
Abstract
Introduction
Risankizumab has demonstrated efficacy and safety in phase 3 studies in patients with moderate to severe plaque psoriasis. This randomized clinical trial assessed the efficacy and safety of risankizumab in patients with moderate to severe plaque psoriasis in the Russian Federation.
Methods
Patients with moderate to severe plaque psoriasis were randomized 4:1 to 16 weeks of double-blind treatment with risankizumab 150 mg or placebo (period A; dosing at baseline and week 4) followed by an open-label extension (period B) during which all patients received risankizumab 150 mg at weeks 16, 28, and 40 and were followed up to week 52. The primary study endpoint was the proportion of patients achieving ≥ 90% improvement in the Psoriasis Area and Severity Index (PASI 90) at week 16, and secondary endpoints included Static Physician’s Global Assessment scores and the Dermatology Life Quality Index. Treatment-emergent adverse events were monitored throughout the two study periods.
Results
Of the 50 patients who entered period A, 41 were randomized to receive risankizumab and 9 to receive placebo. Forty-eight patients entered period B, and 47 completed the study. A significantly larger proportion of risankizumab-treated patients achieved PASI 90 at week 16 compared with placebo-treated patients [response rate difference: 38.8% (95% CI 7.8–69.7%; P = 0.035)]. Consistently higher proportions of risankizumab-treated patients achieved secondary endpoints compared with the placebo-treated patients. Safety profiles were similar between the treatment groups, and no patients discontinued the study drug owing to adverse events.
Conclusion
Risankizumab was efficacious and well tolerated in patients with moderate to severe plaque psoriasis in the Russian Federation.
Trial Registration
ClinicalTrials.gov NCT03518047.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Why carry out this study? |
Risankizumab has been shown to be well tolerated and efficacious in patients with moderate to severe plaque psoriasis and is approved in more than 75 countries, including the United States, Canada, Europe, and Japan. |
The IMMpress study (NCT03518047) was a phase 3, randomized, double-blind, placebo-controlled study that evaluated the efficacy and safety of risankizumab in patients with moderate to severe plaque psoriasis in the Russian Federation. |
What was learned from the study? |
A significantly greater proportion of patients receiving risankizumab (vs. placebo) achieved the primary study endpoint of ≥ 90% improvement in the Psoriasis Area and Severity Index at week 16, and safety profiles were similar for the treatment groups. |
The findings of this phase 3, randomized, double-blind, placebo-controlled, clinical study in patients with moderate to severe plaque psoriasis in the Russian Federation demonstrate that risankizumab was highly effective, with a safety profile consistent with other phase 3 risankizumab studies in patients with plaque psoriasis carried out in other countries. |
Introduction
Psoriasis, a chronic, systemic inflammatory skin condition, is associated with significant infectious, psychological, arthritic, gastrointestinal, cardiometabolic, and malignant comorbidities [1, 2]. The reported prevalence of psoriasis varies from 0.09 to 11.4% depending on geographic region, the definitions of prevalence and cases of psoriasis used, the age and sex distribution of the population, and the sampling technique [3, 4]. In most developed countries, the prevalence of psoriasis is between 1.2 and 8.5% [3, 5]. The prevalence of psoriasis in the Russian Federation was reported as 233.4 per 100,000 in 2015 [6], and its incidence as between 39.0 and 65.0 per 100,000 person-years in 2016 [7, 8].
The pathology of psoriasis is driven by complex interactions between multiple immune cell populations and is mediated by the release of proinflammatory cytokines, including tumor necrosis factor-α, interleukin (IL)-17, and IL-23. The IL-23/IL-17 signaling pathway has been identified as a key regulatory component in the development and maintenance of psoriatic lesions [9, 10].
Risankizumab is a selective IL-23 inhibitor that binds to the p19 subunit of IL-23, preventing its interaction with its receptor, IL-23R [11]. Risankizumab has been shown to be well tolerated and efficacious in patients with moderate to severe plaque psoriasis [12,13,14,15,16], and is approved in more than 75 countries, including the United States, Canada, Europe, and Japan [17, 18].
The IMMpress study (NCT03518047) was a phase 3, randomized, double-blind study evaluating the efficacy and safety of risankizumab in patients with moderate to severe plaque psoriasis in the Russian Federation.
Methods
The IMMpress study was carried out at 6 locations in the Russian Federation between July 19, 2018, and December 19, 2019.
Eligible patients were randomized in a 4:1 ratio to receive risankizumab 150 mg (2 × 75-mg prefilled syringe [PFS]) subcutaneously (sc) or a matching placebo at baseline and week 4 (period A; Fig. 1).

Study design. Bold indicates study visit ± 3 days. BL baseline, DB double-blind, OL open-label, W week
At the end of the 16-week, double-blind period A, all study patients continued into the open-label period (period B), in which they received open-label risankizumab 150 mg sc (2 × 75-mg PFS) at weeks 16, 28, and 40. Follow-up continued until at least week 52, with a follow-up phone call at week 60.
Patients who discontinued the study drug before the end of the study completed an early termination visit as soon as possible after discontinuation and were followed up at the scheduled visits.
