
Abstract
Introduction
Tildrakizumab 200 mg/2 mL pre-filled syringe is a new preparation of tildrakizumab that is developed to facilitate patients’ compliance. This phase I clinical trial compares the local tolerability, safety, and subjects’ preferred method of administration of tildrakizumab when administered as a new single 200 mg/2 mL subcutaneous injection or as two 100 mg/1 mL subcutaneous injections in healthy subjects.
Methods
Visual analogue scores were used to self-assess injection site pain immediately (< 1 min) after each administration and at 1 h and 48 h after each administration. Treatment injection site reactions were assessed at 1 h and 48 h after each administration. Treatment safety was monitored throughout the study period. Subjects’ preferred method of administration was assessed 4 weeks after the last administration (day 56).
Results
No statistically significant difference in visual analogue scores and injection site reactions was detected between the two treatments. Treatment-emergent adverse events were mild, and there were no deaths or serious adverse events. Most subjects (61.5%) preferred the treatment when administered as a single 200 mg/2 mL subcutaneous injection rather than as two 100 mg/mL subcutaneous injections.
Conclusions
Administration of 200 mg tildrakizumab as a single 2 mL subcutaneous injection was safe, well tolerated, and preferred over two separate 100 mg/1 mL subcutaneous injections by healthy subjects. Eudract No. 2020-000183-37.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Why carry out this study? |
Tildrakizumab (TIL) is a humanized monoclonal antibody approved for the treatment of psoriasis at doses of 100 mg and 200 mg as one or two separate 100 mg/1 mL injections, respectively. |
To improve convenience for patients treated with the 200 mg dose, we assessed the local tolerability, safety, and preferred method of administration (either a 200 mg/2 mL single subcutaneous (SC) injection or two 100 mg/1 mL SC injections) in healthy subjects. |
What was learned from the study? |
We found no statistically significant difference in tolerability and safety assessments between the two treatments. |
Tildrakizumab administration as a single 200 mg/2 mL SC injection was preferred by most subjects. |
Introduction
Psoriasis is a chronic immune-mediated inflammatory skin disease affecting approximately 3% of the population globally [1]. Plaque psoriasis is the most common form, which clinically manifests as skin erythematosquamous plaques that evolve as a consequence of the underlying immunological phenomena [2]. Besides T-helper (Th)-1 differentiation of the naïve T cells, resulting in the production of Th-1 cytokines such as tumor necrosis factor alpha, Th-17 differentiation plays the most prominent role in the immunopathogenesis of the disease [3]. In the latter case, interleukin (IL)-23 promotes naïve T cells to Th-17 differentiation and therefore to release Th-17 cytokines, mainly represented by the IL-17 family [4]. Antibodies and molecules that selectively block these pathways have revolutionized psoriasis therapy. Recently, biologics targeting IL-23 or IL-17 have shown remarkable efficacy along with favorable safety profiles for the long-term management of psoriasis [5, 6]. Tildrakizumab, a humanized monoclonal antibody selectively targeting IL-23 via the p19 subunit, is approved for the treatment of adults with moderate to severe plaque psoriasis who are candidates for systemic therapy [7]. The recommended tildrakizumab dose is 100 mg by subcutaneous (SC) injection at weeks 0 and 4 and every 12 weeks thereafter. However, in Europe, the use of a 200 mg dose is suggested for patients with certain characteristics (e.g., high bodyweight) [7]. In clinical trials, both doses of tildrakizumab have demonstrated a similar sustained long-term efficacy for skin manifestations as well as for patient quality of life, with equal safety profiles [8, 9]. Recently, the effectiveness and safety of tildrakizumab across psoriatic disease manifestations has been confirmed in real-life clinical practice in large prospective cohort studies [10, 11]. Currently, tildrakizumab is available as a single-use pre-filled syringe (PFS) containing 100 mg of product in 1 mL solution (100 mg/1 mL PFS) for SC injection. Thus, the 200 mg dose is administered as two separate 100 mg/1 mL SC injections at different injection sites.
