
Abstract
Introduction
Hidradenitis suppurativa (HS) is a chronic, immune-mediated skin condition characterized by inflammatory lesions that can cause pain, impaired physical activity, and reduced quality of life. This study evaluated the efficacy and safety of risankizumab, a humanized immunoglobulin G1 monoclonal antibody that specifically inhibits interleukin 23 by binding to its p19 subunit, for the treatment of HS.
Methods
This phase II multicenter, randomized, placebo-controlled, double-blind study investigated the efficacy and safety of risankizumab in patients with moderate-to-severe HS. Patients were randomized 1:1:1 to receive subcutaneous risankizumab 180 mg; risankizumab 360 mg; or placebo at weeks 0, 1, 2, 4, and 12. Patients initially randomized to placebo received blinded risankizumab 360 mg at weeks 16, 17, and 18; patients initially randomized to risankizumab received blinded matching placebo at the same time points. From weeks 20–60, all patients received open-label risankizumab 360 mg every 8 weeks. The primary endpoint was the achievement of HS Clinical Response (HiSCR) at week 16. Safety was assessed by monitoring of treatment-emergent adverse events (TEAEs).
Results
A total of 243 patients were randomized (risankizumab 180 mg, n = 80; risankizumab 360 mg, n = 81; placebo, n = 82). HiSCR was achieved by 46.8% of patients with risankizumab 180 mg, 43.4% with risankizumab 360 mg, and 41.5% with placebo at week 16. The primary endpoint was not met, and the study was terminated early. Incidence of TEAEs, severe TEAEs, TEAEs considered possibly related to study drug, and TEAEs leading to discontinuation of study drug were generally low and comparable across treatment groups.
Conclusion
Risankizumab does not appear to be an efficacious treatment for moderate-to-severe HS. Future studies to understand the complex molecular mechanisms underlying HS pathogenesis and develop improved therapies are warranted.
Trial Registration
ClinicalTrials.gov identifier: NCT03926169.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Why carry out this study? |
Hidradenitis suppurativa (HS) is a chronic, immune-mediated, inflammatory skin condition that can cause pain, functional impairments, and diminished quality of life. |
Therapeutic options for HS are currently limited and needed; this study evaluated the efficacy and safety of risankizumab, an interleukin 23 inhibitor, for the treatment of HS. |
What was learned from the study? |
Improvements in HS symptoms, pain, and quality of life were similar across the risankizumab and placebo treatment groups; the primary endpoint was not met, and the study was terminated early. |
No new safety findings were observed in patients with HS; the safety profile was consistent with the safety profiles observed in clinical studies of risankizumab for other indications. |
Risankizumab does not appear to be an efficacious treatment for HS; a better understanding of the molecular mechanisms causing HS development and progression is needed to develop improved therapies. |
Introduction
Hidradenitis suppurativa (HS) is a chronic, immune-mediated, inflammatory skin condition characterized by recurrent, inflamed, painful nodules, abscesses, draining and nondraining fistulas, and scarring [1, 2]. Scarring and symptoms of HS, including pain and malodorous discharge, can cause functional impairments, disability, and diminished quality of life [1, 2]. Treatment options for HS are currently limited, and there is an unmet need for effective therapies to manage the debilitating symptoms of HS and prevent scarring and disability [1,2,3,4].
First-line treatments for HS include topical and systemic antibiotics; however, these approaches are often ineffective in patients with moderate-to-severe HS [1, 2]. Surgical intervention is also used to manage acute lesions, which may provide short-term relief of HS symptoms, but surgery is not often effective for long-term control and is frequently used as an adjunct to medical therapy [1, 2]. Surgical treatment can provide long-term improvement in severe but localized forms of the disease [1]. Adalimumab, an anti-tumor necrosis factor (TNF) antibody, is the only therapy currently approved for the treatment of HS [2,3,4,5,6]. Adalimumab demonstrated superior efficacy over placebo in the phase III PIONEER I and PIONEER II studies [7], with similar results reported in combination with surgery in the phase IV SHARPS trial [8].
