
Abstract
Introduction
We evaluated the anti-interleukin-36 receptor antibody spesolimab in patients with moderate-to-severe palmoplantar pustulosis (PPP).
Methods
This phase IIb trial comprised a loading dose period to week (W) 4, then maintenance dosing to W52. Patients were randomised 2:1:1:1:2 to subcutaneous spesolimab 3000 mg to W4 then 600 mg every 4 weeks (q4w), spesolimab 3000 mg to W4 then 300 mg q4w, spesolimab 1500 mg to W4 then 600 mg q4w, spesolimab 1500 mg to W4, 300 mg q4w to W16 then 300 mg every 8 weeks (q8w), or placebo switching to spesolimab 600 mg q4w at W16. The primary efficacy endpoint was percentage change from baseline in Palmoplantar Pustular Area and Severity Index (PPP ASI) at W16. Secondary endpoints included a Palmoplantar Pustular Physician’s Global Assessment (PPP PGA) score of 0/1. Safety (including adverse events [AEs], local tolerability) was assessed.
Results
152 patients were treated. The primary endpoint was not met; mean differences for spesolimab versus placebo ranged from − 14.6% (95% confidence interval [CI]: − 31.5%, 2.2%) to − 5.3% (95% CI: − 19.1%, 8.6%); none reached significance. At W16, 23 (21.1%) and two (4.7%) patients in the combined spesolimab and placebo groups, respectively, achieved PPP PGA 0/1 (mean difference 16.4%; 95% CI: 3.8%, 25.7%), increasing to 59 (54.1%; combined spesolimab) and 12 (27.9%; placebo switch to spesolimab) patients at W52. Non-Asian patients had significant improvements in the primary endpoint (mean difference − 17.7%; nominal P = 0.0394) and PPP PGA 0/1 at W16 with spesolimab versus placebo. Rates of AEs and AE-related discontinuations were similar for spesolimab and placebo. Local tolerability events and injection-site reactions were more frequent with spesolimab than placebo.
Conclusion
The primary objective to demonstrate a non-flat dose–response relationship and proof-of-concept was not achieved; improvements with spesolimab occurred in secondary endpoints and in non-Asian patients, indicating potential modest benefits. Spesolimab was generally well tolerated (ClinicalTrials.gov NCT04015518).
Plain Language Summary
A clinical trial of spesolimab for patients with palmoplantar pustulosis. Palmoplantar pustulosis (PPP) is a painful, difficult-to-treat skin disease that is found on patients’ palms and the soles of their feet. In this clinical trial, we studied an injected medicine called spesolimab for treating patients with PPP. Patients were split into five groups; four groups received different doses of spesolimab and one received placebo (an injection without spesolimab). After 16 weeks, patients receiving placebo switched to spesolimab. We measured the body area affected by PPP and how severe PPP was at week 16. Patients’ doctors also assessed skin affected by PPP. At 16 weeks of treatment, there was no significant difference between spesolimab and placebo in terms of the PPP-affected area and severity. However, more patients had clear or almost clear skin with spesolimab than placebo. Among non-Asian patients, more showed an improvement in their PPP with spesolimab than with placebo; this was not the case with Asian patients. Patients taking spesolimab or placebo reported side effects, of which the most common were colds, aches and headaches. More patients receiving spesolimab reported a reaction at the injection site compared with placebo. We monitored patients for up to 1 year, and results remained similar. We showed that spesolimab may have a modest effect on the body area affected by PPP, as well as the severity of PPP, and did not seem to cause more side effects than placebo, except for reactions at the injection site.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Why carry out this study? |
Palmoplantar pustulosis (PPP) is difficult to treat and has a high unmet clinical need; interleukin-36 may play a role in PPP pathogenesis. |
Spesolimab is a humanised monoclonal anti-interleukin-36 receptor antibody; this 52-week, phase IIb trial evaluated its efficacy and safety in patients with moderate-to-severe PPP. |
What was learned from the study? |
The primary efficacy endpoint (percentage change in Palmoplantar Pustular Area and Severity Index at week 16) was not met; however, improvements in the secondary endpoint of a Palmoplantar Pustular Physician Global Assessment score of 0/1, and in both these endpoints in the non-Asian subgroup, were observed. |
Spesolimab was generally well tolerated after repeated dosing over 52 weeks, although a higher proportion of patients had localised injection-site reactions with high-dose spesolimab than with placebo. |
Overall, spesolimab showed modest improvements in efficacy endpoints in patients with PPP; further research to investigate the clinical benefit of spesolimab with respect to non-Asian ethnicity is warranted. |
Introduction
Palmoplantar pustulosis (PPP) is a painful, persistent, difficult-to-treat skin disease. It has a higher prevalence in women than in men, with an onset age of 44–65 years [1]. PPP is characterised by persistent macroscopically visible, sterile pustules on the palms or soles [2, 3], and can impair patient quality of life [4]. PPP co-occurs with plaque psoriasis in approximately 16% of patients [1]. Although PPP was previously considered a form of plaque psoriasis [2], it is now recognised as a distinct entity [5]. The 1-year prevalence of PPP has been estimated at < 0.1% in Western countries [6], and a prevalence of > 0.1% has been reported in Japan [7]. Two types of PPP have been identified: Type A is characterised by vesicles preceding pustules and is rarely associated with plaque psoriasis; Type B exhibits pustules without vesicles and is frequently associated with plaque psoriasis [3]. Type A has a higher prevalence than Type B in Japan [3].
