
Abstract
Atopic dermatitis (AD) is a chronic or chronically relapsing inflammatory skin disease which results from a complex, multifaceted interaction between environmental factors in genetically predisposed patients. Epidermal barrier impairment, alteration of the cutaneous microbiota, effect of external antigens, neurosensory dysfunction, and inflammatory and immune dysregulation all play a pivotal role in inducing and maintaining AD lesions. AD significantly impacts the patient’s quality of life and general well-being and is often associated with anxiety and/or depressive symptoms. Classical treatment options include topical corticosteroids and calcineurin inhibitors, phototherapy, and systemic immunosuppression with oral corticosteroids, cyclosporine, methotrexate, and azathioprine in more severe cases. A turning point in facing AD was accomplished when the efficacy and safety of dupilumab, a monoclonal antibody targeting the interleukin (IL)-4 receptor α subunit, led to its approval for the treatment of moderate-to-severe or severe AD in children, adolescents, and adults. Subsequently, a more extensive understanding of AD etiology and pathogenesis has allowed the development of several topical and systemic novel therapy options. Most of these drugs are monoclonal antibodies which interfere with the type 2 inflammatory cascade, especially its key cytokines IL-4 and IL-13, or its downstream Janus kinase signaling pathway. However, considering the relevance of other subtypes of T helper (Th) cells, such as Th1 and Th22, and the important role of specific cytokines (IL-31) in generating pruritus, the horizon of potential therapeutic targets has widened extremely. In this review, we aim to present the most promising systemic agents currently under investigation and illustrate the most significant aspects of their efficacy, safety, and tolerability.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Atopic dermatitis is a complex, multifactorial inflammatory skin disease resulting from the interaction between genetic predisposition, alteration of the skin barrier and cutaneous microbiota, and environmental factors. |
Up till recently, the most effective treatment options included topical and systemic corticosteroids, topical calcineurin inhibitors, phototherapy, and systemic immunosuppressants such as cyclosporine. |
In recent years, the efficacy and safety of dupilumab, a fully human monoclonal antibody which specifically targets the interleukin (IL)-4/IL-13 pathway, have revolutionized the approach to treatment. |
Better understanding of disease etiopathogenesis has led to the development of several drugs, in particular monoclonal antibodies and small molecules, directed against cytokines or specific receptors implicated in atopic dermatitis. |
Additional studies are still needed to further investigate the safety and efficacy of these new drugs and, even more importantly, to compare them with the available treatment options for atopic dermatitis. |
Introduction
Atopic dermatitis (AD) is a common chronic or chronically relapsing, intensely pruritic inflammatory skin disease. Worldwide, it affects up to 20% of children and 2–8% of adults, who often present a personal or familial history of atopic diathesis [1]. AD point prevalence appears to be increasing in all continents and slightly higher in female than male individuals (0.6–24.3% vs. 0.8–17.6%) [2].
In most cases, AD arises during infancy or childhood, but late-onset forms account for 20% of AD cases in adults [3]. Clinically, significant heterogeneity is observed. AD is characterized by cutaneous sensitivity and intensely pruritic eczematous lesions, which can be localized or disseminated. Phenotypes such as juvenile plantar dermatosis, nummular eczema, prurigo nodularis-like, and psoriasiform AD are not infrequent [4, 5].
Quality of life (QoL) is significantly hindered by AD, both in infants-children and adults. About 10% of patients report depressive symptoms and a non-negligible impact on daily social and working activities. Addressing the needs of the patient, family, and caregivers is required to guarantee a comprehensive approach in treating AD [6, 7].
Therapeutic choices are mostly based on disease severity. Conventional treatment options for mild-to-moderate AD include topical corticosteroids (TCSs) and topical calcineurin inhibitors (TCIs) as well as ultraviolet radiation therapy in selected patients. Moderate-to-severe cases require systemic immunosuppression with a short course of systemic corticosteroids or cyclosporine; less commonly, methotrexate, azathioprine, or mycophenolic acid can be prescribed [8]. More recently, the monoclonal antibody dupilumab has proven extremely effective in controlling both clinical signs and AD-related symptoms [9, 10]. The mainstay of baseline therapy rests on topical emollients and non-foaming detergents to reduce the need for active topical treatment and relieve pruritus [8, 11].
