
Abstract
Precision dermatology uses individualized dermatologic disease-directed targeted therapy (D3T2) for the management of dermatoses and for the evaluation and therapy of cutaneous malignancies. Personalized/precision strategies are based on biomarkers that are most frequently derived from tissue transcriptomic expression or genomic sequencing or from circulating cytokines. For instance, the pathologic diagnosis of a pigmented lesion and determining the prognosis of a malignant melanocytic neoplasm can be enhanced by genomic/transcriptomic analysis. In addition to biopsy, innovative techniques have been developed for obtaining transcriptomes in skin conditions; as an example, patches can be applied to a psoriasis plaque for a few minutes to capture the epidermis/upper dermis transcriptome. Atopic dermatitis and prurigo nodularis may also be candidate conditions for precision dermatology. Precision dermatology has a role in managing melanoma and nonmelanoma skin cancers and rare cutaneous tumors—such as perivascular epithelioid cell tumor (PEComa)—that can originate in or metastasize to the skin. For instance, advanced/metastatic basal cell carcinomas can be treated with Hedgehog inhibitors (vismodegib and sonidegib) targeting the smoothened (SMO) or patched 1 (PTCH1) gene alterations that are a hallmark of these cancers and activate the Hedgehog pathway. Advanced/metastatic basal and cutaneous squamous cell cancers often have a high tumor mutational burden (which predicts immunotherapy response); immune checkpoint blockade with cemiplimab, a programmed cell death protein 1 (PD1) inhibitor, is now approved for these malignancies. Gene expression profiling of primary cutaneous squamous cell carcinoma can identify those individuals at high risk for subsequent metastases. In the realm of rare neoplasms, PEComas—which can originate in the skin, albeit uncommonly—have tuberous sclerosis complex 1 (TSC1)/tuberous sclerosis complex 2 (TSC2) gene alterations, which activate mammalian target of rapamycin (mTOR) signaling, and can be suppressed by nab-sirolimus, now approved for this condition. In summary, precision dermatologic techniques/strategies are an important emerging approach for evaluation and management of skin disorders and cutaneous neoplasms, and may serve as a paradigm for the application of precision medicine beyond dermatology.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
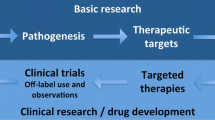
Precision dermatology incorporates individualized dermatologic disease-directed targeted therapy (D3T2) for the management of dermatoses and includes important applications in the evaluation, diagnosis, prognosis, and therapy of patients with cutaneous malignancies. |
Patient-focused specific therapy is based on either biomarker profiles (derived from disease-related skin lesions or plasma) or genomic profiling of the tumor. |
Precision dermatology can be used to determine effective agents for patients with dermatoses such as psoriasis, atopic dermatitis, and prurigo nodularis. |
Precision dermatology can be used to evaluate the diagnosis and/or determine the treatment of not only melanoma and nonmelanoma [such as basal cell carcinoma (BCC) and squamous cell carcinoma (SCC)] cutaneous malignancies, but also rare cancers that originate or metastasize to the skin, such as perivascular epithelioid cell tumor (PEComa). |
Precision dermatology is the next therapeutic frontier for the treatment of patients with dermatologic conditions. |
Introduction
Precision medicine utilizes specific molecular biomarkers to assess a single individual’s condition or neoplasm, so that a precise—and, optimally, most effective—treatment for that person with disease-directed drugs or tumor-targeted agents can be determined [1,2,3]. Disease-directed antibiotic therapy is used to manage patients with infectious conditions [4,5,6,7,8,9]. Similarly, matched targeted therapy is used to treat cancers [10,11,12,13,14]. Precision medicine may also take into account environment and lifestyle. In this review, we will focus on precision dermatology based on molecular biomarkers.
Dermatologic disease-directed targeted therapy (referred to herein as D3T2 or precision dermatology) can be used to predict which drug(s) will be optimal for the management of an individual with a specific cutaneous condition or cancer. Herein, we discuss illustrative examples in which precision dermatology has been applied: dermatoses (such as psoriasis, atopic dermatitis, and prurigo nodularis), cutaneous malignancies [such as basal cell carcinoma (BCC), squamous cell carcinoma (SCC), and melanoma], and rare cancers, such as perivascular epithelioid cell tumor (PEComa) that can originate in the skin.
Methods
The Medline database was searched for terms using PubMed. The terms included “atopic dermatitis,” “BCC,” “dermatology,” “melanoma,” “oncology,” “personalized treatment,” “precision,” “precision dermatology,” “PEComa,” “perivascular epithelioid cell tumor,” “precision medicine,” “pruritus,” “prurigo nodularis,” “psoriasis,” “rare,” “rare tumor,” “SCC,” “targeted therapy,” and “treatment,” and works that were considered relevant were used as the basis for this narrative review.
Compliance with Ethics Guidelines
This article is based on previously conducted studies and does not contain any new studies with humans or animals.
Discussion
Precision dermatology—the application of precision medicine using molecular markers, including transcriptomic assessment, to dermatologic conditions and cancers—is being used for the management of psoriasis; potentially, the same learning-based transcriptomic type test could be applicable in atopic dermatitis. Targeted therapy chosen based on plasma cytokine biomarker profiles may be effective in prurigo nodularis. Precision dermatology (genomic/transcriptomic) is being exploited for the evaluation and/or management of melanoma and nonmelanoma skin cancers (such as BCC and SCC). Finally, genomics has identified precisely targeted new treatments for rare cutaneous tumors, such as PEComas.
Psoriasis
Psoriasis is a T helper type 1 (Th1)-cell-mediated and Th17-cell-mediated inflammatory papulosquamous condition which may be associated with arthritis and other comorbidities. Numerous topical therapies, such as corticosteroids, retinoids, salicylic acid, tar, and vitamin D are available for individuals with limited disease; recently, tapinarof (an aryl hydrocarbon receptor agonist) and roflumilast [a phosphodiesterase 4 (PDE4) inhibitor] have been approved for topical use. In addition, phototherapy, primarily using narrow-band (322 nm) ultraviolet B light or broad-band (290 to 320 nm) ultraviolet B light or using psoralen and ultraviolet A (320 to 400 nm) light, is also a treatment modality. Systemic anti-metabolites (such as methotrexate), cyclosporin, and retinoids (such as acitretin) are also available. Other systemic therapies such as apremilast (a PDE4 inhibitor), tumor necrosis factor (TNF)-alpha inhibitors, and other biologic agents that block interleukins (ILs) (such as inhibitors of IL-17, IL-23, and IL-12/23) are utilized for patients with more extensive psoriasis; in addition, Janus kinase (JAK) inhibitors (tofacitinib and upadacitinib) have recently been added to the pharmacologic armamentarium for psoriatic arthritis [15,16,17,18,19,20].
Currently, one of the factors that strongly influences the selection of systemic therapies for psoriasis is financial coverage of the medication by the patient’s insurance company. Many patients must fail to respond to or not be eligible for methotrexate therapy prior to receiving approval for other treatments; IL inhibitors will often not be approved unless TNF-alpha inhibitors have been unsuccessful. Hence, the management of psoriasis patients is often based on drug cost, and not necessarily potential medication efficacy [21].
A machine-learning-based test for predicting the response of psoriasis to biologic agents has recently been developed; it uses transcriptomes [ribonucleic acids (RNAs)] obtained from the patient’s psoriasis plaques to predict treatment response to IL-23 inhibitors, IL-17 inhibitors, or TNF-alpha inhibitors. Patches were applied to psoriasis plaques for 5 min and captured the whole transcriptome from the epidermis and upper dermis. The messenger ribonucleic acid (mRNA) was extracted from the patch and next-generation sequencing was used to analyze the transcriptome; patient psoriasis area and severity index (PASI) scores were also recorded at baseline and at weeks 12 and 16 after drug exposure. The clinical results from treatment and the transcriptomic information obtained from the psoriasis lesions were used to develop a machine-learning-based classifier for IL-23 inhibitors, IL-17 inhibitors, or TNF-alpha inhibitors [22, 23].
The classifiers were subsequently validated as an accurate predictor of the psoriasis patient’s response to each of the classes of biologic therapies. A study of 242 psoriasis patients was performed; 118 patients were treated with an IL-23 inhibitor, 79 patients were treated with an IL-17 inhibitor, and 35 patients were treated with a TNF-alpha inhibitor. The positive predictive values for IL-23 inhibitors, IL-17 inhibitors, and TNF-alpha inhibitors were 93.1%, 92.3%, and 85.7%, respectively (in contrast to the response rates of about 66% to 86% to IL-23 inhibitors; 60–70% to IL-17 inhibitors, and 25–60% to TNF-alpha inhibitors seen in patients; hence, precision dermatology based on a machine-learning-based test that evaluates baseline mRNA biomarkers can be used to select the biologic drug therapy to which a psoriasis patient is most likely to respond [20, 22,23,24,25,26]. Cost savings may be realized by giving the right drugs to the right patient at the right time: the sine qua non of precision medicine; in psoriasis, these cost savings have been estimated to average $8492 per year when transcriptomics is used to predict responders [21]. Future goals may be to expand the machine learning for classifiers that can also predictively assess the response to PDE4 inhibitors and JAK inhibitors.