Treatment of PPP is challenging. Topical treatment is hampered by the thicker stratum corneum of the palms and soles, resulting in only modest efficacy compared with that for plaque-type psoriasis [8]. In addition, evidence for the effectiveness of PPP treatments such as topical corticosteroids, acitretin, methotrexate, ciclosporin and phototherapy is often of relatively low quality [9]. Several biologics have been investigated in PPP; however, secukinumab showed only a modest benefit versus placebo at 16 weeks in a randomised controlled trial of predominantly white patients [10]. Similarly, smaller studies have reported modest or no clinical benefits with etanercept [11] and ustekinumab [12, 13], and a phase II exploratory study and case studies have shown clinical benefit with apremilast [14, 15]. Guselkumab, a monoclonal anti-interleukin (IL)-23 antibody, has shown efficacy in clinical trials and is approved for PPP treatment in Japan [16,17,18]. However, the difference between guselkumab and placebo with respect to the least-squares mean change from baseline in Palmoplantar Pustular Area and Severity Index (PPP ASI) score at week 16 of a phase III trial was only − 7.7 (guselkumab 100 mg) and − 4.1 (guselkumab 200 mg), suggesting relatively modest efficacy [18]. Therefore, an unmet clinical need exists for efficacious PPP treatments with an acceptable safety profile.
Mutations in the gene encoding the IL-36 receptor (IL-36R) antagonist (IL36RN) have been identified in a minority of patients with PPP [1]; the IL-36R agonist has also been detected in PPP lesions and could potentially drive neutrophil infiltration [19]. Spesolimab is a humanised monoclonal anti-IL-36R antibody that blocks IL-36R signalling [20] and has demonstrated rapid pustular and skin clearance in patients experiencing a generalized pustular psoriasis flare [21].
Here, we evaluate spesolimab efficacy and safety in a phase IIb trial of patients with moderate-to-severe PPP. Also, in light of the higher prevalence of PPP in Japan, differences in PPP characteristics between Japanese and non-Japanese populations, and guselkumab efficacy in Japanese patients, we performed an analysis of spesolimab efficacy stratified by Asian (primarily Japanese) and non-Asian patients. To date, this has not been performed in randomised controlled trials for other biologics such as secukinumab (European patients only) [22] and guselkumab (Japanese patients only) [17, 18].
Methods
Study Design
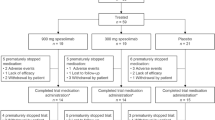
In this phase IIb, randomised, double-blind, placebo-controlled, parallel-design, dose-finding trial, patients were randomised 2:1:1:1:2 at baseline to one of five treatment groups (Fig. 1). Randomisation was performed using Interactive Response Technology with a block size of 7 and was stratified according to region, i.e. Japan versus non-Japan, to ensure sufficient Japanese patients to assess any differences in treatment effects between populations. Each group received a loading dose of subcutaneous (SC) spesolimab 1500 mg or 3000 mg, or placebo, delivered at day 1 and at weeks 1, 2, 3 and 4, followed by spesolimab 300 mg or 600 mg, or placebo. From week 16, patients receiving placebo switched to spesolimab 600 mg every 4 weeks (q4w); patients receiving spesolimab continued maintenance treatment q4w or every 8 weeks (q8w) (Fig. 1). Treatment ended at week 52, and patients underwent follow-up to week 68.

Study design. LD loading dose, q4w every 4 weeks, q8w every 8 weeks, SC subcutaneous, W week
Patients and investigators were blinded to treatment until database lock for the final trial analysis. The primary analysis was performed once patients had completed 16 weeks of treatment; at that time, the database was locked for primary analysis and treatment unblinded. The final analysis was performed once all patients had completed the trial. To confirm the integrity of the treatment blind while the trial was underway, a logistics plan was implemented to ensure that patients, investigators and trial country teams remained blinded to individual patient data and primary analysis results.
Study drugs were administered by the investigator or authorised trial personnel, avoiding areas of psoriasis or tender, bruised, erythematous or indurated skin.
The trial was conducted in compliance with the principles of the Declaration of Helsinki, in accordance with the International Council for Harmonisation Good Clinical Practice guidelines and with applicable regulatory requirements. Applicable independent ethics committees or institutional review boards at each country/participating centre (Table S1 in the electronic supplementary material, ESM) reviewed the trial protocol and informed consent form and granted approval. All patients provided written informed consent prior to participation. The trial is registered on Clinicaltrials.gov: NCT04015518.
Patients
Eligible patients were aged 18–75 years at screening, with a diagnosis of PPP defined as the presence of primary, persistent (> 3 months’ duration), sterile, macroscopically visible pustules on the palms or soles, with or without plaque psoriasis. At screening and baseline, patients were required to have a pustular severity score ≥ 2 in at least one region and ≥ 10 well-demarcated white or yellow pustules across all regions, a Palmoplantar Pustular Physician’s Global Assessment (PPP PGA) score of at least moderate severity (≥ 3) and a minimum PPP ASI score of 12. The main exclusion criteria were a reduction in PPP ASI total score ≥ 5 between screening and baseline visits (2–4 weeks apart); worsening plaque psoriasis within 3 months prior to screening (among those with plaque psoriasis); other skin conditions that affected the ability to score PPP ASI components; other severe, progressive or uncontrolled conditions; or a presence or history of anti-tumour necrosis factor-induced PPP-like disease. Full inclusion and exclusion criteria are listed in Table S2 of the ESM.