While the molecular pathogenesis underlying HS is complex, interleukin (IL)-23 is highly expressed in activated macrophages in skin lesions of patients with HS, and some studies have suggested that IL-23 signaling may play a role in the inflammatory pathways contributing to the development and progression of HS [1,2,3,4, 9]. Risankizumab is a humanized immunoglobulin G1 monoclonal antibody that specifically inhibits IL-23 by binding to its p19 subunit [10]. Risankizumab is approved in multiple countries, including in the USA, European Union, and Japan, for the treatment of moderate-to-severe plaque psoriasis, active psoriatic arthritis, and Crohn’s disease [11,12,13]. Risankizumab is also approved in Japan for the treatment of generalized pustular psoriasis and erythrodermic psoriasis [13].
Whether IL-23 inhibition can reduce the clinical symptoms of HS is not yet understood. Here we report findings from a phase II proof-of-concept study that evaluated the efficacy and safety of risankizumab for the treatment of moderate-to-severe HS.
Methods
Patients
The patient population for this study included adults (aged ≥ 18 years) with moderate-to-severe HS. Eligible patients were required to be diagnosed at least 1 year before the baseline visit and have a total abscess and inflammatory nodule (AN) count ≥ 5 at baseline, HS lesions in ≥ 2 distinct anatomical locations, a draining fistula count ≤ 20 at baseline, and inadequate response to oral antibiotics for the treatment of HS. Patients were ineligible to participate if they had exposure to biologic agents blocking IL-12/23, IL-23, or IL-17 within the past 6 months; prior exposure to anti-TNF therapies (except those for the treatment of HS that demonstrated inadequate response); relevant medical conditions (such as hepatitis B, hepatitis C, HIV, or tuberculosis); or if they were pregnant or breastfeeding.
Study Design and Treatment
This was a phase II, multicenter, randomized, placebo-controlled, double-blind study (ClinicalTrials.gov identifier: NCT03926169). The study included two treatment periods: a randomized, double-blind, placebo-controlled period from weeks 0 to 16 and an open-label risankizumab treatment period from weeks 16 to 68. During the 16-week double-blind treatment period, patients were randomized 1:1:1 to receive subcutaneous risankizumab 180 mg; risankizumab 360 mg; or placebo at weeks 0, 1, 2, 4, and 12. The final efficacy evaluation for the double-blind period was performed at week 16. In the open-label treatment period, patients initially randomized to placebo received blinded risankizumab 360 mg at weeks 16, 17, and 18, while patients initially randomized to risankizumab received blinded matching placebo at the same time points to maintain blinding (administration of placebo at weeks 16, 17, and 18 did not affect the risankizumab dosing schedule). Starting at week 20, all patients received open-label risankizumab 360 mg every 8 weeks through to week 60; the final efficacy evaluation was at week 68.
The study was conducted in accordance with the protocol, International Council for Harmonisation guidelines, and applicable regulations, guidelines, and ethical principles originating from the 1964 Declaration of Helsinki. The study protocol was reviewed and approved by central (Advarra IRB Services, Columbia, MD, USA) and by local independent ethics committees and/or institutional review boards at each study site. Patients provided written informed consent prior to screening or undergoing study-specific procedures.
Assessments
The primary endpoint was the achievement of Hidradenitis Clinical Response (HiSCR) at week 16 (defined as a ≥ 50% reduction in AN count with no increase in abscess count and no increase in draining fistula count relative to baseline [14]). Ranked secondary endpoints included the achievement of ≥ 30% reduction and ≥ 1 unit reduction from baseline in Numerical Rating Scale (NRS30) in Patient’s Global Assessment of Skin Pain (PGA Skin Pain) at week 8 or at week 16 among patients with baseline NRS scores ≥ 3; an experience of ≥ 25% increase in AN counts with a minimum increase of 2 relative to baseline during the double-blind period; change from baseline to week 16 in the Dermatology Life Quality Index (DLQI); and change from baseline to week 16 in HS-related swelling, HS-related odor, and HS-related worst drainage assessed based on Hidradenitis Suppurativa Symptom Assessment (HSSA). Additional efficacy endpoints included change from baseline to week 16 in lesion count by lesion type (AN count, abscess, draining fistula, and inflammatory nodule) and the achievement of a DLQI score of 0/1 (indicating disease has no or almost no effect on patient’s quality of life) at week 16.
Safety assessments in the two study periods included monitoring of treatment-emergent adverse events (TEAEs) during the double-blind period (in all patients who were randomized and received at least 1 dose of study drug from baseline to week 16) and during the open-label period (in all patients who received at least 1 dose of study drug from week 16 through the time of study termination).