More extensive understanding of AD etiology and pathogenesis has led to the development of disease-oriented therapeutic approaches and the study of drugs targeting specific pathogenetic “cornerstones.” This review aimed to illustrate the most significant aspects of AD pathogenesis in light of their potential repercussions on future treatment options, as well as emerging therapies showing a promising profile in terms of clinical efficacy and safety (Table 1). To find eligible treatments, we conducted a search on Pubmed (https://pubmed.ncbi.nlm.nih.gov/) using “atopic dermatitis” and “new/novel/emerging therapies/treatments” as keywords. Ongoing clinical trials are available at https://clinicaltrials.gov. This article is based on previously conducted studies and does not contain any new studies with human participants or animals performed by any of the authors.
Inflammatory and Immune Dysfunction in Atopic Dermatitis
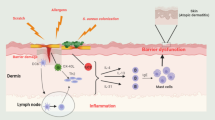
Both innate and adaptive immune system dysregulation are involved in AD pathogenesis. Cutaneous barrier dysfunction promotes transepidermal allergen penetration, therefore triggering skin inflammation. Damaged keratinocytes release proinflammatory cytokines and chemokines known as alarmins, such as interleukin (IL)-25, IL-33, and thymic stromal lymphopoietin (TSLP). In turn, alarmins activate Th2 cells, which results in the production of type 2 cytokines (IL-4, IL-5, and IL-13). Acute AD lesions show a significant Th2, Th22, and Th17 inflammatory infiltrate, while chronic lesions are characterized by reduction of the Th2 component and recruitment of Th1 cells [12,13,14,15].
IL-4 and IL-13 play a pivotal role in AD. They share a common receptor subunit (IL-4Rα), which functions by activation of the downstream JAK/STAT (Janus kinase/signal transducer and activator of transcription) signaling pathway. Overexpression of IL-4 and IL-13 induces immunoglobulin (Ig)E synthesis, promotes skin inflammation and pruritus, and contributes to cutaneous barrier disruption [13, 15].
A neuroimmune dysfunction is critical in inducing itch in patients with AD. Histamine-independent pruritogens include IL-31, TSLP, substance P, and IL-4 itself, which can be directly released by both keratinocytes and immune cells and activate cutaneous non-myelinated C fibers, resulting in itch [16, 17]. In particular, IL-31 induces sensory nerve elongation and branching both in vitro and in vivo, thus resulting in increased nervous density and “hyperinnervation” of AD skin [18].
Emerging Therapy Options for Atopic Dermatitis
Anti-IL-13 and Anti-IL-4 Agents
Dupilumab
Dupilumab is the first biologic drug approved for the treatment of moderate-to-severe AD in adults and adolescents by the US Food and Drug Administration and the European Medicines Agency. It blocks IL-4Rα, thus hindering further downstream signaling of IL-4 and IL-13. Two phase III trials demonstrated its efficacy in inducing an Eczema Area and Severity Index (EASI)-75 response after 16 weeks of treatment in 44–51% of adult patients (vs. 12–15% of participants in the placebo group, P < 0.001) without concomitant TCS use [19]. It also proved to be effective in controlling pruritus and depression-anxiety symptoms and improving the patients’ QoL when compared with placebo [20, 21].
A post hoc analysis on four phase III, randomized, double-blind, placebo-controlled trials investigated the safety and efficacy of dupilumab in 2444 adults with moderate-to-severe AD. Least squares (LS) mean total SCORing Atopic Dermatitis (SCORAD) decreased in all dupilumab-treated groups, with significantly lower results for dupilumab versus control in each study. The percentage of patients achieving SCORAD-50 was greater with dupilumab versus control in each study as well and became statistically significant as early as at week 1 [22].
An ongoing phase III multicenter study (NCT01949311) including approximately 550 patients proved efficacy of dupilumab administration up to 4 years, with 64.4% of patients achieving a 0–1 Investigator Global Assessment (IGA) score. EASI-50, 75, and 90 were achieved in 94.9%, 90.9%, and 75.8%, respectively, with a mean EASI improvement from 3.15 at week 52 to 2.10 at week 204. The percentage of patients who achieved a reduction of at least three points in weekly average pruritus numerical rating scale (NRS) remained stable at week 52 (78.2%) and at week 204 (78.7%), but 70.8% of patients at week 204 vs. 66.9% at week 52 achieved at least a four-point NRS improvement. A total of 84.9% of participants experienced at least one treatment-emergent adverse event (TEAE), with 10.4% and 9.8% of patients developing a serious or severe AE, respectively. The most common TEAEs were nasopharyngitis, upper respiratory tract infections, oral herpes, conjunctivitis, injection site reactions, and headache [23].