Concomitant topical treatment (including corticosteroids), systemic immunomodulatory treatments and biologics were not allowed during the trial except as rescue medication. Non-steroid anti-inflammatory drugs were allowed. Restricted medications are detailed in Table S3 of the ESM.
Study Endpoints
The primary efficacy endpoint was percentage change in PPP ASI from baseline at week 16. Secondary efficacy endpoints included percentage change in PPP ASI from baseline at week 52, a PPP PGA total score of clear/almost clear (score of 0/1) at week 16 and a PPP PGA pustulation subscore of clear/almost clear (score of 0/1) at week 16. Further efficacy endpoints included a PPP PGA total score of clear/almost clear (score of 0/1) over time and a PPP PGA pustulation subscore of clear/almost clear (score of 0/1) over time. Patient-reported outcomes included pain, recorded on a visual analogue scale (VAS), and Dermatology Life Quality Index (DLQI).
Safety assessments included adverse events (AEs), laboratory values, vital signs, electrocardiogram findings and local tolerability at the injection site. AEs were coded using the Medical Dictionary for Regulatory Activities (MedDRA) version 24.0, and severity was investigator-assessed according to Rheumatology Common Toxicity Criteria, version 2.0. A serious AE (SAE) was defined as an AE that resulted in death, persistent or significant disability or incapacity, was life threatening, required or prolonged hospitalisation, resulted in a congenital anomaly or birth defect, or was deemed serious for any other reason. AEs of special interest (AESIs) flagged by the investigator included systemic hypersensitivity reactions, severe infections, opportunistic and mycobacterium tuberculosis infections and hepatic injury. Local tolerability was assessed by the investigator according to ‘swelling’, ‘induration’, ‘heat’, ‘redness’, ‘pain’ or ‘other’ findings.
Statistical Analysis
The sample size was based on an assumed maximum difference in percentage change from baseline of 30% in PPP ASI for placebo versus spesolimab, with a standard deviation of 35%; a fixed rate of a − 25% PPP ASI change at week 16 was considered for placebo. These estimates were derived from a proof-of-concept study of spesolimab in PPP [23]. A total sample size of 140 evaluable patients was used to estimate the probability of a successful trial (i.e. a non-flat dose–response curve and effect difference versus placebo of ≥ 25%) using simulations based on a multiple comparison procedure with modelling techniques (MCPMod) approach. This approach specifies a set of candidate models and uses two stages: the ‘MCP’ stage looks for evidence of a dose response (the non-flat dose–response curve) and selects significant model(s); the ‘Mod’ stage then estimates the dose–response curve and the optimal dose based on the selected model(s) [24]. MCPMod has an advantage over traditional approaches in that very few, if any, assumptions about the underlying dose–response model are made, and any inference is not confined to the selection of the target dose among the dose levels being assessed [24]. Based on the above assumptions, the success probabilities for a difference of 20% and 25% were approximately 93–96% and 77–84% across dose–response models, respectively.
Efficacy analyses were performed on the full analysis set, which comprised all randomised patients who received ≥ 1 dose of study medication and had a baseline measure for the primary endpoint. Safety analyses were performed on the safety analysis set, which included all randomised patients who received ≥ 1 dose of study medication; patients were analysed according to the actual treatment they received.
The primary analysis comprised a combination of MCPMod-based testing (with respect to achieving a non-flat dose–response curve) and an evaluation of the dose-wise benefit at week 16. As a basis for the MCPMod analysis, a mixed model for repeated measurement was used to generate estimates for each treatment group, where the unstructured covariance structure was used to model within-patient measurements. The dose–response relationship at week 16 was then analysed using MCPMod to identify the best-fitting dose–response model while controlling for type I error at a one-sided 5% level.
Continuous secondary and further endpoints to week 16 were evaluated as per the primary analysis. The 95% confidence interval (CI) for single proportions of binary secondary and further endpoints was calculated using the Wilson method, and the 95% CI for treatment differences was based on the Wilson–Newcombe method. For binary secondary endpoints at week 16, a logistic regression and MCPMod analysis was used to evaluate treatment effects of spesolimab versus placebo. Missing data for continuous efficacy endpoints were handled via a missing at random assumption; for all binary efficacy endpoints, no response imputation was used as the primary imputation approach. Any data collected after the use of rescue therapy or 6 weeks after the last drug administration if a patient discontinued early (to allow for the continuing maximum effect period) were censored.
Data for the primary endpoint are presented for each treatment group up to week 16. As no significant dose–response relationship was identified for the primary endpoint, data were pooled from the spesolimab–low, spesolimab medium–low, spesolimab medium–high and spesolimab high groups to form the ‘combined spesolimab group’ for post-hoc analyses. The placebo group was termed the ‘placebo-to-spesolimab group’ when describing results after week 16.
The pre-specified stratified analyses for Asian and non-Asian subgroups were repeated to compare the combined spesolimab group with the placebo-to-spesolimab group. The Asian subgroup was primarily Japanese, but patients from the Republic of Korea and Taiwan were also included as their characteristics were similar to the Japanese patients.
The occurrence of treatment-emergent AEs was analysed descriptively.