To improve convenience for patients treated with a 200 mg dose, a 200 mg/2 mL PFS allowing for the administration of this dose in one injection was developed. In this context, the phase I study described herein was specifically designed to assess the local tolerability and safety of the tildrakizumab 200 mg dose when administered as a single 200 mg/2 mL SC injection or as two 100 mg/1 mL SC injections in healthy subjects. Additionally, this study assessed the subjects’ preferred method of administration of the tildrakizumab 200 mg treatment between the tested injection regimens.
Methods
Study Design and Subjects
This was a phase I, open-label, randomized, crossover study to assess the local tolerability of a new PFS preparation containing 200 mg/2 mL of tildrakizumab when delivered as a single SC injection in healthy subjects. The trial was conducted in a single center (the Phase I Unit of PRA Health Sciences) in Groningen (Netherlands). Potential participants were identified and recruited via a database of volunteers held by PRA Health Sciences. Healthy male and female subjects aged 18–60 years with a body mass index (BMI) between 18.0 and 30.0 kg/m2 and who were in good general health were eligible for enrollment in this study. Subjects with a history of latent or active tuberculosis or those a positive test for human immunodeficiency virus, hepatitis B, hepatitis C, or severe acute respiratory syndrome coronavirus 2 were excluded.
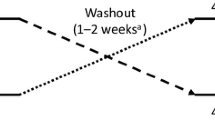
The study consisted of two 200 mg tildrakizumab treatments randomly allocated to subjects in a 1:1 ratio according to a 2 × 2 Williams design: (1) a treatment consisting of one SC injection of 200 mg/2 mL administered in the left or right arm or thigh or in the left or right side of the abdomen or (2) a treatment consisting of two SC injections of 100 mg/1 mL each, which were administered in opposite arms or thighs or in opposite sides of the abdomen [12]. Subjects received either the one SC injection of 2 mL (day 1) followed by the two SC injections of 1 mL (day 28) or the two SC injections of 1 mL at day 1 followed by the one SC injection of 2 mL at day 28 (Fig. 1). Within the treatment arms, subjects were randomized to administration in one of three anatomic sites (arms, thighs, or abdomen). During each treatment period, subjects were admitted to the research site and monitored for 48 h after the study drug administration. The study drug was administered by an experienced, qualified, and trained staff member following the instructions for administration as specified in the Summary of Product Characteristics. Four weeks after the last administration of the study drug (day 56), there was a safety follow-up visit. An additional safety follow-up for adverse events of clinical interest and serious adverse events was performed through a follow-up phone call at day 147. The study (Eudract No. 2020-000183-37) was conducted in conformance with good clinical practice guidelines and the Helsinki Declaration of 1964 and its later amendments. The study protocol was approved by the independent ethics committee Beoordeling Ethiek Biomedisch Onderzoek in Assen, the Netherlands. All participants provided written informed consent. Subjects received reasonable compensation for participation in the study. In addition, travel expenses were reimbursed based on the travel distance.

Study design. PFS pre-filled syringe
Outcome Measures
Local tolerability endpoints assessed injection site pain of treatment using a visual analogue scale (VAS), where 0 refers to no pain and 10 to the strongest pain visualized on a 0–100 mm line, immediately (< 1 min) as well as 1 h and 48 h after administration. Injection site reactions (ISRs), including erythema, swelling, pruritus, hemorrhage, fluid loss at the injection site, and bruising, were assessed and evaluated at 1 h and at 48 h after study drug administration to record all ISRs before participants were discharged. The grading scale of each ISR ranged from 0 = absent, 1 = mild (slightly, barely perceptible), 2 = moderate (distinct presence), to 3 = severe (marked, intense). The location of the ISR was described as at or just around the application area (≤ 2 cm from the application area) or distant (> 2 cm from the application area). For the two 1 mL injections, each administration site (left or right) was assessed and recorded separately. However, only the worst score for each tolerability parameter (worst pain score and worst severity of ISR) was considered as the local tolerability score per subject per time point for descriptive analysis. Other objectives included the monitoring of safety aspects (adverse effects, physical examination, vital signs, and clinical laboratory tests) of the treatments, assessed throughout the study from day 1 to day 56. Moreover, participants’ preference for method of administration between the treatment arms was inquired about 4 weeks after the last administration (day 56).