Lebrikizumab
Lebrikizumab is a humanized monoclonal antibody (mAb) which binds IL-13 with high affinity in a nonreceptor binding domain, thus preventing IL-4Rα dimerization and downstream signaling. It is administered subcutaneously.
In a phase II, randomized, placebo-controlled, double-blind multicenter study, lebrikizumab 125 mg was administered every 4 weeks for 12 weeks with an additional 8-week follow-up period to assess therapeutic safety. The primary end point was the percentage of patients with an EASI-50 response at week 12, which was achieved by 82.4% of participants who received lebrikizumab 125 mg every 4 weeks versus 62.3% of participants who received placebo. Overall, lebrikizumab was well tolerated. Only three patients (2%) who received lebrikizumab and one (2%) patient in the placebo group discontinued participation in the study because of AEs. Injection site reactions occurred in 1.3% of patients administered lebrikizumab and 1.9% of patients administered placebo. Other AEs included conjunctivitis (9.6%), herpes virus infections (7.7%), and eosinophilia (3.2%). No dose–response relationship emerged [24].
Another phase IIb, double-blind, placebo-controlled, randomized trial consisted of a 16-week treatment period with lebrikizumab (125 mg every 4 weeks, 250 mg every 4 weeks, or 250 mg every 2 weeks) followed by a 16-week follow-up period to assess its safety. The primary end point was percentage reduction of baseline EASI at week 16. The study found that higher doses of lebrikizumab resulted in more significant reductions of EASI from baseline. Improvement of EASI occurred in 62.3%, 69.2%, and 72.1% of patients treated with growing doses of lebrikizumab, but only in 41.1% of the placebo group. Rescue medication with TCSs was employed more frequently, earlier, and for longer periods by patients in the placebo group. As far as safety was concerned, the most common AEs reported were upper respiratory tract infections, nasopharyngitis, headache, and injection site reactions. No AE led to trial discontinuation [25].
Tralokinumab
The long-term efficacy and safety of tralokinumab, a fully human mAb directed against IL-13, were evaluated in a phase III, randomized, placebo-controlled trial in 380 adults with moderate-to-severe atopic dermatitis. Tralokinumab 300 mg every 2 weeks was superior to placebo in terms of percentage of patients achieving a 0–1 IGA and EASI-75 after 16 weeks of treatment, and it also significantly improved itch, Dermatology Life Quality Index (DLQI), SCORAD, EASI-50 and 90, and quality of sleep. To assess outcome maintenance, 134 of the 141 participants who achieved a 0–1 IGA and EASI-75 were then re-randomized to receive tralokinumab 300 mg every 2 or 4 weeks in combination with TCSs. At week 32, IGA 0–1 was maintained without the need for any rescue therapy in 89.6% (95% CI 77.8–95.5%) and 77.6% (64.1–87.0%) of patients, respectively [26].
Overall, tralokinumab was well tolerated and safe since most AEs were mild or moderate in severity. The most common AEs were viral upper respiratory tract infections (21.3% vs. 12.2% for placebo), upper respiratory tract infections (7.1% vs. 4.8%), headache (4.2% vs. 3.9%), and conjunctivitis (3.8% vs. 1.9%) [27].
Anti-IL-31 Agents
Nemolizumab
Nemolizumab is a subcutaneously administered fully human mAb directed against the IL-31Rα. Its effects have been studied in AD because of the role of IL-31 in the pathogenesis of itch.
A phase IIb, double-blind, randomized, placebo-controlled multicenter study compared the efficacy of various doses of nemolizumab and placebo in a group of 226 patients suffering from moderate-to-severe AD. Use of mid- or low-potency TCSs was allowed for both arms. At week 24, all nemolizumab dosages were associated with a greater improvement in EASI than in placebo. Furthermore, at week 24, EASI-50, 75, and 90 were achieved by a higher percentage of patients in the nemolizumab group than in the placebo group. All nemolizumab doses induced a rapid improvement of pruritus NRS, which was already statistically significant at week 1, as well as of sleep and QoL. The use of TCSs was greater in the placebo group than in all nemolizumab dose groups at each monthly checkpoint [28, 29].