Results
Study Participants
Between 26 August 2019 and 28 July 2021, 200 patients from 88 sites across 15 countries in Europe, Asia, North America and Australia were enrolled, 152 of whom were randomised to spesolimab high dose (n = 44), spesolimab medium–high dose (n = 22), spesolimab medium–low dose (n = 21), spesolimab low dose (n = 22) and placebo (n = 43). Patient demographics and baseline characteristics were generally well balanced between treatment groups (Table 1); most were female (72.4%), 52.4% were current smokers and the mean time since first PPP diagnosis was 5–10 years. In Asian patients, PPP was less frequently associated with plaque psoriasis than in non-Asian patients (8.3% [5/60] vs 30.4% [28/92]).
Overall, 139 patients (91.4%) completed their week 16 visit, 118 (77.6%) completed 52 weeks of treatment and 134 (88.2%) completed the follow-up at the end of the trial. The main reason for discontinuing treatment was AEs (n = 14; 9.2%) followed by lack of efficacy (n = 6; 3.9%) (Fig. S1 in the ESM).
In the Asian subgroup, 52 patients from Japan, seven from the Republic of Korea and one from Taiwan (a total of 60 patients) were enrolled and randomised.
Efficacy
At week 16, the adjusted mean percentage changes from baseline in PPP ASI were similar in the spesolimab low, medium–low and medium–high groups, and were numerically lower in the spesolimab high and placebo groups (Fig. 2a). The mean difference versus placebo was greatest in the spesolimab medium–low group (− 14.6%; 95% CI − 31.5%, 2.2%; P = 0.0883) and smallest in the spesolimab high group (− 5.3%; 95% CI − 19.1%, 8.6%; P = 0.4514). The difference versus placebo in the spesolimab medium–high group was − 12.6% (95% CI − 29.4%, 4.3%; P = 0.1414), and the spesolimab low group was − 10.5% (95% CI − 27.4%, 6.3%; P = 0.2179). No statistically significant differences were observed, and no significant dose–response model was identified. Therefore, the primary endpoint was not met.

Percentage change from baseline in PPP ASI a up to week 16 for each treatment group in all patients, and up to week 52 for the combined spesolimab group and placebo/placebo-to-spesolimab group in b non-Asian patients and c Asian patients. For panel a, means and CIs were estimated by (REML)-based MMRM including the fixed, categorical effects of treatment at each visit, region and the continuous effect of baseline at each visit as well as random effects of subject. For panels b and c, means and CIs were estimated by (REML)-based MMRM including the fixed, categorical effects of treatment-by-subgroup at each visit and the continuous effect of baseline at each visit as well as random effects of subject. aPatients received placebo up to week 16 before switching to spesolimab. CI confidence interval, MMRM mixed model repeated measures, PPP ASI Palmoplantar Pustular Area and Severity Index, REML restricted maximum likelihood, SE standard error
A post-hoc efficacy analysis was performed for the combined spesolimab groups versus placebo up to week 16 and for the combined spesolimab groups versus the placebo-to-spesolimab group after week 16. PPP ASI scores continued to decrease up to week 52 (Fig. S2 in the ESM), at which point the mean difference from baseline in PPP ASI score between the combined spesolimab group and placebo was − 15.0% (95% CI − 28.3%, − 1.8%).
When patients were stratified into Asian and non-Asian subgroups, the non-Asian combined spesolimab group showed a greater percentage decrease in PPP ASI from baseline to weeks 16 and 52 (Fig. 2b, c). At week 16 in non-Asian patients, the percentage decrease from baseline in PPP ASI was greater in the combined spesolimab group versus the placebo group (mean difference − 17.7%; 95% CI − 34.6%, − 0.9%; nominal P value = 0.0394; Fig. 2b). By contrast, there was no significant difference between the combined spesolimab and placebo groups in the Asian subgroup at week 16 (mean difference 2.7%; 95% CI − 12.6%, 18.0%; nominal P value = 0.7269; Fig. 2c). At week 52, the difference between the combined spesolimab and placebo-to-spesolimab groups increased to − 25.7% (95% CI − 45.4%, − 6.1%) in non-Asian patients, whereas the difference between these groups in Asian patients was only − 3.2% (95% CI − 20.8%, 14.3%).
At both weeks 16 and 52, a greater proportion of patients had a PPP PGA total score of 0/1 (clear/almost clear) in the combined spesolimab group versus the placebo (up to week 16) and placebo-to-spesolimab (after week 16) groups (Fig. 3a); the 95% CI of the mean difference between the groups does not include zero, which indicates that this difference reached nominal statistical significance. Patients were then stratified by Asian ethnicity; in the non-Asian subgroup, a greater proportion of patients achieved a PPP PGA total score of 0/1 in the combined spesolimab group versus the placebo-to-spesolimab group at week 52 (Fig. 3b); this difference was not observed in Asian patients (Fig. 3c).