Statistical Analysis
The primary endpoint was evaluated between each risankizumab dose and placebo using the Cochran–Mantel–Haenszel test, adjusting for stratification factors (prior exposure to anti-TNF therapies and baseline worst Hurley stage). Missing efficacy values during the double-blind period were handled using nonresponder imputation incorporating multiple imputation to handle missing data due to COVID-19 as the primary approach for categorical endpoints and mixed-effect model repeat measures for continuous endpoints. All safety analyses were performed on the safety populations, defined as all patients who received at least one dose of study drug in the respective study period. The number and percentage of patients experiencing TEAEs were tabulated using the Medical Dictionary For Regulatory Activities (version 24.0) system organ class and preferred terms; severity and relationship to the study drug were assessed by the investigator.
Results
Patients
A total of 243 patients were randomized (risankizumab 180 mg, n = 80; risankizumab 360 mg, n = 81; placebo, n = 82; Fig. 1). One patient randomized to receive risankizumab 360 mg did not receive the study drug as the patient withdrew from the trial for logistical reasons before the first dose was administered. Baseline demographics and characteristics were generally balanced across all treatment groups (Table 1). The mean (standard deviation [SD]) age was 38.1 (11.8) years, and a majority of patients were female (62.6%) and White (79.4%). Baseline disease activity scores were similar across treatment groups, and 28.8% of patients had previous exposure to anti-TNF biologic therapy. The mean (SD) duration of HS was 11.1 (9.5) years.

Flow chart of patient disposition. aOne patient was randomized to RZB 360 mg but did not receive treatment. bOne patient completed week 16 but did not enter the open-label period. “Other” includes COVID-19 infection, logistical restrictions due to COVID-19, and study termination. RZB Risankizumab
Among the 243 patients randomized, 219 (90.1%) completed the double-blind period (risankizumab 180 mg, n = 70; risankizumab 360 mg, n = 75; placebo, n = 74), and all but one patient (in the risankizumab 360 mg group) entered the open-label period (n = 218). The primary analysis was performed once all patients had completed the double-blind treatment period; following the primary analysis, the study was terminated because the primary endpoint was not met. Most patients (n = 181, 77.0%) discontinued the study during the open-label period due to early termination of the study.
Efficacy
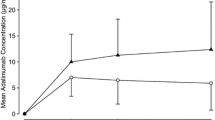
At the end of the double-blind treatment period, the primary endpoint (HiSCR) was achieved by 46.8% of patients with risankizumab 180 mg, 43.4% with risankizumab 360 mg, and 41.5% with placebo (Fig. 2). Secondary endpoint outcomes were similar across all treatment groups (Table 2). Among patients with baseline PGA Skin Pain NRS ≥ 3, achievement of NRS30 at weeks 8 and/or 16 ranged from 27.9% to 40.0%. The proportion of patients who experienced a ≥ 25% increase in AN counts with a minimum increase of 2 relative to baseline during the double-blind period ranged from 18.5 to 29.3%. Mean changes from baseline in HS-related swelling, HS-related odor, HS-related worst drainage assessed based on HSSA scores were generally similar among all treatment groups. The mean change from baseline in DLQI score was numerically greater with risankizumab treatment (− 3.5 with 180 mg, − 3.7 with 360 mg) than with placebo (− 2.1). A similar proportion of patients in each treatment group (ranging from 5.1% to 7.9%) achieved DLQI 0/1 at week 16 (Fig. 3). Reductions in AN count, abscesses, draining fistulas, and inflammatory nodules were also similar across treatment groups at week 16 (Fig. 4).

Patients achieving Hidradenitis Suppurativa Clinical Response (HiSCR) at week 16. Nonresponder imputation incorporating multiple imputation was used to handle missing data due to COVID-19. CI Confidence interval

Patients achieving Dermatology Life Quality Index (DLQI) score 0/1 (indicating disease has no/almost no effect on patient’s quality of life)) at week 16. Nonresponder imputation incorporating multiple imputation was used to handle missing data due to COVID-19

Change from baseline to week 16 in lesion count by lesion type using mixed-effect model repeated measures analysis. AN Abscess and inflammatory nodule, LS least squares, SE standard error
Safety
Overall, the incidence of TEAEs, severe TEAEs, TEAEs considered possibly related to study drug, and TEAEs leading to discontinuation of study drug were low and comparable across treatment groups during both the double-blind period and the open-label period (Table 3). The median (range) of study drug exposure was 112 (29, 140) days in the double-blind period and 140 (23, 385) days in the open-label period. During the double-blind period, the most common TEAEs (> 3%) among patients who received risankizumab were headache (10.6%), nasopharyngitis (8.1%), back pain (4.4%), urinary tract infection (4.4%), fatigue (3.8%), nausea (3.1%), upper respiratory tract infection (3.1%), and worsening of hidradenitis suppurativa (3.1%). During the open-label period, TEAE rates were numerically higher among patients in the placebo/risankizumab 360 mg group than in the risankizumab 180 mg/risankizumab 360 mg group or continuous risankizumab 360 mg group; the most common TEAEs among patients who received risankizumab were worsening of hidradenitis suppurativa (11.0%), headache (5.5%), and diarrhea (3.7%). No deaths were reported during the study.