Long-term efficacy and safety were evaluated in two phase III, multicenter studies. Nemolizumab proved to reduce pruritus in a continuous manner over time. In the first study, the percent change in the weekly mean pruritus visual analogue scale score from baseline to week 68 was 65.9% for patients who received nemolizumab, and it only slightly decreased after 12 weeks from the last injection. The improvement in patient-reported itch was similar in the second study. Improvement of EASI was maintained after the end of treatment as well, indicating the good long-term efficacy of nemolizumab. Similar results were obtained when sleep and QoL were analyzed. TEAEs occurred in the majority of patients who received nemolizumab in both studies but were severe in only less than 5% of cases. The most common AE was nasopharyngitis (33.9%) [30].
JAK Inhibitors
Upadacitinib
Upadacitinib is an orally administered selective JAK-1 inhibitor. A phase III, multicenter, randomized, placebo-controlled trial (AD Up, NCT03568318) proved its efficacy in combination with TCSs in 901 adults with moderate-to-severe AD. After 16 weeks of daily treatment, EASI-75 was achieved by 77% and 65% of patients receiving upadacitinib 30 mg and 15 mg, respectively, versus 26% of placebo-treated participants. A validated IGA of AD (vIGA-AD) of 0–1 was achieved by 59%, 40%, and 11% of patients, respectively. Overall, upadacitinib was well tolerated, as the most frequent AEs were mild or moderate in severity (acne, nasopharyngitis, upper respiratory tract infections, oral herpes). One percent of patients in the upadacitinib 30 mg and 15 mg groups and 2% of patients in the placebo group discontinued treatment because of AEs [31].
The AD Up trial was then extended to investigate the long-term efficacy of upadacitinib (30 mg or 15 mg) plus TCSs versus placebo plus TCSs in patients with moderate-to-severe AD. At week 52, the LS mean percent change from baseline EASI was 277.4% for upadacitinib 30 mg, 267.7% for upadacitinib 15 mg, and the LS mean percent change from baseline weekly pruritus NRS was 254.5% and 239.0%, respectively. Overall, only 2.9% of patients treated with upadacitinib 30 mg and 7.0% of patients treated with upadacitinib 15 mg who had achieved response at week 16 lost that response at week 52, indicating a good long-term efficacy. At week 52, both upadacitinib dosages were well tolerated [32].
Baricitinib
Baricitinib is an oral selective inhibitor of JAK-1 and JAK-2. A randomized, double-blind, placebo-controlled study compared the efficacy of baricitinib 2 mg or 4 mg with placebo in 124 patients with moderate-to-severe AD. Patients were allowed to use TCSs when needed. The primary end point was the achievement of EASI-50 at week 16. The number of patients who achieved EASI-50, when compared with placebo, was higher in the 4-mg baricitinib group (P = 0.027) and in the 2-mg baricitinib, even though the latter did not reach statistical significance. Moreover, patients who were administered baricitinib used 30% less TCSs than patients who received placebo. AEs mostly included abnormal white cell count (one case of neutropenia, two of leukocyte count decrease, and one with abnormal lymphocyte count) as well as one case of headache and one case of eczema [33].
Two independent phase III, multicenter, double-blind, monotherapy trials (BREEZE-AD1 and BREEZEAD2) tested the efficacy of once-daily baricitinib 4 mg, 2 mg, or 1 mg against placebo. A total of 1239 patients with moderate-to-severe AD were enrolled. The primary end point was the proportion of patients that achieved a vIGA-AD score of 0–1 at week 16, which was reached by 4.8% participants in the placebo group, 11.8% for baricitinib 1 mg, 11.4% for baricitinib 2 mg, and 16.8% for baricitinib 4 mg in BREEZE-AD1 and 4.5% for placebo, 8.8% for baricitinib 1 mg, 10.6% for baricitinib 2 mg, and 13.8% for baricitinib 4 mg in BREEZE-AD2. Most patients required topical or, less frequently, systemic therapy with corticosteroids, even though the need for rescue therapy was higher in patients who received placebo versus baricitinib [34].
Both studies were prolonged up to week 68. The primary end point was the percentage of patients with a vIGA-AD of 0–1 at week 32, 52, and 68 of continuous therapy, and both dosages of baricitinib provided long-term efficacy [35]. Baricitinib plus TCSs has also proven its efficacy in controlling disease-related sleep and itch and positively impacted the QoL of patients with moderate-to-severe AD [36,37,38].