Statistical Analyses
The 95% confidence interval for the between-treatment difference in VAS score of pain at each time point was analyzed by an analysis of variance (ANOVA) model with treatment, period, and treatment sequence as fixed effects and subject nested within treatment sequence as a random effect.
The odds of having an ISR under the two treatments at each time point was derived using a genmod model with treatment and period as fixed effects. The distribution of ISR severity (absent, mild, moderate, or severe) between the two treatments was provided per type of ISR, overall and by treatment sequence, and by time point of assessment. All local tolerability and safety outcomes were analyzed in the safety analysis set (all randomized subjects who received at least one dose of the study drug). The subjects’ preferred method of administration was analyzed using the per protocol analysis set (all randomized subjects who completed the two study treatments without a major protocol deviation).
Results
Study Population Disposition and Characteristics
After screening a total of 99 subjects, 39 subjects were evaluated as the per protocol analysis set and 42 subjects as the safety analysis set. A total of 26 male subjects and 16 female subjects with a mean age [standard deviation (SD)] of 32.8 (13.5) years and a mean BMI (SD) of 24.1 (2.7) kg/m2 were enrolled into the study, randomized, and received at least one tildrakizumab dose. Three subjects discontinued due to noncompliance with the protocol. The demographic and baseline characteristics of the subjects are shown in Table 1.
Pain Intensity According to VAS
Immediately (< 1 min) after study drug administration, 25 subjects (59.5%) reported a VAS > 0 mm after treatment with the new 200 mg/2 mL PFS, and 26 subjects (66.7%) did so after treatment with the two separate 100 mg/1 mL SC injections. At 1 h after study drug administration, the number of subjects reporting a VAS > 0 mm was approximately halved to 13 subjects (31.0%) with the new 200 mg/2 mL PFS and 12 subjects (30.8%) with the two separate 100 mg/1 mL SC injections. At 48 h after study drug administration, only 2 subjects (5.1%) reported a VAS > 0 mm (with a highest VAS score of 2 mm on a scale of 0 to 100 mm), both after the two separate 100 mg/1 mL SC injections. The last reported injection site pain was on day 4, by only 1 subject after administration of the two separate 100 mg/1 mL SC injections. Immediately after administration of the new 200 mg/2 mL PFS, the mean (SD) VAS score was 6.6 mm (10.7). After 1 h, the score had already reduced to 1.2 mm (2.5), and 48 h after drug administration there was no reported injection site pain [mean VAS score (SD) 0.0 mm (0.0)]. For subjects receiving two separate 100 mg/1 mL SC injections, the mean (SD) VAS score was 4.5 mm (5.2) immediately after drug administration, 0.8 mm (1.4) after 1 h, and 0.1 mm (0.4) at 48 h after drug administration. Overall, mean VAS scores were low for both treatments at each of the scheduled time points (Table 2). Based on the applied ANOVA model, no statistically significant difference in VAS scores was detected between the two treatments at any of the scheduled time points.
Injection Site Reactions
Two subjects (4.8%) receiving the new 200 mg/2 mL PFS and two subjects (5.1%) receiving the two separate 100 mg/1 mL SC injections experienced at least one ISR at 1 h after study drug administration. At 48 h after study drug administration, 5 subjects (11.9%) receiving the new 200 mg/2 mL PFS and 4 subjects (10.3%) receiving the two separate 100 mg/1 mL SC injections had at least one ISR. Overall, the number of subjects with ISRs was low for both treatments at each of the scheduled time points (Table 3). The observed ISRs included erythema, swelling, and bruising. None of the subjects reported pruritus, hemorrhage, or fluid loss at the injection site. All ISRs were mild and ≤ 2 cm from the injection site. Based on the applied genmod model, no statistically significant difference in the odds of having an ISR was detected between the two treatments at any of the scheduled time points. The few ISRs reported at 48 h were reported as being resolved within 1 week after study drug administration (n = 4), within 2 weeks (n = 3), or within 3 weeks (n = 2).