Atopic Dermatitis
Atopic dermatitis is predominantly a Th2-cell-mediated inflammatory skin condition. In atopic individuals, in addition to elevated immunoglobulin E and eosinophilic responses, the Th2 cytokines IL-4, IL-5, and IL-13 have a pathogenic role; also, IL-31 participates in dermatitis-associated pruritus. Non-immunologic components in atopic dermatitis pathogenesis include epidermal barrier dysfunction secondary to filaggrin deficiency and skin microbiome alterations [27,28,29,30,31,32].
The treatment of atopic dermatitis is suggested based on the level of disease (mild, moderate, or severe) and whether the therapy is for an acute flare or maintenance. Numerous topical therapies are available: corticosteroids, calcineurin inhibitors (such as pimecrolimus and tacrolimus), PDE4 inhibitors (such as crisaborole and roflumilast), and JAK inhibitors (such as ruxolitinib). In addition, for more severe disease, phototherapy and systemic immunosuppressants (such as azathioprine, corticosteroids, cyclosporin, methotrexate, and mycophenolate mofetil) are also available. Dupilumab is another systemic therapy for atopic dermatitis; it binds to the IL-4 receptor alpha, inhibiting IL-4 and IL-13 signaling; in addition, drugs targeting IL-13 have either recently been approved (tralokinumab) or are being evaluated in clinical trials (lebrikizumab). Finally, oral JAK inhibitors (abrocitinib and upadacitinib) are approved for atopic dermatitis treatment in the United States [27,28,29,30, 33].
Researchers have suggested that stratification of atopic dermatitis patients should be performed. Categorization based upon biomarker endotypes could differentiate newborn, children, and adult atopic dermatitis patients into groups characterized by different biochemical forms of skin barrier dysfunction, immune dysfunction, and microbial dysbiosis. Thereafter, individualized therapy directed towards the specific abnormalities that each patient exhibits could be initiated [32].
In summary, atopic dermatitis has several immunologic pathways that contribute to its pathogenesis; hence, it should be an excellent candidate for the application of precision dermatology to provide targeted therapy for the individual patient. Similar to psoriasis, a machine-learning-based test could potentially be developed to predict the response to topical or systemic agents such as inhibitors of PDE4, IL-4, IL-13, IL-31, and JAK [25, 32].
Prurigo Nodularis
Prurigo nodularis is a chronic inflammatory dermatosis characterized by single or multiple intensely pruritic, often symmetrically distributed, hyperkeratotic nodules; typically, they are located on the extremities and trunk. Several inflammatory and pruritic cytokines are elevated in patients with prurigo nodularis: IL-4, IL-13, and IL-31. Systemic—established and investigational—treatment modalities used in the management of this condition include a calcineurin inhibitor (such as cyclosporine), an IL-4 inhibitor (such as dipulimab), IL-31 inhibitors [such as nemolizumab and the oncostatin-M (OSM)-specific beta receptor antagonist vixarelimab], immunosuppressants (such as azathioprine, corticosteroids, and methotrexate), JAK inhibitors (such as abrocitinib and tofacitinib), and receptor tyrosine kinase (KIT) inhibitors [34, 35].
Researchers were able to clinically identify response predictors of pruritus resolution in prurigo nodularis patients treated with dupilumab (which inhibits IL-4 and IL-13 signaling). Patients who experienced at least a 50% decrease in itch within 8 weeks of treatment were likely to eventually experience 100% resolution of their pruritus; however, those who did not reach this therapeutic endpoint usually did not respond to the medication. Also, a therapeutic response to dupilumab took longer to occur in prurigo nodularis with versus without atopic dermatitis [36].
Other investigators evaluated circulating plasma cytokines in patients with prurigo nodularis and discovered two distinct groups of patients: cluster 1 (consisting of fewer African Americans, a noninflammatory plasma profile, and a higher rate of myelopathy) and cluster 2 [consisting of more African Americans, an inflammatory plasma profile with higher levels of IL-1 alpha, IL-4, IL-5, IL-6, IL-10, IL-17A, IL-22, IL-25, and interferon (IFN)-alpha, higher itch scores, and lower quality-of-life scores]. Also, in comparison to Caucasian prurigo nodularis patients, African American prurigo nodularis patients had lower transferrin and higher acute-phase reactants [such as C-reactive protein (CRP) and erythrocyte sedimentation rate (ESR)], eosinophils, and ferritin. The investigators concluded that patients with prurigo nodularis could be differentiated into one of two groups—each characterized by distinctive demographic and clinical features—based on the results of a plasma biomarker profile evaluation [37].
Hence, it has been postulated that it may be possible to establish individualized and specifically targeted treatments for prurigo nodularis patients based upon their plasma cytokine profiles [35, 37]. Therefore, serologic evaluation—with or without the additional observation of a clinical drug challenge—may help to differentiate subsets of prurigo nodularis patients and predict optimal therapeutic interventions. Indeed, similar methods of evaluation may potentially be useful for other patients with systemic conditions characterized by severe pruritus, such as chronic renal failure or end-stage liver disease.
Melanoma
Patients with earlier-stage melanoma have a better 5-year survival rate than those with later-stage disease. Herein, we will focus on advances in molecular technology that can potentially aid in the diagnosis and in evaluating the prognosis of early-stage cutaneous melanomas (with metastatic disease having been addressed extensively in the oncology literature) [38,39,40,41,42].
Tests are commercially available that utilize gene expression profiles (GEPs) to assist in several areas: (i) the decision to biopsy a clinically and/or dermatoscopically equivocal pigmented lesion; (ii) the dermatopathologist’s final diagnosis when histologic features do not clearly differentiate the lesion as being benign versus malignant; and (iii) the determination of the prognosis for patients with early-stage melanoma. Indeed, a melanoma GEP consensus-based recommendation panel proposed appropriate use criteria for the integration of diagnostic and prognostic GEP assays into the management of cutaneous melanoma [38,39,40,41,42,43,44,45,46,47,48,49,50,51,52,53,54,55,56,57,58,59,60,61,62,63,64,65,66,67,68,69].
2-GEP and 3-GEP Testing: Clinical Diagnosis
A noninvasive molecular test has been developed to provide additional information for clinicians when deciding whether to biopsy a pigmented lesion that is equivocal based on its clinical appearance, dermatoscopic features, or both. This pigmented lesion assay (PLA) analyzes the levels of RNA expression of two melanoma-associated biomarker genes: long intergenic non-protein coding RNA 518 (LINC) (also referred to as LINC00518) and preferentially expressed antigen in melanoma (PRAME). The former is involved in melanoma proliferation as part of a cluster of regulatory RNA molecules, and the latter interferes with retinoic acid receptor (RAR) signaling, thereby promoting tumor progression (Table 1) [43,44,45,46,47].
An adhesive patch is applied to the pigmented lesion; RNA-containing stratum corneum that is adherent to the patch is collected when the patch is removed. Quantitative reverse-transcriptase polymerase chain reaction (qRT-PCR) is used to both reverse transcribe the RNA to deoxyribonucleic acid (DNA) and to amplify the DNA in order to determine the level of the specific gene expression being evaluated. If LINC or PRAME or both genes are detected, the PLA is reported as positive (and clinicians are likely to biopsy the lesion); however, if neither gene is present, the PLA is reported as negative and clinicians might consider monitoring the lesion with periodic clinical evaluation [43,44,45,46,47].
The sensitivity, specificity, and negative predictive value of the PLA were 91–95%, 69–91%, and greater than 99%, respectively. Additional evaluation was performed for telomerase reverse transcriptase (TERT; a high-risk DNA driver mutation commonly found in early-stage melanoma) from the pigmented lesion sample, and this was recently incorporated as an additional diagnostic test to enhance the PLA’s sensitivity. The 3-GEP test (Table 1), by combining DNA risk factors (TERT mutation analysis) and melanoma-associated RNA gene expression (LINC and/or PRAME detection), increased the test sensitivity to rule out melanoma from 93 to 97% [48, 49].
23-GEP and 35-GEP Testing: Pathology Diagnosis
There are circumstances when the diagnosis of the pigmented lesion is histologically challenging for the dermatopathologist because the lesion has equivocal features under light microscopy. Noninvasive molecular tests have been developed to provide additional information for pathologists in their assessment of a primary cutaneous melanocytic lesion for which malignant potential is uncertain. One of the tests assesses the expression of 23 genes to determine whether the lesion is benign or malignant; the genes, which are involved in cell differentiation, cell–cell signaling, and immune response, vary in a predictable manner between nevi and malignant melanoma (Table 1) [50,51,52,53,54,55,56,57,58].