Abrocitinib
Abrocitinib is an oral selective JAK-1 inhibitor. Safety and efficacy of abrocitinib 100 mg or 200 mg versus placebo once daily in adolescents and adults with moderate-to-severe AD were evaluated by a phase III, multicenter, double-blind, randomized trial. At week 12, the percentage of patients who achieved an IGA of 0–1 was significantly higher in the abrocitinib 100 mg and 200 mg groups (24% and 44%, respectively) when compared with placebo (8%). EASI-75 was achieved by 40% of participants receiving abrocitinib 100 mg and 63% receiving abrocitinib 200 mg (vs. 12% for placebo). Serious TEAEs were reported in 3% of patients in both abrocitinib groups and 4% in the placebo group. Most AEs consisted of nausea, nasopharyngitis and upper respiratory tract infections, and headache [39].
Abrocitinib also confirmed its efficacy when compared with dupilumab and placebo. After 12 weeks of treatment, an IGA of 0–1 was achieved by 48.4% of patients in the abrocitinib 200 mg group, 36.6% in the abrocitinib 100 mg group, 36.5% in the dupilumab group, and 14.0% in the placebo group, while EASI-75 was achieved by 70.3%, 58.7%, 58.1%, and 27.1% of participants, respectively. Abrocitinib 200 mg proved to be more effective than dupilumab in controlling itch after 2 weeks of therapy, but at week 16, no significant difference emerged between dupilumab and both abrocitinib dosages in terms of most key secondary end points. The most frequent AEs in the abrocitinib groups were nausea and acne [40, 41].
Interestingly, abrocitinib also seems effective in adults with moderate-to-severe AD who have previously undergone treatment with dupilumab [42]. Abrocitinib also impacted favorably on patient-oriented eczema measure, patient global assessment, pruritus and nighttime itch, DLQI, and hospital anxiety and depression scale [43].
Anti-OX40 Agents
GBR 830
GBR 830 is the first humanized IgG1 mAb against OX40. It is administered intravenously. OX40 is a co-stimulatory molecule mainly expressed by T cells which induces TCR activation, inhibits apoptosis, and promotes T cell function, thus sustaining the immune response. Therefore, GBR 830 seems a promising option to target chronic inflammation.
A phase IIa, randomized, double-blind, placebo-controlled trial evaluated its efficacy and safety in adults with moderate-to-severe AD. When compared to placebo, EASI improved gradually and continuously throughout the study; at day 71, the percent change from baseline was 56% in the GBR 830 group vs. 38% in the placebo group. IGA improved accordingly. A total of 62.9% of patients experienced at least one AE, most of which were mild. The most reported TEAE was headache [44].
A smaller phase I, single-center, open-label, repeated-dose study investigated the safety of GBR 830. TEAEs occurred in 77.3% of patients and were mostly mild or moderate (fever, chills, malaise, nasopharyngitis, and aphthous ulcers). At day 155, 65.0% of patients had achieved EASI-50, 55.0% had achieved EASI-75, and 20.0% had achieved EASI-90. At day 155, a minimum one-point improvement of IGA had occurred in 80.0% of patients [45].
Conclusions
AD is a chronic inflammatory skin disease which significantly hinders the QoL of patients, their families, and caregivers. Therefore, effective therapy is mandatory to reduce signs and symptoms and to avoid repercussions of the anxiety and depression spectrum.
The most recent advances in AD treatment have allowed clinicians to improve therapeutic efficacy and safety, especially when newer drugs are compared to “traditional” immunosuppressants such as cyclosporine, methotrexate, and azathioprine, which are burdened by more frequent and more serious AEs. In clinical trials and real-life experience available to date, these agents have demonstrated a positive short- and long-term clinical impact on AD manifestations in terms of both efficacy and tolerability. As far as mAbs are concerned, they are safer and do not require any specific screening before or during treatment. However, they are administered subcutaneously, which may reduce the patient’s compliance, especially in infants-children and adolescents. JAK inhibitors, on the other hand, are administered orally, which may increase acceptance from the patient, but their use demands a thorough pre-treatment screening and constant monitoring to assess potential AEs.