Proportion of patients achieving PPP PGA of 0/1 at weeks 16 and 52 in a all patients, b non-Asian patients and c Asian patients. aPatients received placebo up to week 16 before switching to spesolimab. CI confidence interval, n number of patients with a PPP PGA of 0/1, N number of patients evaluated, PPP PGA Palmoplantar Pustular Physician Global Assessment
A greater proportion of patients in the combined spesolimab group achieved a PPP PGA pustulation subscore of 0/1 (clear/almost clear) at week 16 (32.1% [35/109]) versus the placebo group (11.6% [5/43]; difference 20.5%; 95% CI: 5.3%, 31.8%; Fig. S3a in the ESM). At week 52, this increased to 57.8% (63/109) and 32.6% (14/43) of patients in the combined spesolimab and placebo groups, respectively (difference 25.2%; 95% CI: 7.6%, 40.2%; Fig. S3a in the ESM). Non-Asian patients showed the greatest differences in PPP PGA pustulation subscore between the combined spesolimab and placebo-to-spesolimab groups; the differences, 30.3% (95% CI: 11.7%, 42.7%) and 40.2% (95% CI: 17.4%, 56.5%) at weeks 16 and 52, respectively, were statistically significant (nominal significance indicated by the lack of inclusion of zero in the 95% CI of the difference; Fig. S3b in the ESM). By contrast, no notable difference between the treatment groups was observed in Asian patients (difference was 6.3% [95% CI − 19.3%, 26.3%] at week 16 and 3.2% [95% CI − 22.9%, 27.9%] at week 52; Fig. S3c in the ESM).
Patient-Reported Outcomes
Patients reported improvements in their pain VAS at week 16 in both the combined spesolimab and the placebo groups; pain VAS scores continued to improve to week 52 in both groups (Fig. 4a). A non-significant difference was observed between the combined spesolimab and placebo groups at week 16, as the 95% CI of the mean difference between groups included zero.

Mean absolute change from baseline in a pain VAS and b DLQI total score at weeks 16 and 52. Means and CIs were estimated by (REML)-based MMRM including the fixed, categorical effects of treatment at each visit, region and the continuous effect of baseline at each visit as well as random effects of subject. aPatients received placebo up to week 16 before switching to spesolimab. CI confidence interval, DLQI Dermatology Life Quality Index, MMRM mixed model repeated measures, REML restricted maximum likelihood, VAS visual analogue scale
DLQI total scores improved over 52 weeks in the combined spesolimab and placebo-to-spesolimab groups (Fig. 4b). However, no notable difference between these groups was observed at week 16.
Safety
The overall AE rate was similar across all treatment groups up to week 16, and most AEs were of mild or moderate intensity (Table 2). The most common AEs were a localised injection-site reaction, injection-site erythema, nasopharyngitis and arthralgia (Table 3). The rates of infections/infestations were similar in patients treated with spesolimab (27 [24.8%]) and placebo (14 [32.6%]). In contrast, more patients with spesolimab reported localised injection-site reactions (such as erythema, pain, swelling or pruritus; n = 37; 33.9%) compared with placebo (n = 4; 9.3%). The incidence of these reactions appeared highest in the spesolimab high dose group, and they were the most common investigator-defined treatment-related AEs (Table 3).
Up to week 16, five (4.6%) patients receiving spesolimab and two (4.7%) receiving placebo experienced an SAE (Table 2). Each SAE preferred term was reported in only one patient, and none were considered treatment-related. Five (4.6%) patients receiving spesolimab and five (11.6%) receiving placebo discontinued treatment because of AEs (Table 2); worsening PPP was the most common AE leading to discontinuation (n = 1 with spesolimab medium–high, n = 2 with spesolimab high; n = 3 with placebo); all others were reported in one patient each. Two patients discontinued treatment due to SAEs: one due to retinal artery embolism (spesolimab low) and one due to prostate cancer (placebo). The rates of severe AEs were similar between the combined spesolimab and placebo groups (6.4% and 7.0%, respectively; Table 2); worsening PPP was reported in the spesolimab medium–high and high groups (n = 1 each); all others were reported in one patient each. One patient receiving placebo had AESIs of herpes zoster and mycobacterium tuberculosis. Local tolerability signs and symptoms occurred in eight (18.2%) patients in the spesolimab high group and in five (11.6%) in the placebo group (Table 2); all were grade 1 or 2. No deaths occurred up to week 16.
The overall rates of AEs reported after week 16 are summarised in Table S4 in the ESM. The types of AEs experienced were similar to those up to week 16 and, with the exception of localised injection-site reactions, the frequencies for most AEs were similar across the treatment groups (Table S5 in the ESM). The most common investigator-defined treatment-related AEs were localised injection-site reaction, injection-site erythema and injection-site pain (Table S5 in the ESM).
After week 16, 13 (9.4%) patients experienced SAEs (Table S4 in the ESM); SAE preferred terms were reported in one patient each. Two patients in the spesolimab high group experienced SAEs that were considered treatment-related: basal cell carcinoma (n = 1), and dyshidrotic eczema, worsening PPP and pustular psoriasis (n = 1). Four patients had AEs leading to treatment discontinuation. Three were SAEs: colon cancer in the spesolimab medium–high group (n = 1), psoriatic arthropathy in the placebo-to-spesolimab group (n = 1), and dyshidrotic eczema, worsening PPP and pustular psoriasis in the spesolimab high group (n = 1). The non-serious AE leading to treatment discontinuation was pustulotic arthro-osteitis in the spesolimab medium–high group (n = 1). Fourteen (10.1%) patients experienced severe AEs (Table S4 in the ESM); all preferred terms were reported in one patient each. All AESIs after week 16 were infections. In patients receiving spesolimab, 22.7–52.3% experienced a local tolerability sign or symptom versus 18.6% who switched from placebo to spesolimab. Nearly all were grade 1 or 2 (Table S4 in the ESM). No deaths were reported after week 16.