Tissue from the biopsy specimen which has been fixed in formalin and embedded in paraffin is used for the test. RNA is extracted from the processed biopsy specimen, and the expression of the 23 genes is determined by qRT-PCR. A numeric score (ranging from − 16.7 to 11.7) is determined; the results are reported as benign (with a numeric range from − 16.7 to − 2.1), intermediate (with a numeric range from − 2.0 to − 0.1), or malignant (with a numeric range from 0 to 11.1) [50,51,52,53,54,55,56,57,58].
The 23-GEP test has a sensitivity and specificity of 91.5 to 94.0% and 90.0 to 92.5%, respectively. However, depending on the study, the test resulted in an indeterminate (intermediate) diagnosis for 2.9 to 16.2% of the lesions. Subsequently, a 35-GEP test was developed to improve the classification of the pigmented lesions that were diagnosed as intermediate using the 23-GEP test [50,51,52,53,54,55,56,57,58].
The 35-GEP test is only recommended for individuals 18 years of age or older, and classifies the melanocytic lesion as either benign, having an intermediate risk of malignancy, or malignant (Table 1) [59, 60]. The sensitivity and specificity (99.1% and 96.2%, respectively) of the 35-GEP test are superior to those of the 23-GEP test. In addition, when the 35-GEP test is used, only 3.5% of the pigmented lesions are classified as indeterminate [59, 60].
10-GEP and 31-GEP Testing: Prognosis
Noninvasive molecular tests have been developed to provide prognostic information for the patient who has been diagnosed with a melanoma. The 10-GEP test was used to identify patients with primary cutaneous melanoma at low risk for nodal metastases (Table 1) [42, 61]. The 31-GEP test is primarily useful for risk stratification in patients with early-stage melanoma, where individuals who might benefit from heightened surveillance and closer follow-up (even though they may have previously been designated as being low risk) are identified (Table 1) [62,63,64,65,66,67,68,69].
Similar to the 23-GEP and 35-GEP tests, RNA is extracted from the formalin-fixed, paraffin-embedded tissue biopsy specimen in the 31-GEP test; qRT-PCR is then used to quantify the expression of the 31 genes and to classify the lesion regarding risk for metastasis. There are four categories: class 1A (0 to 0.41, lowest risk), class 1B (0.42 to 0.49, low risk), class 2A (0.50 to 0.58, high risk), and class 2B (0.59 to 1, highest risk). Hence, lesions that are either class 1B or class 2A are considered to present increased (or intermediate) risk [62,63,64,65,66,67,68,69].
The 31-GEP test also provides the percentage likelihood of sentinel lymph node biopsy positivity. For example, a 55-year-old patient with a T1 tumor (Breslow depth of less than 1 mm) to T2 tumor (Breslow depth of between 1 and 2 mm) and a 31-GEP class 1A result would have a less than 5% risk of sentinel lymph node positivity and a more than a 99% melanoma-specific survival. However, there are no definitive management recommendations regarding sentinel lymph node biopsy when there is discordance between the tumor depth and 31-GEP class result. In addition, the 31-GEP test results are not currently incorporated in the staging criteria of the American Joint Committee on Cancer (AJCC) or the National Comprehensive Cancer Network (NCCN) [62,63,64,65,66,67,68,69].
Basal Cell Carcinoma (BCC)
The most frequently occurring skin cancer is BCC. Although some of these tumors may be locally invasive, they can be successfully managed using localized treatment such as a nonsurgical therapy (such as cryotherapy, photodynamic therapy, radiation therapy, or topical therapies including 5-fluorouracil and imiquimod) or a surgical intervention (such as curettage and desiccation, standard excision, or Mohs micrographic surgery). In contrast, locally advanced and metastatic BCCs are uncommon and are not amenable to localized treatment [70,71,72].
Most BCCs have a mutation in the Sonic Hedgehog signaling pathway. Genomic alteration of the transmembrane tumor suppressor protein patched 1 (PTCH1), the receptor for the Hedgehog protein, can activate the pathway by releasing its inhibition of the smoothened (SMO) proto-oncoprotein; also, albeit less commonly, mutations in SMO can result in pathway activation. Once SMO is no longer inhibited, downstream signaling continues, involving not only suppressor of fused homolog (SUFU) but also activation of the glioma-associated oncogene (GLI) family of transcription factors [72, 73].
Precision dermatology—targeted therapy based on the tumor’s molecular profile—is already being applied for the treatment of advanced and metastatic BCCs. Vismodegib and sonidegib are Hedgehog inhibitors that specifically target SMO. Indeed, they are approved by the Food and Drug Administration (FDA) for the management of advanced/metastatic BCCs [73,74,75].
In addition to PTCH1 abnormalities, several other genomic aberrations have been observed that involve the SMO, SUFU, and tumor protein (TP)53 genes. Indeed, in BCC patients whose tumors are resistant to either vismodegib or sonidegib, some of these gene mutations may in the future be targetable [72, 75,76,77]. Other agents can also theoretically be utilized in a genomically targeted approach. For instance, itraconazole inhibits the Hedgehog pathway at the level of SMO. Arsenic trioxide inhibits GLI transcriptional activity [74].
Advanced and metastatic BCCs have several unique features. In particular, they often have high tumor mutational burdens; there are also case reports of programmed death-ligand 1 (PD-L1) gene amplification [73,74,75]. These molecular features sensitize to immune checkpoint blockade with programmed cell death protein 1 (PD1)/PD-L1 inhibitors. Therefore, immunotherapy is also a treatment option for patients with advanced and metastatic BCCs [73, 75, 76, 78]. Currently, cemiplimab—a PD1 inhibitor—is the only FDA-approved immunotherapy agent for Hedgehog inhibitor-resistant advanced and metastatic BCCs [74, 78, 79]. However, there are several reports of other anti-PD1 agents (such as nivolumab or pembrolizumab) that have been successfully used to treat advanced (Figs. 1, 2, 3) or metastatic (Fig. 4) BCCs, either as monotherapy or as part of a combination utilizing both a Hedgehog inhibitor and an immunotherapy agent [73, 75, 80, 81]. Immunotherapy is an especially effective therapeutic intervention for advanced and metastatic BCC patients with late disease (characterized by many molecular alterations and an increased tumor mutational burden), in contrast to those individuals in which there are fewer genomic aberrations and a lower tumor mutational burden (Fig. 5) [76, 77].