The growing knowledge regarding AD etiology and pathogenesis has allowed the expansion of the arsenal of potential immunological targets and treatment options to approach the disease. On the other hand, proof-of-concept demonstration of the efficacy of new therapies in patients suffering from AD has recently given new perspectives regarding new aspects of its etiopathogenesis. At the moment, clinicians can prescribe agents targeting the IL-4/IL-13 pathway and small molecules such as JAK inhibitors (Table 1), but further studies will we essential to identify clinical and/or laboratory biomarkers to predict the potential efficacy of a specific drug, in order to achieve a true patient-tailored personalized therapeutic approach to AD.
References
Wollenberg A, Barbarot S, Bieber T, et al. Consensus-based European guidelines for treatment of atopic eczema (atopic dermatitis) in adults and children: part I. J Eur Acad Dermatol Venereol. 2018;32(5):657–682.
Bylund S, Kobyletzki LB, Svalstedt M, Svensson Å. Prevalence and incidence of atopic dermatitis: a systematic review. Acta Derm Venereol. 2020;100(12):adv00160.
Bieber T. Atopic dermatitis: an expanding therapeutic pipeline for a complex disease. Nat Rev Drug Discov. 2022;21(1):21–40.
Lyons JJ, Milner JD, Stone KD. Atopic dermatitis in children: clinical features, pathophysiology, and treatment. Immunol Allergy Clin N Am. 2015;35(1):161–83.
Silverberg NB. Typical and atypical clinical appearance of atopic dermatitis. Clin Dermatol. 2017;35(4):354–9.
Ali F, Vyas J, Finlay AY. Counting the burden: atopic dermatitis and health-related quality of life. Acta Derm Venereol. 2020;100(12):adv00161.
Koszorú K, Borza J, Gulácsi L, Sárdy M. Quality of life in patients with atopic dermatitis. Cutis. 2019;104(3):174–7.
Wollenberg A, Barbarot S, Bieber T, et al. Consensus-based European guidelines for treatment of atopic eczema (atopic dermatitis) in adults and children: part II. J Eur Acad Dermatol Venereol. 2018;32(6):850–878.
Silverberg JI, Thyssen JP, Fahrbach K, et al. Comparative efficacy and safety of systemic therapies used in moderate-to-severe atopic dermatitis: a systematic literature review and network meta-analysis. J Eur Acad Dermatol Venereol. 2021;35(9):1797–810.
Siegels D, Heratizadeh A, Abraham S, et al. Systemic treatments in the management of atopic dermatitis: a systematic review and meta-analysis. Allergy. 2021;76(4):1053–1076.
van Zuuren EJ, Fedorowicz Z, Christensen R, Lavrijsen A, Arents BWM. Emollients and moisturisers for eczema. Cochrane Database Syst Rev. 2017;2(2):CD012119.
Weidinger S, Beck LA, Bieber T, Kabashima K, Irvine AD. Atopic dermatitis. Nat Rev Dis Primers. 2018;4(1):1.
Li H, Zhang Z, Zhang H, Guo Y, Yao Z. Update on the pathogenesis and therapy of atopic dermatitis. Clin Rev Allergy Immunol. 2021;61(3):324–38.
David Boothe W, Tarbox JA, Tarbox MB. Atopic dermatitis: pathophysiology. Adv Exp Med Biol. 2017;1027:21–37.
Malik K, Heitmiller KD, Czarnowicki T. An update on the pathophysiology of atopic dermatitis. Dermatol Clin. 2017;35(3):317–26.
Wong LS, Yen YT, Lee CH. The implications of pruritogens in the pathogenesis of atopic dermatitis. Int J Mol Sci. 2021;22(13):7227.
Oetjen LK, Mack MR, Feng J, et al. Sensory neurons co-opt classical immune signaling pathways to mediate chronic itch. Cell. 2017;171(1):217–228.e13.
Feld M, Garcia R, Buddenkotte J, et al. The pruritus- and TH2-associated cytokine IL-31 promotes growth of sensory nerves. J Allergy Clin Immunol. 2016;138(2):500–508.e24.
Simpson EL, Bieber T, Guttman-Yassky E, et al. Two phase 3 trials of dupilumab versus placebo in atopic dermatitis. N Engl J Med. 2016;375(24):2335–2348.
Silverberg JI, Yosipovitch G, Simpson EL, et al. Dupilumab treatment results in early and sustained improvements in itch in adolescents and adults with moderate to severe atopic dermatitis: Analysis of the randomized phase 3 studies SOLO 1 and SOLO 2, AD ADOL, and CHRONOS. J Am Acad Dermatol. 2020;82(6):1328–36.