Subjects’ Preferred Method of Administration
At 4 weeks after the last study drug administration, most subjects (61.5%) indicated that they preferred the new 200 mg/2 mL SC injection over the two separate 100 mg/1 mL SC injections. The remaining subjects (38.5%) indicated that they preferred the two separate 100 mg/1 mL SC injections over the single 200 mg/2 mL SC injection (Fig. 2).

Subjects’ preferred method of administration (per protocol population). PFS pre-filled syringe
Safety
Thirteen out of 42 subjects (31.0%) reported 26 treatment-emergent adverse events (TEAEs) after administration of the new 200 mg/2 mL PFS. Fourteen subjects (35.9%) reported 23 TEAEs after administration of the two separate 100 mg/1 mL SC injections. Based on a bivariate distribution analysis, 7 out of 42 subjects (16.7%) reported at least one TEAE after administration of the new 200 mg/2 mL PFS but not after the two separate 100 mg/1 mL SC injections. Eight subjects (19.0%) reported at least one TEAE after administration of the two separate 100 mg/1 mL SC injections but not after the new 200 mg/2 mL PFS. Six subjects (14.3%) reported at least one TEAE after administration of both treatments. Thus, there was no meaningful difference in the percentage of reported TEAEs between treatments. Moreover, all TEAEs were assessed as mild and were resolved by the end of the study. The most common TEAEs were injection site pain, injection site bruising, headache, injection site erythema, injection site swelling, abdominal pain, diarrhea, and flatulence. A summary of all TEAEs reported in the study is provided in Supplementary Table 1.
Discussion
Tildrakizumab is a biologic humanized monoclonal antibody against the p19 subunit of IL-23 that is approved at doses of 100 mg and 200 mg for patients with moderate-to-severe plaque psoriasis. Individual variables such as body weight can affect the efficacy of biologic treatments; thus, a higher dosing should be considered for patients with certain characteristics [13]. Tildrakizumab is the only IL-23p19 inhibitor with approved dose individualization for adaptation to such real-life clinical challenges [7]. However, the 200 mg dose of tildrakizumab is administered in the form of two 100 mg/1 mL SC injections. To facilitate the administration of the dose of 200 mg, increasing patient convenience and treatment adherence, a PFS of 200 mg/mL has been developed. The primary objective of this phase I, open-label, randomized, crossover study was to verify if the injection of a larger volume of the newly developed tildrakizumab, a 200 mg/2 mL PFS (2 mL versus 1 mL), at a single site would influence tolerability and safety when compared with the approved 100 mg/1 mL PFS.
We found that the mean VAS scores used to assess pain in the injection site and the number of subjects with ISRs were low for both treatments at each of the scheduled time points. There were no statistically significant differences between the two treatments in the injection site pain, self-assessed by subjects or based on the odds of having an ISR, showing that the tolerability of the new PFS was comparable to that of two separate 100 mg/1 mL SC injections. Indeed, all ISRs were mild and ≤ 2 cm from the injection site and quickly resolved spontaneously. Delivery volume is a factor contributing to SC injection site pain [14], and high injection volumes are typically associated with increased patient discomfort and sometimes pain at the site of administration, with less injection site pain reported where reduced volume is possible [14]. However, in line with our findings, other approved biological treatments have shown good tolerability when delivered using a single 2 mL injection. A single 2 mL SC injection of tralokinumab IL-13-neutralising human IgG4 monoclonal antibody used in patients with asthma was also well tolerated by healthy adults and presented a lower overall incidence of ISRs (9.6% through week 16) compared with the two separate SC injections [15, 16].