Advanced basal cell carcinoma (BCC): clinical presentation. Posterior (A) and lateral (B) views of the left upper back of a 62-year-old man with an advanced BCC without metastatic disease. The mass had been increasing in size during the previous 13 months and presented as a large (10 × 8 × 2.5 cm) exophytic tumor. The erythema surrounding the tumor was caused by adhesive-bandage-associated allergic contact dermatitis. Republished from [73] with permission from Elsevier

Advanced basal cell carcinoma (BCC): pathology presentation. Lower (A) and higher (B) magnification views of the microscopic examination of the advanced BCC from the 62-year-old man’s left upper back show nodular aggregates of basaloid tumor cells extending from the overlying epidermis and invading the underlying dermis. Next-generation sequencing of the tumor demonstrated a high tumor mutational burden of 53 mutations per megabase (10 or more mutations per megabase is considered to be a high tumor mutational burden) and 11 deleterious genomic variants, including PTCH1 (splice site 1504-1G > T), ASXL1 Q760, INPP4B W521, KEL R130Q, PIK3R1 R534, PTEN (splice site 210 2A > T), RAC1 P29S, TERT promoter-124C > T, TP53 R196, TP53 Q100, and WT1 C350R (hematoxylin and eosin: A, × 4; B, × 20). Republished from [73] with permission from Elsevier

Advanced basal cell carcinoma (BCC): complete and sustained tumor clearance. Posterior (A) and lateral (B) views of the left upper back of a 62-year-old man demonstrates clearance of the advanced BCC. He concurrently received vismodegib and a total of four doses of nivolumab; the latter was discontinued because of a skin rash and recurrent transaminitis, and the former was stopped after 8.5 months because of appetite loss. Multiple skin biopsies confirmed complete and sustained remission after 5 months of treatment. He has remained in complete remission for 68.5 months (though therapy was stopped ~ 60 months earlier). Republished from [73] with permission from Elsevier

Metastatic basal cell carcinoma (BCC) with liver metastases in a 56-year-old man: liver biopsy. At age 54 years, a man developed a BCC on his left posterior shoulder; the excised tumor recurred; the subsequent positive postoperative margins were treated with radiotherapy. Two years later, at 56 years old, an evaluation of back pain demonstrated metastatic BCC; lower (A) and higher (B) magnification views show aggregates of basaloid tumor cells with large pleomorphic nuclei on the liver biopsy tissue specimen. In addition to liver metastases, his BCC had also metastasized to his axial skeleton and lungs. The metastatic BCC was refractory not only to chemotherapy (cisplatin and paclitaxel), but also Hedgehog inhibitors (vismodegib and sonidegib; the latter had been combined with buparlisib, a pan-class I PI3K inhibitor). Next-generation sequencing of his liver metastasis showed a tumor mutational burden of 103 mutations per megabase and multiple genomic alterations including programmed death ligand 1 (PD-L1) amplification. Based on these results, which correlate with a better response to immunotherapy, he was treated with the anti-programmed death (anti-PD1) checkpoint inhibitor nivolimab and achieved a near-complete remission of his widely metastatic BCC within 4 months after starting therapy (hematoxylin and eosin: A, × 10; B, × 40). Within 1 year he achieved complete remission, which has been durable for over 6 years and is ongoing. Republished from [76] with permission from Springer Nature

New primary cutaneous superficial basal cell carcinomas (BCCs) during successful nivolumab treatment of widely metastatic BCC (including liver metastases) in a 58-year-old man: clinical lesions and pathology presentation of skin tumors. Distant (A) and closer (B, C) views of new primary cutaneous BCCs that presented as erythematous plaques on the left anterior shoulder (6 × 6 mm, and labeled A with ink on his skin in images A and B) and left chest (8 × 6 mm, and labeled B with ink on his skin in images A and C) and developed 9 months after nivolumab was initiated; this corresponded to 5 months after achieving near-complete remission of his widely metastatic BCC, when he was still receiving immunotherapy treatment with the checkpoint inhibitor. Microscopic examination of both skin biopsy tissue specimens showed similar pathologic changes: superficial buds of basaloid tumor cells extending from the epidermis into the papillary dermis (which contained solar elastosis, small telangiectasias, and a sparse lymphocytic inflammatory infiltrate); in addition to palisading of the tumor keratinocytes at the periphery of the carcinoma aggregates, retraction of the surrounding dermal stroma resulted in cleft formation (D). In contrast to the tissue specimen from his liver biopsy, next-generation sequencing of the new primary cutaneous superficial BCCs showed a tumor mutational burden of 45 mutations per megabase and fewer genomic alterations; in addition, it did not demonstrate programmed death ligand 1 (PD-L1) amplification. Both of the new primary cutaneous superficial BCCs were treated with electrodessication and curettage; there was complete healing of the skin cancer treatment sites without tumor recurrence at an 8-month follow-up examination (hematoxylin and eosin: D, × 10). Republished from [77] with permission from MDPI (Multidisciplinary Digital Publishing Institute), Barcelona, Spain
Squamous Cell Carcinoma (SCC)
Cutaneous SCC has significantly increased in incidence. Indeed, metastatic disease can develop in individuals whose tumors have high-risk factors. Once metastases develop, survival rates drop [82,83,84].
Three staging systems have been proposed for cutaneous SCC. The AJCC is limited to tumors of the head and neck; although it includes all histologic subtypes of malignancy, it primarily focuses on cutaneous SCC. The Brigham and Woman’s Hospital (BWH) system assesses outcome and provides prognostication based upon whether specific clinical and pathologic risk factors are present. The NCCN provides clinical guidance for the treatment of cutaneous SCC by stratifying the tumor as high risk or low risk [82,83,84].
The BWH system categorizes tumors as either T0 (in situ SCC), T1 (no risk factors), T2a (one risk factor), T2b (two or three risk factors), or T3 (four risk factors or bone invasion). The clinical risk factor is a tumor diameter of 2 cm or larger; pathologic risk factors include a poorly differentiated histology, perineural invasion, and tumor invasion beyond the subcutaneous fat (however, if there is bone invasion, the tumor is categorized as T3). The NCCN stratification incorporates additional clinical (such as location, poorly defined or well-defined borders, primary or recurrent, presence or absence of immunosuppression, rapid or slow growth rate, and presence or absence of neurologic symptoms) and pathologic (such as histologic subtype, depth, and lymphatic or vascular involvement) tumor parameters to those included by the BWH system [82,83,84].
However, these clinicopathologic staging systems have not only low sensitivity but also low positive predictive value. For example, some of the cutaneous SCC patients who were initially classified as low risk developed metastatic disease. In addition, other patients with cutaneous SCC who are classified as high-risk do not develop metastases [82,83,84,85,86,87,88,89,90,91,92].
A 40-GEP test has been developed to improve identification of individuals with primary cutaneous SCC who have a high risk of subsequently developing metastatic disease and to guide the decisions made by physicians regarding the possible performance of sentinel lymph node biopsy, potential management (such as radiation, chemotherapy, or immunotherapy), and follow-up intervals (ranging from every 3 to 6 to 12 months). The test can be used for a cutaneous SCC patient with one or more risk factors (Table 2) [82, 85,86,87,88,89,90,91,92]. Indeed, when it is used in combination with traditional risk factors, the result provides additional risk prediction of metastasis and death for the patient [82,83,84,85,86,87,88,89,90,91,92].
The method for genomic evaluation of the cutaneous SCC using the 40-GEP test is similar to that used to evaluate melanocytic lesions. RNA is extracted after macrodissection of the cutaneous SCC tumor tissue (including tumor stroma) from the formalin-fixed, paraffin-embedded tissue biopsy specimen. Subsequently, qRT-PCR is used to generate complementary DNA so that the expression of the 40 genes can be quantified and the tumor’s risk for metastasis can be classified [82,83,84,85,86,87,88,89,90,91,92].
The biological risk of metastasis in a SCC patient is classified as either low risk (class 1, with a 93.9% 3-year metastasis-free survival), moderate risk (class 2A, with an 80.5% 3-year metastasis-free survival), or high risk (class 2B with a 47.8% 3-year metastasis-free survival), based only on the results of the 40-GEP test. Overall, the risk of metastasis is less than half of that for the general study population for class 1 patients. A risk of 20% aligns with that of high-risk staging (BWH T2b and T3) for class 2A patients, and a risk of greater than 50% for class 2B patients is significantly higher than the risk of less than 7% for patients with class 1 results [82,83,84,85,86,87,88,89,90,91,92].
The metastasis rate was also determined when the result of the 40-GEP test in a patient with primary cutaneous SCC was assessed in combination with the additional history of whether the patient had only one or more than one associated high-risk factor (defined by combining those used by the BWH and NCCN staging systems); 60 of the 63 metastases (in the cohort group of 420 patients) occurred within 3 years. The metastasis rate for primary cutaneous SCC patients with one high-risk factor was 4%, 10.8%, and 60.0% for individuals classified as class 1, class 2A, and class 2B, respectively. Also, the metastasis rate for primary cutaneous SCC patients with two or more high-risk factors was 9%, 25.0%, and 50.0% for individuals classified as class 1, class 2A, and class 2B, respectively [82,83,84,85,86,87,88,89,90,91,92].
Finally, like advanced basal cell carcinomas, advanced cutaneous squamous cell carcinomas may have high mutational burdens, a feature that correlates with response to immunotherapy. The anti-PD1 agent cemiplimab is active in advanced cutaneous squamous cell cancers and is FDA approved for this indication [79, 93, 94].
Rare Cutaneous Tumors: Perivascular Epithelioid Cutaneous Tumor (PEComa)
Rare visceral tumors provide a unique opportunity for precision medicine utilizing genomically-based treatment. Similarly, the management of rare cutaneous tumors may also be optimized by incorporating precision dermatology with cancer-directed therapy based upon next-generation sequencing of the malignant neoplasm [95].
PEComa is a rare tumor that is composed of cells that demonstrate both melanocytic and smooth muscle differentiation. Most PEComas are systemic (originating from various visceral organs or soft tissue) and predominantly occur in women (with the female-to-male ratio ranging from 4:1 to 7:1). Benign and malignant variants of the tumor exist [96, 97].
Cutaneous PEComa is extraordinarily rare; only 67 patients have been described: 60 with primary benign PEComa, 5 with primary malignant PEComa, and 2 with skin metastases from a malignant visceral PEComa. In contrast to systemic PEComa, the female-to-male ratio was only 2:1. The clinical presentation of a cutaneous PEComa is most commonly an asymptomatic, slowly enlarging nodule (Fig. 6). Similar to systemic PEComa, cutaneous neoplasm consists of an admixture of tumor cells (either epithelioid or spindle or both) and blood vessels (Figs. 6, 7) [96, 98].