Cork MJ, Eckert L, Simpson EL, et al. Dupilumab improves patient-reported symptoms of atopic dermatitis, symptoms of anxiety and depression, and health-related quality of life in moderate-to-severe atopic dermatitis: analysis of pooled data from the randomized trials SOLO 1 and SOLO 2. J Dermatolog Treat. 2020;31(6):606–14.
Barbarot S, Wollenberg A, Silverberg JI, et al. Dupilumab provides rapid and sustained improvement in SCORAD outcomes in adults with moderate-to-severe atopic dermatitis: combined results of four randomized phase 3 trials. J Dermatolog Treat. 2022;33(1):266–77.
Beck LA, Deleuran M, Bissonnette R, et al. Dupilumab provides acceptable safety and sustained efficacy for up to 4 years in an open-label study of adults with moderate-to-severe atopic dermatitis. Am J Clin Dermatol. 2022;23(3):393–408.
Simpson EL, Flohr C, Eichenfield LF, et al. Efficacy and safety of lebrikizumab (an anti-IL-13 monoclonal antibody) in adults with moderate-to-severe atopic dermatitis inadequately controlled by topical corticosteroids: a randomized, placebo-controlled phase II trial (TREBLE). J Am Acad Dermatol. 2018;78(5):863–871.e11.
Guttman-Yassky E, Blauvelt A, Eichenfield LF, et al. Efficacy and safety of lebrikizumab, a high-affinity interleukin 13 inhibitor, in adults with moderate to severe atopic dermatitis: a phase 2b randomized clinical trial. JAMA Dermatol. 2020;156(4):411–20.
Silverberg JI, Toth D, Bieber T, et al. Tralokinumab plus topical corticosteroids for the treatment of moderate-to-severe atopic dermatitis: results from the doubleblind, randomized, multicentre, placebo-controlled phase III ECZTRA 3 trial. Br J Dermatol. 2021;184(3):450–463.
Blauvelt A, Langley RG, Lacour JP, et al. Long-term 2-year safety and efficacy of tralokinumab in adults with moderate-to-severe atopic dermatitis: Interim analysis of the ECZTEND open-label extension trial. J Am Acad Dermatol. 2022;87(4):815–24.
Silverberg JI, Pinter A, Pulka G, et al. Phase 2B randomized study of nemolizumab in adults with moderate-to-severe atopic dermatitis and severe pruritus. J Allergy Clin Immunol. 2020;145(1):173–82.
Silverberg JI, Pinter A, Alavi A, et al. Nemolizumab is associated with a rapid improvement in atopic dermatitis signs and symptoms: subpopulation (EASI ≥ 16) analysis of randomized phase 2B study. J Eur Acad Dermatol Venereol. 2021;35(7):1562–8.
Kabashima K, Matsumura T, Komazaki H, Kawashima M, Nemolizumab JP01 and JP02 Study Group. Nemolizumab plus topical agents in patients with atopic dermatitis (AD) and moderate-to-severe pruritus provide improvement in pruritus and signs of AD for up to 68 weeks: results from two phase III, long-term studies. Br J Dermatol. 2022;186(4):642–651.
Reich K, Teixeira HD, de Bruin-Weller M, et al. Safety and efficacy of upadacitinib in combination with topical corticosteroids in adolescents and adults with moderate-to-severe atopic dermatitis (AD Up): results from a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet. 2021;397(10290):2169–81.
Silverberg JI, de Bruin-Weller M, Bieber T, et al. Upadacitinib plus topical corticosteroids in atopic dermatitis: week 52 AD up study results. J Allergy Clin Immunol. 2022;149(3):977–987.e14.
Guttman-Yassky E, Silverberg JI, Nemoto O, et al. Baricitinib in adult patients with moderate-to-severe atopic dermatitis: a phase 2 parallel, double-blinded, randomized placebo-controlled multiple-dose study. J Am Acad Dermatol. 2019;80(4):913–921.e9.
Simpson EL, Forman S, Silverberg JI, et al. Baricitinib in patients with moderate-to-severe atopic dermatitis: results from a randomized monotherapy phase 3 trial in the United States and Canada (BREEZE-AD5). J Am Acad Dermatol. 2021;85(1):62–70.
Silverberg JI, Simpson EL, Wollenberg A, et al. Long-term efficacy of baricitinib in adults with moderate to severe atopic dermatitis who were treatment responders or partial responders: an extension study of 2 randomized clinical trials. JAMA Dermatol. 2021;157(6):691–9.