Incidence of TEAEs of safety interest were low and generally similar across treatment groups during both study periods (Table 4). Incidence of serious infections and herpes zoster was also low in both study periods; there were no events of opportunistic infection. One patient experienced COVID-19 pneumonia during the double-blind period, which led to discontinuation of risankizumab, but the event of COVID-19 pneumonia was not considered related to risankizumab. No serious infections during the open-label period led to discontinuation of risankizumab. There was one event of malignancy (breast cancer and considered serious) in one patient during the open-label period, which led to study drug discontinuation but was assessed as having no reasonable possible relationship to risankizumab. There were no reports of adjudicated major adverse cardiovascular events, adjudicated anaphylactic reactions, tuberculosis, or serious hypersensitivity during the study. The incidence of nonserious hypersensitivity events was higher in the risankizumab 360 mg group (7.5%) than in the risankizumab 180 mg group (2.5%) or placebo group (2.4%) during the double-blind period. All events were mild to moderate in severity, and the majority of cases were related to dermatitis, contact dermatitis, and eczema, and were considered likely attributed to the required use of daily antiseptic washes throughout the study. A higher incidence of hepatic events was observed in the risankizumab 180 mg/360 mg group (5.7%) than in the continuous risankizumab 360 mg group (0%) or placebo/risankizumab 360 mg group (1.4%) during the open-label period; all hepatic events were indicative of laboratory abnormalities and were mild to moderate in severity. There was no potential Hy’s law laboratory findings. No hepatic events were considered to be serious, the total number of hepatic events were numerically low, and none led to study drug discontinuation. No clinically meaningful trends or apparent dose effects were observed for laboratory values and vital signs.
Discussion
In this phase II study evaluating the efficacy and safety of risankizumab for the treatment of HS, the proportion of patients achieving key efficacy endpoints after 16 weeks of treatment was similar in patients who received risankizumab 180 mg or 360 mg versus patients who received placebo. As the primary endpoint was not met, the study was terminated early. A high placebo effect was observed in this study, which has also been reported in other randomized clinical trials of HS [15] and may be influenced by the waxing and waning course commonly observed in HS. There were no new safety findings reported in patients with HS relative to those observed in previous risankizumab studies for other indications [16,17,18,19,20].
There is a great unmet need for HS treatment options. Currently, the only treatment approved in most jurisdictions including the USA and European Union for patients with HS is adalimumab. Variable responses to adalimumab in patients with HS have been reported, with one study reporting 42–59% of patients achieving HiSCR after 12 weeks of treatment [7]. Our findings demonstrate that risankizumab therapy (at the evaluated doses of 180 mg and 360 mg) does not improve the signs and symptoms of HS. Guselkumab, another anti-IL-23 monoclonal antibody, also failed to demonstrate significant clinical improvements over placebo after 16 weeks of treatment in a recent phase II study of patients with moderate-to-severe HS (ClinicalTrials.gov identifier: NCT03628924) [21]. These results suggest that IL-23 may not be a relevant therapeutic target for HS and that inhibition of IL-23 in the doses evaluated in clinical trials thus far is not an effective strategy to treat HS.