Primary malignant cutaneous perivascular epithelioid cell tumor (PEComa): clinical presentation and hematoxylin-and-eosin-stained pathology presentation. A 43-year-old man who had worked as a welder for several years presented with a tumor of 5 months’ duration (that had grown from a small, raised area to its current size over a period of 3 months) on the extensor surface of his distal left forearm just proximal to the wrist (A). The neoplasm was a painless, flesh-colored, exophytic scaly nodule measuring 10 × 10 × 5 mm, with a surrounding collarette of epithelium and central ulceration; the initial clinical impression was a keratoacanthoma (a variant of squamous cell carcinoma). Lower (B) and higher (C) magnification views of the microscopic examination of the excisional biopsy show ulceration of the epidermis, crust, and a collarette of epithelium extending from the epidermis into the dermis and surrounding a dermal tumor predominantly consisting of epithelioid cells (most with clear cytoplasm and some with foamy cytoplasm) and numerous capillaries; in addition, spindle tumor cells are present at the tumor periphery, and there are some multinucleated tumor cells. The tumor is classified as malignant based upon the presence of two high-risk, worrisome features: increased mitotic activity (with three mitoses per ten high-power fields) and scattered nuclear pleomorphism (demonstrated by high-grade nuclear atypia) (hematoxylin and eosin: B, × 2; C, × 20). Republished from [96] with permission from the University of California, Davis Department of Dermatology, Sacramento, California

Primary malignant cutaneous perivascular epithelioid cell tumor (PEComa): pathology presentation of smooth muscle and melanocyte markers using immunoperoxidase stain. The tumor cells show strong and diffuse staining with the smooth muscle marker caldesmon (A) and the melanocytic marker microphthalmia transcription factor (MiTF) (B); however, the melanocytic marker human melanoma black 45 (HMB45) shows weak and diffuse staining (C). Genomic analysis of the man’s blood showed a Fanconi anemia complementation group C (FANCC) germline mutation. Next-generation sequencing of his tumor showed four actionable pathogenic aberrations: baculoviral IAP (inhibitor of apoptosis) repeat containing 3 (BIRC3) splice site 1622-27_1631del37, FANCC R185*, tumor protein 53 (TP53) R248W, and tuberous sclerosis complex 1 (TSC1) T4151. His systemic workup was negative for metastases and a wide local excision of the tumor site was performed. There had been no recurrence or metastasis of the tumor after 15 months of follow-up; however, if the tumor was to recur or metastasize, based on the genomic profile of his PEComa, sirolimus would be considered for first-line therapy (caldesmon: A, × 20; MiTF: B, × 20; HMB45: C, × 20). Republished from [96] with permission from the University of California, Davis Department of Dermatology, Sacramento, California
Next-generation sequencing of PEComas has been performed in a small number of visceral tumors [97, 99,100,101,102]. Analysis of the genomics of 31 advanced or metastatic PEComas showed a mean of 3.2 alterations per tumor. Mutually exclusively occurring mammalian target of rapamycin (mTOR) pathway gene abnormalities—based on next-generation sequencing—were observed in 20 of the 31 patients; the altered genes (in order of decreasing frequency) were tuberous sclerosis complex 2 (TSC2), transcription factor binding to immunoglobulin heavy contrast Mu (IGHM) enhancer 3 (TFE3) fusions, tuberous sclerosis complex 1 (TSC1), and folliculin (FLCN) (Table 3) [102].
Next-generation sequencing of cutaneous PEComa has only been reported, to our knowledge, in two individuals. The first patient was included in the summarized results of 31 patients with advanced or metastatic PEComa [102]. The second patient was a 43-year-old man with a primary cutaneous malignant PEComa on his left upper extremity and with four pathogenic aberrations in genes, as follows: baculoviral inhibitor of apoptosis (IAP) repeat containing 3 (BIRC3), Fanconi anemia complementation group C (FANCC), TP53, and TSC1 (Figs. 6, 7) [96]. His tumor had been removed during the biopsy; subsequently, a wide local excision of the site was performed. There had been no recurrence or metastasis of the tumor after 15 months of follow-up; however, if his tumor was to recur or metastasize, based on the genomic profile of his PEComa, nab-sirolimus [an mTOR inhibitor, with mTOR being activated in patients with tuberous sclerosis complex (TSC) alterations] would be considered for first-line therapy [96].
On November 23, 2021, the FDA approved the mTOR inhibitor nab-sirolimus (nanoparticle albumin-bound sirolimus) for the treatment of PEComas [99, 103]. The mTOR pathway is activated by alterations in a variety of genes, including TSC1 and TSC2; PEComa patients with a genomic aberration of TSC2 may respond better to sirolimus-type therapy than those with a gene abnormality of TSC1 [97, 104]. In addition to sirolimus-type monotherapy, concurrent palliative radiation therapy has also been used in the management of a 67-year-old man with PEComa of the lung [100]. Surprisingly, a woman with a malignant pelvic TFE3-associated PEComa (which typically lacks a mutation in the TSC gene) whose tumor responded to the mTOR inhibitor everolimus was recently described [97, 105].
However, not all patients with PEComa respond to mTOR inhibitors [106]. Based on the genomic landscape of aberrations observed in PEComa, alternative targeted therapies may potentially be used in PEComa patients with a tumor resistant to mTOR inhibitors, perhaps because of the presence of co-existing oncogenic driver abnormalities. For example, a group of investigators successfully used the checkpoint inhibitor pembrolizumab in a 69-year-old woman whose recurrent PEComa was associated with elevated PD-L1 expression [107].
Conclusion
Precision dermatology is the next therapeutic frontier for the treatment of patients with dermatologic conditions. It incorporates dermatologic disease-directed targeted therapy (D3T2) for the management of dermatoses such as psoriasis, atopic dermatitis, and prurigo nodularis. In addition, precision dermatology will also play an essential role in the evaluation, diagnosis, prognosis, and therapy of individuals with skin cancer, including not only common primary cutaneous tumors (such as melanoma, BCC, and SCC), but also rare cutaneous neoplasms (such as PEComa).
References
Tsimberidou AM, Fountzilas E, Nikanjam M, Kurzrock R. Review of precision cancer medicine: evolution of the treatment paradigm. Cancer Treat Rev. 2020;86: 102019. https://doi.org/10.1016/j.ctrv.2020.102019.
Adashek JJ, Subbiah V, Kurzrock R. From tissue-agnostic to N-of-one therapies: (r)evolution of the precision paradigm. Trends Cancer. 2021;7(1):15–28. https://doi.org/10.1016/j.trecan.2020.08.009.
Adashek JJ, Goloubev A, Kato S, Kurzrock R. Missing the target in cancer therapy. Nat Cancer. 2021;2:369–71. https://doi.org/10.1038/s43018-021-00204-w.
Moser C, Lerche CJ, Thomsen K, et al. Antibiotic therapy as personalized medicine—general considerations and complicating factors. APMIS. 2019;127(5):361–71. https://doi.org/10.1111/apm.12951.
Rosenblatt JE. Laboratory tests used to guide antimicrobial therapy. Mayo Clin Proc. 1991;66(9):942–8. https://doi.org/10.1016/s0025-6196(12)61583-3.
Araj GF. Available laboratory tests to guide antimicrobial therapy. J Med Liban. 2000;48(4):199–202.
Peterson LR, Shanholtzer CJ. Tests for bactericidal effects of antimicrobial agents: technical performance and clinical relevance. Clin Microbiol Rev. 1992;5(4):420–32. https://doi.org/10.1128/CMR.5.4.420.
Laverdiere M, Sabath LD. Historical survey of tests to determine bacterial susceptibility to antimicrobial agents. Mt Sinai J Med. 1977;44(1):73–88.
Gumbo T. Integrating pharmacokinetics, pharmacodynamics and pharmacogenomics to predict outcomes in antibacterial therapy. Curr Opin Drug Discov Dev. 2008;11(1):32–42.
Cortes J, Pavlovsky C, SalBele S. Chronic myeloid leukaemia. Lancet. 2021;398(10314):1914–26. https://doi.org/10.1016/S0140-6736(21)0124-6.
Sicklick JK, Kato S, Okamura R, et al. Molecular profiling of cancer patients enables personalized combination therapy: the I-PREDICT study. Nat Med. 2019;25(5):744–50. https://doi.org/10.1038/s41591-019-0407-5.
Goodman AM, Piccioni D, Kato S, et al. Prevalence of PDL1 amplification and preliminary response to immune checkpoint blockade in solid tumors. JAMA Oncol. 2018;4(9):1237–44. https://doi.org/10.1001/jamaoncol.2018.1701.
Patel SP, Kurzrock R. PD-L1 expression as a predictive biomarker in cancer immunotherapy. Mol Cancer Ther. 2015;14(4):847–57. https://doi.org/10.1158/1535-7163.MCT-14-0983.
Jardin DL, Goodman A, de Melo GD, Kurzrock R. The challenges of tumor mutational burden as an immunotherapy biomarker. Cancer Cell. 2021;39(2):154–73. https://doi.org/10.1016/j.ccell.2020.10.001.