In this study, the incidence of TEAEs was low, and no substantial differences were found between the two treatments. Our results are in agreement with the results from a 5-year efficacy and safety outcome based on the long-term extension of the reSURFACE 1 and 2 trials, which demonstrated a reassuring safety profile of the 200 mg tildrakizumab dose, equal to that of the 100 mg dose [17]. The safety profile of tildrakizumab delivered by the new single 200 mg/2 mL PFS in this study showed no new or unexpected safety signals over 147 days, consistent with the safety reported previously [17,18,19]. No deaths occurred during the study, and all TEAEs were assessed as mild. The use of the 2 mL PFS did not result in any specific safety findings; in particular, 19.0% of subjects reported at least one TEAE after administration of the two separate 100 mg/1 mL SC injections, but not after the new 200 mg/2 mL PFS.
Moreover, this study shows that the new single PFS containing 200 mg/2 mL tildrakizumab was also rated as the preferred method of administration over the two separate 100 mg/1 mL SC injections by most subjects (61.5%). This result is plausibly a reflection of the enhanced convenience and ease of use of the new 200 mg/2 mL PFS compared to having two injections of 100 mg/1 mL, which may enhance patient adherence [20]. Also, patient preferences when choosing treatments for psoriasis include dosing- and frequency-related variables among the most important treatment attributes [21]. However, data also show that smaller volumes of subcutaneously administered methotrexate in rheumatoid arthritis patients resulted in a higher preference and improved local tolerability [22]. Thus, the lower injection volume of 2 × 100 mg/mL may explain the fact that a minority of the participants in this study preferred the two injections over the new single PFS injection.
Limitations of the study include the involvement of healthy subjects, which may have affected the results. However, the inclusion of healthy subjects in the trial as opposed to psoriasis subjects might have allowed a clearer interpretation of the trial results, since psoriasis subjects have confounding factors resulting from changes in disease state and/or the use of concomitant medications, all of which must be considered in the data analysis and interpretation. Published phase I tolerability data for other biologics such as the human monoclonal antibody tralokinumab given as single 2 mL injection or 2 × 1 mL injections were also collected in healthy subjects [15]. Moreover, the mean BMI of healthy subjects in this study was lower (24.1) compared to that reported in psoriasis studies (29.8–30.0) [23], which may have an impact on the transferability of the data.
Conclusions
Taken together, the administration of tildrakizumab as a single 200 mg/2 mL SC injection appears to be well tolerated and safe. Furthermore, these findings suggest that a single tildrakizumab SC injection of 200 mg/2 mL is the favored method of administration and thus may improve patient convenience and adherence to treatment.
References
Michalek IM, Loring B, John SM. A systematic review of worldwide epidemiology of psoriasis. J Eur Acad Dermatol Venereol. 2017;31(2):205–12.
Armstrong AW, Read C. Pathophysiology, clinical presentation, and treatment of psoriasis: a review. JAMA. 2020;323(19):1945–60.
Hawkes JE, Chan TC, Krueger JG. Psoriasis pathogenesis and the development of novel targeted immune therapies. J Allergy Clin Immunol. 2017;140(3):645–53.
Gooderham MJ, Papp KA, Lynde CW. Shifting the focus—the primary role of IL-23 in psoriasis and other inflammatory disorders. J Eur Acad Dermatol Venereol. 2018;32(7):1111–9.
Ghoreschi K, Balato A, Enerbäck C, Sabat R. Therapeutics targeting the IL-23 and IL-17 pathway in psoriasis. Lancet. 2021;397(10275):754–66.
Sabat R, Wolk K, Loyal L, Döcke WD, Ghoreschi K. T cell pathology in skin inflammation. Semin Immunopathol. 2019;41(3):359–77.
EMA. Ilumetri [Internet]. European Medicines Agency. 2018. https://www.ema.europa.eu/en/medicines/human/EPAR/ilumetri. Accessed 10 Dec 2021.
