papp K, Leonardi C, Pariser D, Green L, Bhatia N, et al. Efficacy and safety of apremilast in patients with mild to moderate plaque psoriasis: results of a phase 3, multicenter, randomized, double-blind, placebo-controlled trial. J Am Acad Dermatol. 2022;86:77–85.
Mrowietz U, Barker J, Conrad C, Jullien D, Gisondi P, Flower A, et al. Efficacy and safety of apremilast in patients with limited skin involvement, plaque psoriasis in special areas, and impaired quality of life: results from the EMBRACE randomized trial. J Eur Acad Dermatol Venereol. 2023;37:348–55.
Otezla Japan [package insert]. Tokyo, Japan: Celgene; 2019 September 2019.
Okubo Y, Takahashi H, Hino R, Endo K, Kikuchi S, Ozeki Y, et al. Efficacy and safety of apremilast in the treatment of patients with mild-to-moderate psoriasis in Japan: results from PROMINENT, a phase 3b, open-label, single-arm study. Dermatol Ther (Heidelb). 2022;12:1469–80.
Strober B, Ryan C, van de Kerkhof P, van der Walt J, Kimball AB, Barker J, et al. Recategorization of psoriasis severity: Delphi consensus from the International Psoriasis Council. J Am Acad Dermatol. 2020;82:117–22.
Stein Gold L, Papp K, Pariser D, Bhatia N, Sofen H, Albrecht L, et al. Efficacy and safety of apremilast in patients with mild-to-moderate psoriasis up to 32 weeks: Results from the extension phase of the randomized, phase 3 ADVANCE trial. J Am Acad Dermatol. 2023;88:430–3.
Acknowledgements
We thank the patients and physicians who participated in the survey.
Medical Writing/Editorial Assistance
Writing support was funded by Amgen Inc. and provided by Kristin Carlin, BSPharm, MBA, of Peloton Advantage, LLC, an OPEN Health company, and Dawn Nicewarner, PhD, employee of and stockholder in Amgen Inc.
Funding
This study was sponsored by Amgen Inc., Thousand Oaks, CA, USA. Amgen is funding the Rapid Service Fee.
Author information
Authors and Affiliations
Contributions
All authors participated in drafting the work or revising it critically for important intellectual content and provided final approval. Conceptualization: Siddharth Chaudhari, Cynthia Deignan. Formal analysis and investigation [patient data collection/acquisition of data, and/or analysis and interpretation of data]: Masatoshi Abe, Yukari Okubo, Hidetoshi Takahashi, Koki Endo, Siddharth Chaudhari, Cynthia Deignan, Hamid Amouzadeh, Ryosuke Hino.
Corresponding author
Ethics declarations
Conflict of Interest
Masatoshi Abe: Celgene and Maruho – consultant, paid speaker, research grant, and/or participated in clinical trials. Yukari Okubo: Eisai, Maruho, Shiseido, and Torii – research grants; AbbVie, Amgen Inc., Boehringer Ingelheim, Bristol Myers Squibb, Celgene, Eisai, Eli Lilly, Janssen Pharma, Jimro, Kyowa Kirin, LEO Pharma, Maruho, Novartis Pharma, Pfizer, Sanofi, Sun Pharma, Taiho, Tanabe-Mitsubishi, Torii, and UCB – honoraria; AbbVie, Amgen, Boehringer Ingelheim, Bristol Myers Squibb, Celgene, Eli Lilly, Janssen Pharma, LEO Pharma, Maruho, Pfizer, Sun Pharma, and UCB – clinical trials. Hidetoshi Takahashi: AbbVie, Amgen Inc., Eli Lilly, Janssen, Kaken Pharmaceutical, Kyowa Kirin, LEO Pharma, Maruho, Mitsubishi Tanabe, Novartis, Sanofi, Sato Pharmaceutical, Sun Pharma, Taiho Pharmaceutical, Torii, and UCB – honoraria. Koki Endo: Nothing to disclose. Siddharth Chaudhari: Amgen K.K. – employment. Cynthia Deignan and Hamid Amouzadeh: Amgen Inc. – employees and stockholders. Ryosuke Hino: AbbVie, Amgen, Celgene, Eli Lilly, Janssen Pharma, Kyowa Kirin, LEO Pharma, Maruho, Novartis Pharma, Sanofi, Sun Pharma, Taiho, Tanabe-Mitsubishi, Torii Pharma, and UCB Pharma – honoraria; Amgen, Celgene, Eli Lilly, LEO Pharma, Maruho, Otsuka Pharma, and Sanofi – clinical trials.
Ethical Approval
The study was approved by the institutional review board/ethics committee before commencement and conducted in compliance with Good Clinical Practice, the International Council for Harmonisation Guideline E6, the Declaration of Helsinki, and applicable regulatory requirements. Patients provided written informed consent before study-related procedures.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Abe, M., Okubo, Y., Takahashi, H. et al. Consistent Efficacy of Apremilast in Patients with Psoriasis Regardless of Baseline Disease Severity or Special Area Involvement: Subgroup Analysis from PROMINENT. Dermatol Ther (Heidelb) 14, 1587–1597 (2024). https://doi.org/10.1007/s13555-024-01179-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13555-024-01179-z