No relevant differences in laboratory parameters between treatment groups, nor any marked changes in mean values for any parameter or vital signs over time, were observed. No relevant differences in neutrophil counts, eosinophil counts or liver enzyme levels were observed between treatment groups, and very few patients developed abnormalities of potential clinical significance in these parameters (Table S6 in the ESM).
Discussion
The primary endpoint of this trial, the percentage change in PPP ASI from baseline at week 16, has also been used in other clinical trials of biologics [10, 11, 13, 18, 23, 25], although it is not validated and scores may differ between trials. Here, the primary endpoint was not met, and no dose–response relationship was identified between spesolimab doses and placebo. However, clinical benefit was observed in binary secondary endpoints; furthermore, numerical differences in PPP ASI from baseline over 52 weeks favoured spesolimab, as observed in other trials in PPP [10, 18], which indicates that responses to biologics develop over time. PPP ASI scores decreased from baseline to week 16 with placebo, suggesting a considerable placebo response, as seen in other PPP clinical trials [10, 23]. The fluctuant nature of and spontaneous improvements in PPP may have masked any effects of spesolimab during the trial, although patients demonstrating a PPP ASI total score reduction ≥ 5 between screening and baseline were excluded to minimise a placebo response. By contrast, a higher proportion of patients in the combined spesolimab group achieved a PPP PGA total score or pustulation subscore of clear/almost clear compared with the placebo-to-spesolimab group, with the greatest difference observed at week 52. Modest benefits were observed with spesolimab in pain and health-related quality of life at weeks 16 and 52. Therefore, we cannot exclude the possibility that spesolimab has a modest effect on PPP, with a benefit that continues to accumulate after the 16-week placebo-controlled period. Further studies with active comparators, the collection of skin biopsies and longer placebo-controlled periods would provide greater insights into the effects of spesolimab versus natural resolution or placebo.
Our results revealed that ethnicity may affect spesolimab efficacy. Non-Asian patients in the combined spesolimab group showed improvements in PPP ASI scores, and higher proportions achieved PPP PGA and PPP PGA pustulation scores of clear/almost clear, versus the placebo-to-spesolimab group. This was not observed in the Asian (primarily Japanese) subgroup, so it is possible that the spesolimab efficacy seen in the overall trial population was driven by the non-Asian patients. The high placebo response for PPP ASI observed in Asian patients differed from that in non-Asian patients and could partially explain the lack of nominal statistical significance. The reason behind this apparent difference between Asian and non-Asian populations is unknown and warrants further investigation in a sufficiently powered trial, including a potential subanalysis of responders and non-responders in the Asian population to understand any factors contributing to efficacy. Variations in gene expression profiles (e.g. IL36RN and IL-36 alpha, beta and gamma [IL36A, IL36B, IL36G]) represent one possibility, or Type A (more prevalent in Japan) and Type B PPP might respond differently to spesolimab. Alternatively, cultural differences regarding recruitment could have played a role.
For the primary analysis, the four spesolimab dose regimens were evaluated separately versus placebo, as it was anticipated that the different drug exposures would lead to differences in the primary efficacy endpoint. However, the smallest difference versus placebo in the primary endpoint was seen with the highest spesolimab dose, indicating no spesolimab dose–response relationship. Subsequent combining of spesolimab dose groups to perform post-hoc analyses of the secondary endpoints was considered a conservative way to assess efficacy compared with using the best-performing spesolimab dose group.
Spesolimab was well tolerated. In a phase IIa study of spesolimab in PPP, AE and SAE incidence rates were 89.5% and 2.6%, respectively [23]. These rates of AEs and SAEs were in line with those observed in the combined spesolimab group at week 16 (73.4% and 4.6%, respectively). Local tolerability symptoms and injection-site reactions in the present trial were more frequent with spesolimab than with placebo.
A strength of this trial is that it is the first global clinical trial in PPP to compare efficacy outcomes in Asian and non-Asian patients. Although the primary efficacy endpoint was not met, a notable clinical benefit was evident with other efficacy measures, particularly in non-Asian patients. The 1-year duration of the trial expanded our knowledge of the spesolimab safety profile; no significant toxicity or compromised immunity was observed when the drug was administered repeatedly over a prolonged period. The trial was limited by the small sample size in each dose group and the primary endpoint (PPP ASI) not being a validated measure. In addition, there was no subgroup analysis conducted to see if there was any difference in spesolimab efficacy in patients with Type A or Type B PPP. However, as the presentation of PPP in Japan is consistent with Type A PPP [3], and as 52/60 Asian patients were Japanese, the Asian/non-Asian analysis suggests that there are two types of PPP, and the association between Type A/B phenotypes and Asian/non-Asian patients with PPP requires further investigation. Also, skin biopsies were not available for many patients, precluding any gene expression analysis.
Conclusion
In this trial of spesolimab in patients with moderate-to-severe PPP, the primary endpoint (percentage change in PPP ASI from baseline at week 16) for comparison with placebo was not met and no dose–response relationship was identified. However, non-significant improvements from baseline in PPP ASI and nominally significant improvements in PPP PGA scores were observed with spesolimab. This trial provides valuable long-term evidence in addition to previous reports that spesolimab is generally well tolerated following repeated administration over 1 year, albeit with a higher rate of localised injection-site reactions. These results are consistent with two previous studies of anti-IL-36R biologics in PPP, suggesting modest potential benefits of this treatment approach. The apparent difference between non-Asian and Asian populations warrants further investigation.