Papp K, Thaçi D, Reich K, Riedl E, Langley RG, Krueger JG, et al. Tildrakizumab (MK-3222), an anti-interleukin-23p19 monoclonal antibody, improves psoriasis in a phase IIb randomized placebo-controlled trial. Br J Dermatol. 2015;173(4):930–9.
Blauvelt A, Sofen H, Papp K, Gooderham M, Tyring S, Zhao Y, et al. Tildrakizumab efficacy and impact on quality of life up to 52 weeks in patients with moderate-to-severe psoriasis: a pooled analysis of two randomized controlled trials. J Eur Acad Dermatol Venereol. 2019;33(12):2305–12.
Burlando M, Castelli R, Cozzani E, Parodi A. Treatment of moderate-to-severe plaque psoriasis with tildrakizumab in the real-life setting. Drugs Context. 2021;10:2–6.
Drerup KA, Seemann C, Gerdes S, Mrowietz U. Effective and safe treatment of psoriatic disease with the anti-IL-23p19 biologic tildrakizumab: results of a real-world prospective cohort study in nonselected patients. Dermatology. 2022;238(4):615–9.
Smith CH, Yiu ZZN, Bale T, Burden AD, Coates LC, Edwards W, et al. British Association of Dermatologists guidelines for biologic therapy for psoriasis 2020: a rapid update. Br J Dermatol. 2020;183(4):628–37.
Schwarz CW, Loft N, Rasmussen MK, Nissen CV, Dam TN, Ajgeiy KK, et al. Predictors of response to biologics in patients with moderate-to-severe psoriasis: a Danish Nationwide Cohort Study. Acta Derm Venereol. 2021;101(10):adv00579.
St Clair-Jones A, Prignano F, Goncalves J, Paul M, Sewerin P. Understanding and minimising injection-site pain following subcutaneous administration of biologics: a narrative review. Rheumatol Ther. 2020;7(4):741–57.
Jain M, Doughty D, Clawson C, Li X, White N, Agoram B, et al. Tralokinumab pharmacokinetics and tolerability when administered by different subcutaneous injection methods and rates. Int J Clin Pharmacol Ther. 2017;55(7):606–20.
Sanofi–Aventis. Product monograph: Dupixent. 2020 [Internet]. https://pdf.hres.ca/dpd_pm/00058817.PDF.
Thaci D, Piaserico S, Warren RB, Gupta AK, Cantrell W, Draelos Z, et al. Five-year efficacy and safety of tildrakizumab in patients with moderate-to-severe psoriasis who respond at week 28: pooled analyses of two randomized phase III clinical trials (reSURFACE 1 and reSURFACE 2). Br J Dermatol. 2021;185(2):323–34.
Reich K, Griffiths CEM, Gordon KB, Papp KA, Song M, Randazzo B, et al. Maintenance of clinical response and consistent safety profile with up to 3 years of continuous treatment with guselkumab: results from the VOYAGE 1 and VOYAGE 2 trials. J Am Acad Dermatol. 2020;82(4):936–45.
Blauvelt A, Leonardi CL, Gooderham M, Papp KA, Philipp S, Wu JJ, et al. Efficacy and safety of continuous risankizumab therapy vs treatment withdrawal in patients with moderate to severe plaque psoriasis: a phase 3 randomized clinical trial. JAMA Dermatol. 2020;156(6):649–58.
Belinchón I, Rivera R, Blanch C, Comellas M, Lizán L. Adherence, satisfaction and preferences for treatment in patients with psoriasis in the European Union: a systematic review of the literature. Patient Prefer Adherence. 2016;10:2357–67.
Alcusky M, Lee S, Lau G, Chiu GR, Hadker N, Deshpande A, et al. Dermatologist and patient preferences in choosing treatments for moderate to severe psoriasis. Dermatol Ther (Heidelb). 2017;7(4):463–83.