Several other investigational treatments are also being evaluated for the treatment of HS; agents targeting IL-17 and Janus kinase 1 have recently demonstrated promise in placebo-controlled studies, and evaluation of other novel therapies are also underway. Secukinumab (an anti-IL-17α monoclonal antibody) demonstrated superiority over placebo after 16 weeks of treatment in patients with moderate-to-severe HS in two recent phase III studies (SUNSHINE [ClinicalTrials.gov identifier NCT03713619] and SUNRISE [ClinicalTrials.gov identifier NCT03713632]) [22]. Clinically meaningful improvements have also been reported from open-label cohort studies for brodalumab (an IL-17α receptor antagonist) [23, 24] and from phase II randomized controlled trials for HS with bimekizumab (an anti-IL-17α /IL-17F monoclonal antibody) [25] and povorcitinib (an oral, small-molecule Janus kinase 1 inhibitor) [26]. Clinical responses reported with anti-IL-17 agents, but not anti-IL-23 antibodies might suggest that IL-17-producing cells other than T helper 17 cells may be involved in the pathophysiology of HS.
The limitations of this study include the early study termination, which presents challenges for interpreting safety and efficacy findings from the open-label period based on limited data collected before the study termination. Additional limitations include a relatively high proportion of White participants, which may not be fully representative of the diverse population of patients with HS. Nevertheless, it is important to publish results from negative clinical trials to help elucidate the mechanisms underlying the complicated biology of HS and guide research toward the development of novel therapeutic options.
Conclusion
Our findings suggest that risankizumab at doses of 180 mg or 360 mg does not appear to be an efficacious treatment for HS. A better understanding of the molecular mechanisms contributing to HS development and progression is needed to develop improved therapies.
References
Sabat R, Jemec GBE, Matusiak L, Kimball AB, Prens E, Wolk K. Hidradenitis suppurativa. Nat Rev Dis Primers. 2020;6:18.
Amat-Samaranch V, Agut-Busquet E, Vilarrasa E, Puig L. New perspectives on the treatment of hidradenitis suppurativa. Ther Adv Chronic Dis. 2021;12:20406223211055920.
Aarts P, Dudink K, Vossen A, et al. Clinical implementation of biologics and small molecules in the treatment of hidradenitis suppurativa. Drugs. 2021;81:1397–410.
Frew JW, Marzano AV, Wolk K, et al. A systematic review of promising therapeutic targets in hidradenitis suppurativa: a critical evaluation of mechanistic and clinical relevance. J Investig Dermatol. 2021;141:316-324.e312.
AbbVie Inc. Humira (adalimumab). Prescribing information. 2022. https://www.rxabbvie.com/pdf/humira.pdf. Accessed 28 Nov 2022.
Marzano AV, Genovese G, Casazza G, et al. Evidence for a ‘window of opportunity’ in hidradenitis suppurativa treated with adalimumab: a retrospective, real-life multicentre cohort study. Br J Dermatol. 2021;184:133–40.
Kimball AB, Okun MM, Williams DA, et al. Two phase 3 trials of adalimumab for hidradenitis suppurativa. N Engl J Med. 2016;375:422–34.
Bechara FG, Podda M, Prens EP, et al. Efficacy and safety of adalimumab in conjunction with surgery in moderate to severe hidradenitis suppurativa: the SHARPS randomized clinical trial. JAMA Surg. 2021;156:1001–9.
Schlapbach C, Hänni T, Yawalkar N, Hunger RE. Expression of the IL-23/Th17 pathway in lesions of hidradenitis suppurativa. J Am Acad Dermatol. 2011;65:790–8.
Singh S, Kroe-Barrett RR, Canada KA, et al. Selective targeting of the IL23 pathway: generation and characterization of a novel high-affinity humanized anti-IL23A antibody. MAbs. 2015;7:778–91.
AbbVie Inc. Skyrizi (risankizumab-rzaa). Prescribing information. 2022. https://www.rxabbvie.com/pdf/skyrizi_pi.pdf. Accessed 30 Nov 2022.
AbbVie Ltd. Skyrizi (risankizumab). Summary of product characteristics. 2022. https://www.ema.europa.eu/en/documents/product-information/skyrizi-epar-product-information_en.pdf. Accessed 30 Nov 2022.
AbbVie GK. Skyrizi (risankizumab). Full Japanese prescribing information. 2021. https://a-connect.abbvie.co.jp/-/media/assets/pdf/products/skyrizi/Skyrizi_tmpDocument.pdf. Accessed 7 Dec 2022.
Kimball AB, Sobell JM, Zouboulis CC, et al. HiSCR (Hidradenitis Suppurativa Clinical Response): a novel clinical endpoint to evaluate therapeutic outcomes in patients with hidradenitis suppurativa from the placebo-controlled portion of a phase 2 adalimumab study. J Eur Acad Dermatol Venereol. 2016;30:989–94.