References
Twelves S, Mostafa A, Dand N, et al. Clinical and genetic differences between pustular psoriasis subtypes. J Allergy Clin Immunol. 2019;143(3):1021–6.
Navarini AA, Burden AD, Capon F, et al. European consensus statement on phenotypes of pustular psoriasis. J Eur Acad Dermatol Venereol. 2017;31(11):1792–9.
Murakami M, Terui T. Palmoplantar pustulosis: current understanding of disease definition and pathomechanism. J Dermatol Sci. 2020;98(1):13–9.
Trattner H, Bluml S, Steiner I, et al. Quality of life and comorbidities in palmoplantar pustulosis—a cross-sectional study on 102 patients. J Eur Acad Dermatol Venereol. 2017;31(10):1681–5.
Bissonnette R, Suarez-Farinas M, Li X, et al. Based on molecular profiling of gene expression, palmoplantar pustulosis and palmoplantar pustular psoriasis are highly related diseases that appear to be distinct from psoriasis vulgaris. PLoS One. 2016;11(5):e0155215.
Andersen YMF, Augustin M, Petersen J, et al. Characteristics and prevalence of plaque psoriasis in patients with palmoplantar pustulosis. Br J Dermatol. 2019;181(5):976–82.
Kubota K, Kamijima Y, Sato T, et al. Epidemiology of psoriasis and palmoplantar pustulosis: a nationwide study using the Japanese national claims database. BMJ Open. 2015;5(1):e006450.
Raposo I, Torres T. Palmoplantar psoriasis and palmoplantar pustulosis: current treatment and future prospects. Am J Clin Dermatol. 2016;17(4):349–58.
Obeid G, Do G, Kirby L, et al. Interventions for chronic palmoplantar pustulosis. Cochrane Database Syst Rev. 2020;1:CD011628.
Mrowietz U, Bachelez H, Burden AD, et al. Secukinumab for moderate-to-severe palmoplantar pustular psoriasis: results of the 2PRECISE study. J Am Acad Dermatol. 2019;80(5):1344–52.
Bissonnette R, Poulin Y, Bolduc C, et al. Etanercept in the treatment of palmoplantar pustulosis. J Drugs Dermatol. 2008;7(10):940–6.
Bertelsen T, Kragballe K, Johansen C, et al. Efficacy of ustekinumab in palmoplantar pustulosis and palmoplantar pustular psoriasis. Int J Dermatol. 2014;53(10):e464–6.
Bissonnette R, Nigen S, Langley RG, et al. Increased expression of IL-17A and limited involvement of IL-23 in patients with palmo-plantar (PP) pustular psoriasis or PP pustulosis; results from a randomised controlled trial. J Eur Acad Dermatol Venereol. 2014;28(10):1298–305.
Dorgham N, Crasto D, Skopit S. Successful treatment of palmoplantar pustulosis with apremilast. J Drugs Dermatol. 2021;20(11):1255–6.
Wilsmann-Theis D, Kromer C, Gerdes S, et al. A multicentre open-label study of apremilast in palmoplantar pustulosis (APLANTUS). J Eur Acad Dermatol Venereol. 2021;35(10):2045–50.
Yamamoto T. Guselkumab for the treatment of palmoplantar pustulosis: a Japanese perspective. Clin Pharmacol. 2021;13:135–43.
Terui T, Kobayashi S, Okubo Y, et al. Efficacy and safety of guselkumab, an anti-interleukin 23 monoclonal antibody, for palmoplantar pustulosis: a randomized clinical trial. JAMA Dermatol. 2018;154(3):309–16.
Terui T, Kobayashi S, Okubo Y, et al. Efficacy and safety of guselkumab in Japanese patients with palmoplantar pustulosis: a phase 3 randomized clinical trial. JAMA Dermatol. 2019;155(10):1153–61.
Johnston A, Xing X, Wolterink L, et al. IL-1 and IL-36 are the dominant cytokines in palmar plantar pustulosis. J Dermatol Sci. 2016;84:e99.
Ganesan R, Raymond EL, Mennerich D, et al. Generation and functional characterization of anti-human and anti-mouse IL-36R antagonist monoclonal antibodies. MAbs. 2017;9(7):1143–54.
Bachelez H, Choon SE, Marrakchi S, et al. Trial of spesolimab for generalized pustular psoriasis. N Engl J Med. 2021;385(26):2431–40.
ClinicalTrials.gov. NCT02008890. Palmoplantar pustular psoriasis efficacy and safety with secukinumab. 2019. https://clinicaltrials.gov/ct2/show/NCT02008890. Accessed 23 June 2023.
Mrowietz U, Burden AD, Pinter A, et al. Spesolimab, an anti-interleukin-36 receptor antibody, in patients with palmoplantar pustulosis: results of a phase IIa, multicenter, double-blind, randomized, placebo-controlled pilot study. Dermatol Ther (Heidelb). 2021;11(2):571–85.
Bretz F, Pinheiro JC, Branson M. Combining multiple comparisons and modeling techniques in dose-response studies. Biometrics. 2005;61(3):738–48.
AnaptysBio. AnaptysBio reports imsidolimab POPLAR phase 2 clinical trial in moderate-to-severe palmoplantar pustulosis (PPP) did not meet primary endpoint. San Diego: AnaptysBio, Inc.; 2021. https://ir.anaptysbio.com/news-releases/news-release-details/anaptysbio-reports-imsidolimab-poplar-phase-2-clinical-trial. Accessed 23 June 2023.