Marushchak O, Yakubov R, Yakubov R, Goldenberg G. Review on novel oral therapies for psoriasis. J Clin Aesthet Dermatol. 2021;14(12):55–63.
Buechler CR, Veenstra J, Stein GL. Psoriasis therapy beyond biologics. SKIN J Cutan Med. 2021;5(6):568–78. https://doi.org/10.25251/skin.5.6.1.
McInnes IB, Sawyer LM, Markus K, LeReun C, Sabry-Grant C, Helliwell PS. Targeted systemic therapies for psoriatic arthritis: a systematic review and comparative synthesis of short-term articular, dermatological, enthesitis and dactylitis outcomes. RMD Open. 2022;8(1): e002074. https://doi.org/10.1136/rmdopen-2021-002074.
Armstrong AW, Read C. Pathophysiology, clinical presentation, and treatment of psoriasis: a review. JAMA. 2020;323(19):1945–60. https://doi.org/10.1001/jama.2020.4006.
Thatiparthi A, Martin A, Liu J, Egeberg A, Wu JJ. Biologic treatment algorithms for moderate-to-severe psoriasis with comorbid conditions and special populations: a review. Am J Clin Dermatol. 2021;22(4):425–42. https://doi.org/10.1007/s40257-021-00603-w.
Krishnan VS, Koks S. Transcriptional basis of psoriasis from large scale gene expression studies: the importance of moving towards a precision medicine approach. Int J Mol Sci. 2022;23(11):6130. https://doi.org/10.3390/ijms23116130.
Wu JJ, Montgomery PW III, Long B, et al. An economic evaluation of the budget impact of precision medicine testing for the treatment of psoriasis. SKIN J Cutan Med. 2021;5(4):372–87. https://doi.org/10.25251/skin.5.4.6.
Dickerson T, Lee B-I, Montgomery P III, et al. Mind.Px—personalized medicine for psoriasis biologic treatment. SKIN J Cutan Med. 2021;5(6): s46. https://doi.org/10.25251/skin.5.supp.46.
Strober B, Fox J, Jekowsky E, Pariser D, Schaecher K. Evidence threshold for a precision medicine test that predicts optimal response to a biologic agent in patients with psoriasis: a consensus panel. J Drugs Dermatol. 2022;21(6):630–6.
Bagel J, Wang Y, Montgomery P III, et al. A machine learning-based test for predicting response to psoriasis biologics. SKIN J Cutan Med. 2021;5(6):621–38. https://doi.org/10.25251/skin.5.6.5.
Jung SM, Kim W-U. Targeted immunotherapy for autoimmune disease. Immune Netw. 2022;22(1): e9. https://doi.org/10.4110/in.2022.22.e9.
Corbett M, Ramessur R, Marshall D, et al. Biomarkers of systemic treatment response in people with psoriasis: a scoping review. Br J Dermatol. 2022. https://doi.org/10.1111/bjd.21677.
Naik PP. Treatment-resistant atopic dermatitis: novel therapeutics, digital tools, and precision medicine. Asia Pac Allergy. 2022;12(2):e20. https://doi.org/10.5415/apallergy.2022.12.e20.
Goh MSY, Yun JSW, Su JC. Management of atopic dermatitis: a narrative review. Med J Aust. 2022. https://doi.org/10.5694/mja2.51560.
Appiah MM, Haft MA, Kleinman E, et al. Atopic dermatitis: review of comorbidities and therapeutics. Ann Allergy Asthma Immunol. 2022. https://doi.org/10.1016/j.anai.2022.05.015.
Beiber T, Paller AS, Kabashima K, et al. Atopic dermatitis: pathomechanisms and lessons learned from novel systemic therapeutic options. J Eur Acad Dermatol Venereol. 2022. https://doi.org/10.1111/jdv.18225.
Stander S. Atopic dermatitis. N Engl J Med. 2021;384(12):1136–43. https://doi.org/10.1056/MEJMra2023911.
Maintz L, Bieber T, Simpson MD, Demessant-Flavigny AL. From skin barrier dysfunction to systemic impact of atopic dermatitis: implications for a precision approach in dermocosmetics and medicine. J Per Med. 2022;12(6):893. https://doi.org/10.3390/jpm12060893.
Bieber T, Simpson EL, Thaci SD, et al. Abrocitinib versus placebo or duilumab for atopic dermatitis. N Engl J Med. 2021;384(12):1101–12. https://doi.org/10.1056/NEJMoa2019380.
Labib A, Ju T, Does AV, Yosipovitch G. Immunotargets and therapy for prurigo nodularis. Immunotargets Ther. 2022;11:11–21. https://doi.org/10.2147/ITT.S316602.
Williams KA, Roh YS, Brown I, et al. Pathophysiology, diagnosis, and pharmacological treatment of prurigo nodularis. Expert Rev Clin Pharmacol. 2021;14(1):67–77. https://doi.org/10.1080/17512433.2021.1852080.
Husein-ElAhmed H, Steinhoff M. Dupilumab in prurigo nodularis: a systematic review of concurrent evidence and analysis of predictive factors to response. J Dermatol Treat. 2022;33(3):1547–53. https://doi.org/10.1080/09546634.2020.185024.
Sutaria N, Aphonse MP, Marani M, et al. Cluster analysis of circulating plasma biomarkers in prurigo nodularis reveals a distinct systemic inflammatory signature in African Americans. J Invest Dermatol. 2022;142(5):1300-1308.e3. https://doi.org/10.1016/j/jid.2021.10.011.
Brownstone N, Zakria D, Dirr MA, Lu Y, Rigel DS. Integrating genomic testing for melanoma into your practice. SKIN J Cutan Med. 2022;6(3):189–94. https://doi.org/10.2525/skin.6.3.2.
Fried L, Tan A, Bajaj S, Liebman TN, Polsky D, Stein JA. Technological advances for the detection of melanoma: advances in molecular techniques. J Am Acad Dermatol. 2020;83(4):996–1044. https://doi.org/10.1016/jaad.2020.03.122.
Timar J, Ladanyi A. Molecular pathology of skin melanoma: epidemiology, differential diagnosis, prognosis and therapy prediction. Int J Mol Sci. 2022;23(10):5384. https://doi.org/10.3390/ijms23105384.
Berman B, Ceilley R, Cockerell C, et al. Appropriate use criteria for the integration of diagnostic and prognostic gene expression profile assays into the management of cutaneous malignant melanoma: an expert panel consensus-based modified Delphi process assessment. SKIN J Cutan Med. 2019;3(5):291–306. https://doi.org/10.25251/skin.3.5.1.
Johansson I, Tempel D, Dwarkasing JT, et al. Validation of a clinicopathological and gene expression profile model to identify patients with cutaneous melanoma with sentinel lymph node biopsy is unnecessary. Eur J Surg Oncol. 2022;48(2):320–5. https://doi.org/10.1016/j.ejso.2021.11.010.
Skelsey MK, Brouha B, Rock J, et al. Non-invasive detection of genomic atypia increases real-world NPV and PPV of the melanoma diagnostic pathway and reduces biopsy burden. SKIN J Cutan Med. 2021;5(5):512–23. https://doi.org/10.2521/skin.5.5.9.
Brouha B, Ferris LK, Skelsey MK, et al. Genomic atypia to enrich melanoma positivity in biopsied lesions: gene expression and pathology findings from a large U.S. registry study. Skin J Cutan Med. 2021;5(1):13–8. https://doi.org/10.25251/skin.5.1.3.
Brouha B, Ferris LK, Skelsey MK, et al. Real-world utility of a non-invasive gene expression test to rule out primary cutaneous melanoma: a large US registry study. J Drugs Dermatol. 2020;19(3):257–62.
Ferris LK, Rigel DS, Siegel DM, et al. Impact on clinical practice of a non-invasive gene expression melanoma rule-out test: 12-month follow-up of negative test results and utility data from a large US registry study. Dermatol Online J. 2019. https://doi.org/10.5070/D3255044059.
Ferris LK, Jansen B, Ho J, et al. Utility of a noninvasive 2-gene molecular assay for cutaneous melanoma and effect on the decision to biopsy. JAMA Dermatol. 2017;153(7):675–80. https://doi.org/10.1001/jamadermatol.2017.0473.
Ferris LK, Moy RL, Gerami P, et al. Noninvasive analysis of high-risk driver mutations and gene expression profiles in primary cutaneous melanoma. J Invest Dermatol. 2019;139(5):1127–34. https://doi.org/10.1016/j.jid.2018.10.041.
Jackson SR, Jansen B, Yao Z, Ferris LK. Risk stratification of severely dysplastic nevi by non-invasive gene expression and mutation analyses. SKIN J Cutan Med. 2020;4(2):124–9. https://doi.org/10.25251/skin.4.2.5.