Amir Ali A, Seng EK, Alavi A, Lowes MA. Exploring changes in placebo treatment arms in hidradenitis suppurativa randomized clinical trials: a systematic review. J Am Acad Dermatol. 2020;82:45–53.
Gordon KB, Strober B, Lebwohl M, et al. Efficacy and safety of risankizumab in moderate-to-severe plaque psoriasis (UltIMMa-1 and UltIMMa-2): results from two double-blind, randomised, placebo-controlled and ustekinumab-controlled phase 3 trials. Lancet. 2018;392:650–61.
Kristensen LE, Keiserman M, Papp K, et al. Efficacy and safety of risankizumab for active psoriatic arthritis: 24-week results from the randomised, double-blind, phase 3 KEEPsAKE 1 trial. Ann Rheum Dis. 2022;81:225–31.
Östör A, Van den Bosch F, Papp K, et al. Efficacy and safety of risankizumab for active psoriatic arthritis: 24-week results from the randomised, double-blind, phase 3 KEEPsAKE 2 trial. Ann Rheum Dis. 2022;81:351–8.
D’Haens G, Panaccione R, Baert F, et al. Risankizumab as induction therapy for Crohn’s disease: results from the phase 3 ADVANCE and MOTIVATE induction trials. Lancet. 2022;399:2015–30.
Ferrante M, Panaccione R, Baert F, et al. Risankizumab as maintenance therapy for moderately to severely active Crohn’s disease: results from the multicentre, randomised, double-blind, placebo-controlled, withdrawal phase 3 FORTIFY maintenance trial. Lancet. 2022;399:2031–46.
ClinicalTrials.gov. A study to evaluate the efficacy, safety, and tolerability of guselkumab for the treatment of participants with moderate to severe hidradenitis suppurativa (HS) (NOVA). https://clinicaltrials.gov/ct2/show/results/NCT03628924. Accessed 28 Nov 2022.
Kimball AB, Alavi A, Jemec GBE, et al. Secukinumab in moderate to severe hidradenitis suppurativa: primary endpoint analysis from the SUNSHINE and SUNRISE phase 3 trials. In: 31st European Academy of Dermatology and Venereology Congress, September 7–10, 2022, Milan.
Frew JW, Navrazhina K, Grand D, et al. The effect of subcutaneous brodalumab on clinical disease activity in hidradenitis suppurativa: an open-label cohort study. J Am Acad Dermatol. 2020;83:1341–8.
Frew JW, Navrazhina K, Sullivan-Whalen M, Gilleaudeau P, Garcet S, Krueger JG. Weekly administration of brodalumab in hidradenitis suppurativa: an open-label cohort study. Br J Dermatol. 2021;184:350–2.
Glatt S, Jemec GBE, Forman S, et al. Efficacy and safety of bimekizumab in moderate to severe hidradenitis suppurativa: a phase 2, double-blind, placebo-controlled randomized clinical trial. JAMA Dermatol. 2021;157:1279–88.
Alavi A, Hamzavi I, Brown K, et al. Janus kinase 1 inhibitor INCB054707 for patients with moderate-to-severe hidradenitis suppurativa: results from two phase II studies. Br J Dermatol. 2022;186:803–13.
Acknowledgements
AbbVie and authors thank all the trial investigators and patients who participated in this clinical trial
Funding
AbbVie funded this study and participated in the study design, research, analysis, data collection, interpretation of data, reviewing, and approval of the publication. All authors had access to relevant data and participated in the drafting, review, and approval of this publication. No honoraria or payments were made for authorship. AbbVie also provided funding for the journal’s Rapid Service Fee.
Medical Writing and Editorial Assistance
Medical writing support was provided by Callie AS Corsa, PhD, of JB Ashtin and funded by AbbVie. JB Ashtin adheres to Good Publication Practice (GPP3) guidelines and International Committee of Medical Journal Editors (ICMJE) recommendations.
Author Contributions
Alexa B Kimball, Errol P Prens, Stefan Beeck, Ziqian Geng, Izabella Messina, and Falk G Bechara contributed to the study concept and design. Alexa B Kimball, Errol P Prens, Thierry Passeron, Emanual Maverakis, Irina Turchin, and Falk G. Bechara contributed to data acquisition. Stefan Beeck, Leonidas Drogaris, Ziqian Geng, Tianyu Zhan, and Izabella Messina contributed to the statistical analysis. Alexa B Kimball, Stefan Beeck, Leonidas Drogaris, Ziqian Geng, Tianyu Zhan, and Izabella Messina contributed to data acquisition. All authors reviewed and critiqued the manuscript and approved the publication of this manuscript.