Acknowledgements
The authors would like to thank the study participants for their involvement in the study.
Medical Writing/Editorial Assistance
Kate Silverthorne, PhD, of OPEN Health Communications (London, UK) provided writing, editorial and formatting support, which was contracted and funded by Boehringer Ingelheim.
Author Contributions
A. David Burden, Binqi Ye, Frank Baehner and Tadashi Terui participated in the design and conduct of the trial. Robert Bissonnette, Alexander A. Navarini, Masamoto Murakami, Akimichi Morita and Tadashi Terui participated in data acquisition. Binqi Ye planned and performed the statistical analysis of the data. All authors participated in the interpretation of the trial data. All authors contributed to the writing and reviewing of the manuscript and all authors approved the final manuscript.
Funding
The trial was supported and funded by Boehringer Ingelheim GmbH, Ingelheim, Germany, along with the rapid service fee to publish in Dermatology and Therapy.
Data Availability
The datasets generated during and/or analysed during the current study are available from Boehringer Ingelheim on reasonable request. To ensure independent interpretation of clinical study results and enable authors to fulfil their role and obligations under the International Committee of Medical Journal Editors (ICMJE) criteria, Boehringer Ingelheim grants all external authors access to clinical study data pertinent to the development of the publication. In adherence with the Boehringer Ingelheim Policy on Transparency and Publication of Clinical Study Data, scientific and medical researchers can request access to clinical study data when it becomes available on Vivli—Center for Global Clinical Research Data (https://vivli.org/), and earliest after publication of the primary manuscript in a peer-reviewed journal, regulatory activities are complete and other criteria are met. Please visit Medical & Clinical Trials|Clinical Research|MyStudyWindow (https://www.mystudywindow.com/msw/datasharing) for further information.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
ADB reports receiving consulting fees from AbbVie, Almirall, BMS, Boehringer Ingelheim, Celgene, Bristol Myers Squibb, Eli Lilly, Janssen, LEO Pharma, Novartis and Union Chimique Belge (UCB) and payment or honoraria for lectures and presentations from Almirall, Boehringer Ingelheim, Eli Lilly and Janssen. RB is an advisory board member, consultant, speaker or investigator and/or received honoraria/grants from AbbVie, Almirall, AnaptysBio, Arcutis, Aristea, Bausch Health/Valeant, Boehringer Ingelheim, Boston Pharma, BMS, Dermavant, Eli Lilly, Escalier, Janssen, Kineta, Kyowa Kirin, LEO Pharma, Pfizer, Regeneron, Sienna and UCB and is also an employee and shareholder of Innovaderm Research. AAN declares being a consultant and advisor and/or receiving speaking fees/grants and/or served as an investigator in clinical trials for AbbVie, Almirall, Amgen, BMS, Boehringer Ingelheim, Celgene, Eli Lilly, Galderma, GlaxoSmithKline, LEO Pharma, Janssen-Cilag, Merck Sharp & Dohme, Novartis, Pfizer, Sandoz, Sanofi, Serono and UCB. MM reports receiving research grants and/or honoraria and/or serving as an investigator in clinical trials for AbbVie, Amgen, Aristea Therapeutics, BMS, Boehringer Ingelheim, Celgene, Eisai, Eli Lilly, Janssen, Kyowa Kirin, Maruho, Novartis, Taiho and Torii. AM reports receiving research grants, consulting fees and/or speaker’s fees from AbbVie, Boehringer Ingelheim, Celgene, Eisai, Eli Lilly, Janssen, Kyowa Hakko Kirin, LEO Pharma, Maruho, Mitsubishi Tanabe, Nichi-Iko, Nippon Kayaku, Novartis, Sun Pharmaceutical Industries, Taiho Pharmaceutical, Torii Pharmaceutical and Ushio. TT has received honoraria for speaking and consultancy from AbbVie, Eisai, Novartis, Janssen Pharmaceutical, Maruho, Taiho, Eli Lilly, BMS and Mitsubishi Tanabe Pharma. TH, BY and FB are employees of Boehringer Ingelheim. The authors met criteria for authorship as recommended by the ICMJE. The authors did not receive payment related to the development of this manuscript. Boehringer Ingelheim was given the opportunity to review the manuscript for medical and scientific accuracy as well as intellectual property considerations.
Ethical Approval
The trial was conducted in compliance with the principles of the Declaration of Helsinki, in accordance with the International Council for Harmonisation Good Clinical Practice guidelines and with applicable regulatory requirements. Applicable independent ethics committees or institutional review boards at each country/participating centre (Table S1 in the ESM) reviewed the trial protocol and informed consent form and granted approval. All patients provided written informed consent prior to participation. The trial is registered on Clinicaltrials.gov: NCT04015518.
Additional information
Prior publication/presentation: this study was previously presented as a poster at the American Academy of Dermatology Annual Meeting, Boston, MA, USA, on 25–29 March 2022; presentation number 32923.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Burden, A.D., Bissonnette, R., Navarini, A.A. et al. Spesolimab Efficacy and Safety in Patients with Moderate-to-Severe Palmoplantar Pustulosis: A Multicentre, Double-Blind, Randomised, Placebo-Controlled, Phase IIb, Dose-Finding Study. Dermatol Ther (Heidelb) 13, 2279–2297 (2023). https://doi.org/10.1007/s13555-023-01002-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13555-023-01002-1