Wollenberg A, Nakahara T, Maari C, et al. Impact of baricitinib in combination with topical steroids on atopic dermatitis symptoms, quality of life and functioning in adult patients with moderate-to-severe atopic dermatitis from the BREEZE-AD7 phase 3 randomized trial. J Eur Acad Dermatol Venereol. 2021;35(7):1543–52.
Lio PA, Simpson EL, Han G, et al. Improvement in sleep and itch and enhanced quality of life in adult patients with moderate-to-severe atopic dermatitis: results from a phase 3 trial of baricitinib therapy. J Dermatolog Treat. 2022;33(4):2057–62.
Rosmarin D, Casillas M, Chen S, et al. Onset of symptom relief reported in daily diaries of patients with atopic dermatitis treated with baricitinib in a United States clinical trial (BREEZE-AD5). J Cutan Med Surg. 2022;26(3):262–266.
Simpson EL, Sinclair R, Forman S, et al. Efficacy and safety of abrocitinib in adults and adolescents with moderate-to-severe atopic dermatitis (JADE MONO-1): a multicentre, double-blind, randomised, placebo-controlled, phase 3 trial. Lancet. 2020;396(10246):255–66.
Bieber T, Simpson EL, Silverberg JI, et al. Abrocitinib versus placebo or dupilumab for atopic dermatitis. N Engl J Med. 2021;384(12):1101–1112.
Reich K, Thyssen JP, Blauvelt A, et al. Efficacy and safety of abrocitinib versus dupilumab in adults with moderate-to-severe atopic dermatitis: a randomised, double-blind, multicentre phase 3 trial. Lancet. 2022;400(10348):273–82.
Shi VY, Bhutani T, Fonacier L, et al. Phase 3 efficacy and safety of abrocitinib in adults with moderate-to-severe atopic dermatitis after switching from dupilumab (JADE EXTEND). J Am Acad Dermatol. 2022;87(2):351–8.
Thyssen JP, Yosipovitch G, Paul C, et al. Patient-reported outcomes from the JADE COMPARE randomized phase 3 study of abrocitinib in adults with moderate-to-severe atopic dermatitis. J Eur Acad Dermatol Venereol. 2022;36(3):434–43.
Guttman-Yassky E, Pavel AB, Zhou L, et al. GBR 830, an anti-OX40, improves skin gene signatures and clinical scores in patients with atopic dermatitis. J Allergy Clin Immunol. 2019;144(2):482–493.e7.
Nakagawa H, Iizuka H, Nemoto O, et al. Safety, tolerability and efficacy of repeated intravenous infusions of KHK4083, a fully human anti-OX40 monoclonal antibody, in Japanese patients with moderate to severe atopic dermatitis. J Dermatol Sci. 2020;99(2):82–9.
Acknowledgements
Funding
No funding or sponsorship was received for this study or publication of this article.
Medical Writing and/or Editorial Assistance
Simona Tavecchio and Stefano Buffon were responsible for medical writing of this manuscript. No editorial assistance was employed.
Author Contributions
Silvia M. Ferrucci: concept and design. Simona Tavecchio: concept and design, drafting the manuscript, checking data. Angelo V. Marzano: supervising. Stefano Buffon: drafting the manuscript.
Disclosures
Silvia M. Ferrucci has been principal investigator and/or speaker to Almirall, Amgen, Abbvie, Sanofi-Genzyme, Lilly, Novartis, Leo Pharma. Participation in Advisory board of Sanofi Genzyme, Abbvie, Almirall. Simona Tavecchio received honoraria as a speaker from Sanofi Genzyme, Leo Pharma, Abbvie. Angelo V. Marzano reports consultancy/advisory boards disease-relevant honoraria from Abbvie, Boehringer-Ingelheim, Novartis, Pfizer, Sanofi and UCB. Stefano Buffon has nothing to disclose.
Compliance with Ethics Guidelines
This article is based on previously conducted studies and does not contain any new studies with human participants or animals performed by any of the authors.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Ferrucci, S.M., Tavecchio, S., Marzano, A.V. et al. Emerging Systemic Treatments for Atopic Dermatitis. Dermatol Ther (Heidelb) 13, 1071–1081 (2023). https://doi.org/10.1007/s13555-023-00920-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13555-023-00920-4