Müller-Ladner U, Rockwitz K, Brandt-Jürgens J, Haux R, Kästner P, Braun J, et al. Tolerability and patient/physician satisfaction with subcutaneously administered methotrexate provided in two formulations of different drug concentrations in patients with rheumatoid arthritis. Open Rheumatol J. 2010;4:15–22.
Reich K, Warren RB, Iversen L, Puig L, Pau-Charles I, Igarashi A, et al. Long-term efficacy and safety of tildrakizumab for moderate-to-severe psoriasis: pooled analyses of two randomized phase III clinical trials (reSURFACE 1 and reSURFACE 2) through 148 weeks. Br J Dermatol. 2020;182(3):605–17.
Acknowledgements
We thank the participants of the study. We would also like to thank Almirall members who participated in the clinical trial design and/or trial oversight: Raquel Otero, Laura Vidal, María Teresa Sanz, Bernabé Proto, Álvaro Rodríguez, Natalia Fernández, Natalie Ivanoff, and Marie Pierre Malice.
Funding
The study was sponsored by Almirall R&D, Barcelona, Spain. Almirall R&D funded the journal’s Rapid Service Fee.
Medical Writing and/or Editorial Assistance
Medical writing support was provided by Stefania Ippati, PhD, TFS HealthScience and Eva Mateu, PhD, TFS HealthScience, and was funded by Almirall R&D, in accordance with Good Publication Practice (GPP3) guidelines.
Author Contributions
Georgios Kokolakis, German Kreis, Meritxell Falqués, Mònica Aparici, and Wiebke Sondermann conceived and designed the analysis and contributed to the writing of the manuscript.
List of Investigators
The principal investigator for this study was Salah Hadi, MD, PRA Health Sciences—Early Development Services, Groningen, the Netherlands.
Prior Presentation
Partial results presented in this paper were displayed at the 31st EADV Congress, which took place in Milan on September 7–10, 2022.
Disclosures
Georgios Kokolakis has received travel grants or honoraria or has been a consultant member of advisory boards and speakers’ bureaus for one or more of the following: AbbVie Deutschland GmbH & Co. KG, Actelion Pharmaceuticals Ltd., Almirall S.A., Amgen GmbH, Basilea Pharmaceutica Ltd., Bayer Schering Pharma AG, Biogen Idec GmbH, Celgene GmbH, Boehringer Ingelheim Pharma GmbH & Co., Bristol Myers Squibb GmbH & Co. KGaA, Celgene GmbH, Celgene International II Sàrl, CSL Behring, Hexal-Sandoz GmbH, Janssen-Cilag GmbH, Incyte Corporation, Leo Pharma GmbH, Lilly Deutschland GmbH, MSD Sharp & Dohme GmbH, Novartis Pharma GmbH, Pfizer Deutschland GmbH, TFS GmbH, and UCB Pharma GmbH. German Kreis, Meritxell Falqués, and Mònica Aparici are employees of Almirall. Wiebke Sondermann has received grants from Medi GmbH Bayreuth, grants and personal fees from Novartis, grants and personal fees from Almirall, personal fees from Bristol Myers Squibb, AbbVie, Boehringer Ingelheim, Amgen, GSK, Janssen, Lilly, UCB, LEO Pharma, Pfizer, and Sanofi Genzyme.
Compliance with Ethics Guidelines
This study was performed in accordance with good clinical practice guidelines and the Helsinki Declaration of 1964 and its later amendments. The study received approval from the independent ethics committee Beoordeling Ethiek Biomedisch Onderzoek in Assen, the Netherlands. All participants provided written informed consent to participate in the study.
Data Availability
The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.
Author information
Authors and Affiliations
Corresponding author
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Kokolakis, G., Kreis, G., Falqués, M. et al. High Tolerability, Favorable Safety, and Subjects' Preference for a Single 200 mg/2 mL Tildrakizumab Injection: A Phase I, Open-Label, Randomized Crossover Trial in Healthy Volunteers. Dermatol Ther (Heidelb) 12, 2135–2144 (2022). https://doi.org/10.1007/s13555-022-00789-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13555-022-00789-9