Clarke LE, Warf MB, Flake DD II, et al. Clinical validation of a gene expression signature that differentiates benign nevi from malignant melanoma. J Cutan Pathol. 2015;42(4):244–52. https://doi.org/10.1111/cup.12475.
Clarke LE, Flake DD II, Busam K, et al. An independent validation of a gene expression signature to differentiate malignant melanoma from benign melanocytic nevi. Cancer. 2017;123(4):617–28. https://doi.org/10.1002/cncr.30385.
Ko JS, Matharoo-Ball B, Billings SD, et al. Diagnostic distinction of malignant melanoma and benign nevi by a gene expression signature and correlation to clinical outcomes. Cancer Epidemiol Biomarkers Prev. 2017;26(7):1107–13. https://doi.org/10.1158/1055-9965.EPI-16-0958.
Minca EC, Al-Rohil RN, Wang M, et al. Comparison between melanoma gene expression score and fluorescence in situ hybridization for the classification of melanocytic lesions. Mod Pathol. 2016;29(8):832–43. https://doi.org/10.1038/modpathol.2016.84.
Reimann JDR, Salim S, Velazquez EF, et al. Comparison of melanoma gene expression score with histopathology, fluorescence in situ hybridization, and SNP array for the classification of melanocytic neoplasms. Mod Pathol. 2018;31(11):1733–43. https://doi.org/10.1038/s41379-018-0087-6.
Castillo SA, Pham AK, Dagrosa AT, et al. Concordance analysis of the 23-gene expression signature (myPath Melanoma) with fluorescence in situ hybridization assay and single nucleotide pleomorphism array in the analysis of challenging melanocytic lesions: results from an academic medical center. Am J Dermatopathol. 2020;42(12):939–47. https://doi.org/10.1097/DAD.000000000000713.
Clarke LE, Mabey B, Flake DD II, et al. Clinical validity of a gene expression signature in diagnostically uncertain neoplasms. Per Med. 2020;17(5):361–71. https://doi.org/10.2217/pme-2020-0048.
Cockerell C, Tschen J, Billings SD, et al. The influence of a gene-expression signature on the treatment of diagnostically challenging melanocytic lesions. Per Med. 2017;14(2):123–30. https://doi.org/10.2217/pme-2016-0097.
Cockerell CJ, Tschen J, Evans B, et al. The influence of a gene expression signature on the diagnosis and recommended treatment of melanocytic tumors by dermatopathologists. Oncology. 2020;98(12):905–12. https://doi.org/10.1159/000510241.
Farberg AS, Ahmed KL, Bailey CN, et al. A 35-gene profile test for use in suspicious pigmented lesions impacts clinical management decisions of dermatopathologists and dermatologists. SKIN J Cutan Med. 2020;4(6):523–33. https://doi.org/10.25251/skin.4.6.4.
Estrada SI, Shackelton JB, Cleaver NJ, et al. Development and validation or a diagnostic 35-gene expression profile test for ambiguous or difficult-to-diagnose suspicious pigmented skin lesions. SKIN J Cutan Med. 2020;4(6):506–22. https://doi.org/10.25251/skin.4.6.3.
Mulder EEAP, Dwarkasing JT, Tempel D, et al. Validation of a clinicopathological and gene expression profile model for sentinel lymph node metastasis in primary cutaneous melanoma. Br J Dermatol. 2021;184(5):944–51. https://doi.org/10.1111/bjd.19499.
Whitman ED, Koshenkov VP, Gastman BR, et al. Integrating 31-gene expression profiling with clinicopathologic features to optimize cutaneous melanoma sentinel lymph node metastasis prediction. JCO Precis Oncol. 2021;5:1466–79. https://doi.org/10.1200/PO.21.00162.
Kwatra SG, Hines H, Semenov YR, Trotter SC, Holland E, Leachman S. A dermatologist’s guide to implementation of gene expression profiling in the management of melanoma. J Clin Aesthet Dermatol. 2020;13(11):s3–14.
Grossman D, Okwundu N, Batlett EK, et al. Prognostic gene expression in cutaneous melanoma identifying the knowledge gaps and assessing the clinical benefit. JAMA Dermatol. 2020;156(9):1004–11. https://doi.org/10.1001/jamadermatol.2020.1729.
Gerami P, Cook RW, Russell MC, et al. Gene expression profiling for molecular staging of cutaneous melanoma in patients undergoing sentinel lymph node biopsy. J Am Acad Dermatol. 2015;72(5):780-785.e3. https://doi.org/10.1016/j.jaad.2015.01.009.
Gerami P, Cook RW, Wilkinson J, et al. Development of a prognostic genetic signature to predict the metastatic risk associated with cutaneous melanoma. Clin Cancer Res. 2015;21(1):175–83. https://doi.org/10.1158/1078-0432.CCR-13-3316.
Greenhaw BN, Covington KR, Kurley SJ, et al. Molecular risk prediction in cutaneous melanoma: a meta-analysis of the 31-gene expression profile prognostic test in 1,479 patients. J Am Acad Dermatol. 2020;83(3):745–53. https://doi.org/10.1016/j.jaad.2020.03.053.
Greenhaw BN, Covington KR, Kurley SJ, et al. Reply to problematic methodology in a systematic review and meta-analysis of DecisionDX-Melanoma. J Am Acad Dermatol. 2020;83(5):e359-360. https://doi.org/10.1016/j.jaad.2020.06.009.
Marchetti MA, Dusza SW, Bartlett EK. Problematic methodology in a systematic review and meta-analysis of DecisionDX-Melanoma. J Am Acad Dermatol. 2020;83(5):e357-358. https://doi.org/10.1016/j.jaad.2020.04.186.
Cohen BJ, Cohen ES, Cohen PR. Basal cell carcinoma: a patient and physician’s experience. Dermatol Ther (Heidelb). 2018;8(3):329–37. https://doi.org/10.1007/s/3555-018-0245-2.
Cohen PR. Basal cell carcinoma: additional subtypes and therapeutic advances. J Am Acad Dermatol. 2019;81(1): e17. https://doi.org/10.1016/j.jaad.2019.02.046.
Krakowski AC, Hafeez F, Westheim A, Pan EY, Wilson M. Advanced basal cell carcinoma: what dermatologists need to know about diagnosis. J Am Acad Dermatol. 2022;86(6S):S1–13. https://doi.org/10.1016/j.jaad.2022.03.023.
Nikanjam M, Cohen PR, Kato S, Sicklick JK, Kurzrock R. Advanced basal cell cancer: concise review of molecular characteristics and novel targeted and immune therapeutics. Ann Oncol. 2019;30(10):1675. https://doi.org/10.1093/annonc/mdz213.
Wilson M, Johnson RP, Senft SC, Pan EY, Krakowski AC. Advanced basal cell carcinoma: what dermatologists need to know about treatment. J Am Acad Dermatol. 2022;86(6S):S14–24. https://doi.org/10.1016/j/jaad.2022.03.022.
Goodman AM, Kato S, Cohen PR, et al. Genomic landscape of advanced basal cell carcinoma: implications for precision treatment with targeted and immune therapies. Oncoimmunology. 2017;7(3): e1404217. https://doi.org/10.1080/2162402X.2017.1404217.
Ikeda S, Goodman AM, Cohen PR, et al. Metastatic basal cell carcinoma with amplification of PD-L1: exceptional response to anti-PD1 therapy. NPJ Genom Med. 2016;1:16037. https://doi.org/10.1038/npgenmed.2016.37.
Cohen PR, Kato S, Goodman AM, Ikeda S, Kurzrock R. Appearance of new cutaneous superficial basal cell carcinomas during successful nivolumab treatment of refractory metastatic disease: implications for immunotherapy in early versus late disease. Int J Mol Sci. 2017;18(8):1663. https://doi.org/10.3390/ijms18081663.
Cohen PR, Kurzrock R. Crucial conceptual concepts in the evaluation and management of advanced basal cell carcinoma. J Am Acad Dermatol. 2022 (in press).
Villani A, Protestio L, Fabbrocini G, Scalvenzi M. New emerging treatment options for advanced basal cell carcinoma and squamous cell carcinoma. Adv Ther. 2022;39(3):1164–78. https://doi.org/10.1007/s12325-022-02044-1.
Cohen PR, Kurzrock R. Basal cell carcinoma: management of advanced or metastatic cancer with checkpoint inhibitors and concurrent paradoxical development of new superficial tumors. J Am Acad Dermatol. 2020;82(6):e253–4. https://doi.org/10.1016/j.jaad.2020.02.052.
Cohen PR, Kurzrock R. Reply to “Effective therapy for advanced basal cell carcinoma.” J Am Acad Dermatol. 2022;86(3): e109. https://doi.org/10.1016/j.jaad.2020.06.988.