Disclosures
Alexa B Kimball has received royalties or licenses from BIDMC; received consulting fees from AbbVie, Alumis, Bayer, Bristol Myers Squibb, Boehringer Ingelheim, Eli Lilly, FIDE, Janssen, MoonLake, Novartis, Pfizer, Priovant, Sanofi, Sonoma Bio, UCB, and Ventyx; serves on advisory boards for Target RWE; serves as advisory council member to the NIH Director; holds stock in Ventyx; and serves on the board of directors of Almirall. Her institution has received grants from AbbVie, Anapyts Bio, Aristea, Bristol Myers Squibb, ChemoCentryx, Eli Lilly, Incyte, Janssen, MoonLake, Novartis, Pfizer, Sonoma Bio, and UCB, and fellowship funding from AbbVie and Johnson and Johnson. Errol P Prens has received advisory board/speaker fees from AbbVie, Amgen, Celgene, Janssen-Cilag, Galderma, InflaRx, Lilly, Novartis, Pfizer, Regeneron, and UCB. His employer has received investigator-initiated grants from AbbVie, Celgene, Janssen-Cilag, and UCB. Thierry Passeron has received grants and/or honoraria from AbbVie, ACM Pharma, Amgen, Almirall, Boehringer Ingelheim, Bristol Myers Squibb, Celgene, Galderma, Genzyme/Sanofi, GlaxoSmithKline, Incyte, Janssen, LEO Pharma, Lilly, Novartis, Pfizer, Sun Pharma, and UCB. Emanual Maverakis has received consulting fees from AbbVie, E.R. Squibb and Sons, and Kyowa Hakko Kirin. Irina Turchin has served as a consultant, speaker, and investigator for AbbVie, Amgen, Arcutis, Aristea, Bausch Health, Boehringer Ingelheim, Celgene, Galderma, Incyte, Janssen, Kiniksa, LEO Pharma, Lilly, Novartis, Pfizer, Sanofi, and UCB. Stefan Beeck, Leonidas Drogaris, Ziqian Geng, Tianyu Zhan, and Izabella Messina are full-time employees of AbbVie and may own AbbVie stock or stock options. Falk G. Bechara has received honoraria for participation in advisory boards, in clinical trials, and/or as a speaker for AbbVie, AbbVie Deutschland, Boehringer Ingelheim, Incyte, Janssen-Cilag, MoonLake, Novartis, and UCB.
Compliance with Ethics Guidelines
The study was conducted in accordance with the protocol, International Council for Harmonisation guidelines, and applicable regulations, guidelines, and ethical principles originating from the 1964 Declaration of Helsinki. The study protocol was reviewed and approved by central (Advarra IRB Services, Columbia, MD, USA) and by local independent ethics committees and/or institutional review boards at each study site. Patients provided written informed consent prior to screening or undergoing study-specific procedures.
Data Availability
The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request. AbbVie is committed to responsible data sharing regarding the clinical trials we sponsor. This includes access to anonymized individual and trial-level data (analysis data sets), as well as other information (e.g., protocols, clinical study reports, or analysis plans), as long as the trials are not part of an ongoing or planned regulatory submission. This includes requests for clinical trial data for unlicensed products and indications. These clinical trial data can be requested by any qualified researchers who engage in rigorous, independent, scientific research, and will be provided following review and approval of a research proposal, Statistical Analysis Plan (SAP), and execution of a Data Sharing Agreement (DSA). Data requests can be submitted at any time after approval in the US and Europe and after acceptance of this manuscript for publication. The data will be accessible for 12 months, with possible extensions considered. For more information on the process or to submit a request, visit the following link: https://www.abbvieclinicaltrials.com/hcp/data-sharing/.html.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Kimball, A.B., Prens, E.P., Passeron, T. et al. Efficacy and Safety of Risankizumab for the Treatment of Hidradenitis Suppurativa: A Phase 2, Randomized, Placebo-Controlled Trial. Dermatol Ther (Heidelb) 13, 1099–1111 (2023). https://doi.org/10.1007/s13555-023-00913-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13555-023-00913-3