Wysong A, Newman JG, Covington KR, et al. Validation of a 40-gene expression profile test to predict metastatic risk in localized high-risk cutaneous squamous cell carcinoma. J Am Acad Dermatol. 2021;84(2):361–9. https://doi.org/10.1016/j/jaad.2020.04.088.
Farberg AS, Fitzgerald AL, Ibrahim SF, et al. Current methods and caveats to risk factor assessment in cutaneous squamous cell carcinoma (cSCC): a narrative review. Dermatol Ther (Heidelb). 2022;12(2):267–84. https://doi.org/10.1007/s13555-021-00673-y.
Alam M, Armstrong A, Baum C, et al. Guidelines of care for the management of cutaneous squamous cell carcinoma. J Am Acad Dermatol. 2018;78(3):560–78. https://doi.org/10.1016/j.jaad.2017.10.007.
Teplitz R, Prado G, Litchman GH, Rigel DS. Impact of gene expression profiling testing on the management of squamous cell carcinoma by dermatologists. J Drug Dermatol. 2019;18(10):908–84.
Arron ST, Wysong A, Hall MA, et al. Gene expression profiling for metastatic risk in head and neck cutaneous squamous cell carcinoma. Laryngoscope Investig Otolaryngol. 2022;7(1):135–44. https://doi.org/10.1002/lio2.724.
Borman S, Wilkinson J, Meldi-Sholl L, et al. Analytical validity of DecisionDx-SCC, a gene expression profile test to identify risk of metastasis in cutaneous squamous cell carcinoma (SCC) patients. Diagn Pathol. 2022;17(1):32. https://doi.org/10.1186/s13000-022-01211-w.
Arron ST, Blalock TW, Guenther M, et al. Clinical considerations for integrating gene expression profiling into cutaneous squamous cell carcinoma management. J Drugs Dermatol. 2021;20(6):s5–11. https://doi.org/10.36849/JDD.2021.6068.
Au JH, Hooper PB, Fitzgerald AL, Somani A-K. Clinical utility of the 40-gene expression profile (40-GEP) test for improved patient management decisions and disease-related outcomes when combined with current clinicopathological risk factors for cutaneous squamous cell carcinoma (cSCC): case series. Dermatol Ther (Heidelb). 2022;12(2):591–7. https://doi.org/10.1007/s13555-021-00665-y.
Ibrahim SF, Kasprzak JM, Hall MA, et al. Enhanced metastatic risk assessment in cutaneous squamous cell carcinoma with the 40-gene expression profile test. Future Oncol. 2022;18(7):833–47. https://doi.org/10.2217/fon-2021-1277.
Farberg AS, Hall MA, Douglas L, et al. Integrating gene expression profiling into NCCN high-risk cutaneous squamous cell carcinoma management recommendations: impact on patient management. Curr Med Res Opin. 2020;36(8):1301–7. https://doi.org/10.1080/03007995.2020.1763284.
Litchman GH, Fitzgerald AL, Kurley SJ, Cook RW, Rigel DS. Impact of a prognostic 40-gene expression profiling test on clinical management decisions for high-risk cutaneous squamous cell carcinoma. Curr Med Res Opin. 2020;36(8):1295–300. https://doi.org/10.1080/03007995.2020.1763283.
Wang B-C, Xiao B-Y, Kuang B-H, Lin G-H. The efficacy and safety of cemiplimab in locally advanced or metastatic cutaneous squamous cell carcinoma: a comparative analysis of retrospective studies versus prospective studies. Dermatol Ther. 2022. https://doi.org/10.1111/dth.15715.
Aberti A, Bossi P. Immunotherapy for cutaneous squamous cell carcinoma: results and perspectives. Front Oncol. 2022;11: 727027. https://doi.org/10.3389/fonc.2021.727027.
Braiteh F, Kurzrock R. Uncommon tumors and exceptional therapies: paradox or paradigm? Mol Cancer Ther. 2007;6(4):1175–9. https://doi.org/10.1158/1535-7163.MCT-06-0674.
Cohen PR, Kato SM, Erickson CP, Calame A, Kurzrock R. Cutaneous perivascular epithelioid cell tumor (PEComa): case report and world literature review of clinical and molecular characteristics. Dermatol Online J. 2022. https://doi.org/10.5070/D328157058.
Bourgmayer A, Nannini S, Bonjean P, Kurtz J-E, Malouf GG, Gantzer J. Natural history and treatment strategies of advanced PEComas: a systemic review. Cancers (Basel). 2021;13(20):5227. https://doi.org/10.3390/cancers13205227.
Wong J, Mammino J, Seyffert J, Schmits K, Marks E, Rivlin D. Primary cutaneous perivascular epithelioid cell tumors: two cases and a review of the literature. Dermatol Online J. 2021;27(10):10. https://doi.org/10.5070/D3271055626.
Groisberg R, Subbiah V. Sequencing PEComas: viewing unicorns through the molecular looking glass. Oncology. 2021;99(1):62–4. https://doi.org/10.1159/000510650.
Bajaj A, Abazeed ME. Molecularly targeted radiation therapy using mTOR inhibition for the management of malignant perivascular epithelioid cell tumor (PEComa): a case report and review. Adv Radiat Oncol. 2021;6(3): 100657. https://doi.org/10.1016/j.adro.2021.100657.
Bennett JA, Ordulu Z, Pinto A, et al. Uterine PEComas: correlation between melanocytic marker expression and TSC alterations/TFE3 fusions. Mod Pathol. 2022;35(4):515–23. https://doi.org/10.1038/s41379-021-00855-1.
Akummalla S, Madison R, Lin DL, et al. Characterization of clinical cases of malignant PEComa via comprehensive genomic profiling of DNA and RNA. Oncology. 2020;98:905–12. https://doi.org/10.1159/000510241.
Wagner AJ, Ravi V, Riedel RF, et al. nab-Sirolimus for patients with malignant perivascular epitheloid cell tumors. J Clin Oncol. 2021;39(33):3660–70. https://doi.org/10.1200/JCO.21.01728.
Switaj T, Sobiborowicz A, Teterycz P. Efficacy of sirolimus treatment in PEComa—10 years of practice perspective. J Clin Med. 2021;10(16):3705. https://doi.org/10.3390/jcm10163705.
Pruwar R, Soni K, Shukla M, Verma A, Kumar T, Pandey M. TFE3-associated perivascular epithelioid cell tumor with complete response to mTOR inhibitor therapy: report of first case and literature review. World J Surg Oncol. 2022;20(1):62. https://doi.org/10.1186/s12957-021-02462-5.
Subbiah V, Trent JC, Kurzrock R. Resistance to mammalian target of rapamycin inhibitor therapy in perivascular epithelioid cell tumors. J Clin Oncol. 2010;28(24): e415. https://doi.org/10.1200/JCO.2010.29.4678.
McBride A, Garcia AJ, Sanders LJ, et al. Sustained response to pembrolizumab in recurrent perivascular epithelioid cell tumor with elevated expression of programmed death ligand: a case report. J Med Case Rep. 2021;15(1):400. https://doi.org/10.1186/s13256-021-02997-x.
Acknowledgements
Funding
No funding or sponsorship was received for this study or the publication of this article.
Authorship
All named authors meet the International Committee of Medical Journal Editors (ICMJE) criteria for authorship for this article, take responsibility for the integrity of the work as a whole, and have given their approval for this version to be published.
Author Contributions
Critical revisions and editing: PRC, RK. Literature search and original draft composition: PRC, RK. Review, editing, and intellectual contributions: PRC, RK. Idea for article and critical revisions: PRC, RK.
Disclosures
Dr. Cohen is a consultant for ParaPRO. Dr. Kurzrock receives research funding from Genentech, Merck Serono, Pfizer, Boehringer Ingelheim, TopAlliance, Takeda, Incyte, Debiopharm, Medimmune, Sequenom, Foundation Medicine, Konica Minolta, Grifols, Omniseq, and Guardant; receives consultant and/or speaker fees from and/or is on the advisory board for X-Biotech, Neomed, Pfizer, Actuate Therapeutics, and Roche; has an equity interest in IDbyDNA and CureMatch Inc.; serves on the board of CureMatch and CureMetrix; and is a co-founder of CureMatch.
Compliance with Ethics Guidelines
This article is based on previously conducted studies and does not contain any new studies with human participants or animals performed by any of the authors.
Data Availability
Data sharing is not applicable to this article as no datasets were generated or analyzed during the current study.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Cohen, P.R., Kurzrock, R. Dermatologic Disease-Directed Targeted Therapy (D3T2): The Application of Biomarker-Based Precision Medicine for the Personalized Treatment of Skin Conditions—Precision Dermatology. Dermatol Ther (Heidelb) 12, 2249–2271 (2022). https://doi.org/10.1007/s13555-022-00801-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13555-022-00801-2