Patients
Eligible patients were ≥ 18 years old at the screening visit, with a diagnosis of chronic moderate to severe plaque psoriasis (with or without psoriatic arthritis) for ≥ 6 months before first administration of the study drug. Moderate to severe psoriasis was defined as ≥ 10% body surface area involvement, a Psoriasis Area and Severity Index (PASI) score of ≥ 10, and a Static Physician’s Global Assessment (sPGA) score of ≥ 3. In addition, patients were required to be candidates for systemic therapy or phototherapy as assessed by the investigator. Eligible patients had not had any prior therapy with an IL-17, IL-12/23p40, or IL-23p19 inhibitor. Concurrent therapy with a biologic and/or other systemic therapy was prohibited during the study.
Efficacy Assessments
Efficacy in the double-blind (period A) and open-label (period B) phases of the study was evaluated in the intent-to-treat (ITT) population, which included all patients who were randomized at week 0 (n = 50).
Primary Efficacy Endpoint
The primary study endpoint was the proportion of patients achieving PASI 90 (≥ 90% reduction from baseline PASI score) at week 16.
Secondary Efficacy Endpoints
Four secondary endpoints were examined during this study: (i) the proportion of patients achieving an sPGA score of 0 (clear) or 0/1 (clear or almost clear) at week 16, (ii) the proportion of patients achieving PASI 75 at week 16, (iii) the proportion of patients achieving PASI 100 at week 16, and (iv) the proportion of patients achieving a Dermatology Life Quality Index (DLQI) score of 0 or 1 at week 16.
Other Efficacy Endpoints
The following endpoints were assessed for all visits at which data were recorded: (i) the proportion of patients achieving an sPGA score of 0 or 0/1; (ii) the proportion of patients achieving PASI 75/90/100; (iii) the change and percentage change from baseline in PASI score; (iv) the proportion of patients achieving a DLQI score of 0 or 0/1; and (v) the change from baseline in DLQI.
Safety Assessments
Safety was evaluated in all patients in the ITT population who received ≥ 1 dose of study drug. Treatment-emergent adverse events (TEAEs) for the double-blind period A were defined as events with an onset after the first dose of study drug and within 140 days after the last dose of study drug for patients who did not enter the open-label period B or before the first dose of risankizumab in period B for patients who entered period B. TEAEs for period B were defined as events with an onset after the first dose of risankizumab in period B and within 140 days after the last dose of risankizumab in the study. TEAEs in the all-risankizumab-treated safety population (patients randomized to receive risankizumab during period A and all patients in period B) were defined as events with an onset after the first dose of risankizumab and within 140 days after the last dose of risankizumab in the study.
Safety endpoints included all TEAEs, serious adverse events (SAEs; defined as any fatal or life-threatening event, any event requiring hospitalization or prolongation of hospitalization, any congenital anomaly, any event causing persistent/significant incapacity, or any event requiring medical or surgical intervention to prevent a serious outcome), any adverse events (AEs) in areas of safety interest (ASI), and any AEs leading to study discontinuation. ASIs were identified based on their higher prevalence in the moderate to severe psoriasis population, customary concerns with injected immunoglobulin products, or the immunomodulatory activity of risankizumab. ASI for this study included adjudicated cardiovascular events, serious infections, tuberculosis, fungal and opportunistic infections (including herpes zoster), malignancies, hypersensitivity reactions, and hepatic events. AEs were coded using the Medical Dictionary for Regulatory Activities (MedDRA) v21.1 preferred terms.
Statistical Analyses
Based on the weighted averages of PASI 90 response rates with risankizumab and placebo of 75.1% and 3.5%, respectively, in the UltIMMa-1 (NCT02684357) and UltIMMa-2 (NCT02684370) trials, an overall sample size of 50 patients (40 patients in the risankizumab arm; 10 patients in the placebo arm) was selected to provide ≥ 90% power to detect a difference in PASI 90 response rate at week 16 based on a two-sided alpha level of 0.05.
The ITT population was used for the analysis of efficacy in period A (double-blind) and period B (open-label). The primary endpoint was compared between treatment groups used the Cochran–Mantel–Haenszel test, adjusting for pooled site.
For the analysis of categorical variables, nonresponder imputation (NRI) was the primary approach and last observation carried forward (LOCF) was the secondary approach. Continuous variables were analyzed using the LOCF approach. Analyses were performed using SAS® version 9.4 (SAS Institute, Inc., Cary, NC, USA) for the UNIX operating system.
Ethics
The study was conducted in accordance with the International Council for Harmonisation guidelines, the applicable regulations and guidelines governing clinical study conduct, and the Declaration of Helsinki. The protocol was approved by a local independent ethics committee at each location as well as a central ethics committee (Ethics Counsel at the Ministry of Health of the Russian Federation, Moscow, Russian Federation; Supplementary Table S1), and all patients provided written informed consent before the initiation of any screening or study-specific procedures.
Results
Study Population
Fifty patients were randomized 4:1 to risankizumab:placebo and entered period A of the study (Fig. 2). Period A was completed by all 9 patients receiving placebo and 39 of 41 patients receiving risankizumab. Of the 2 risankizumab-treated patients who did not complete period A, 1 was lost to follow-up and 1 withdrew consent. Of the 48 patients who entered period B and received risankizumab, 47 patients completed the study. The patient who did not complete period B was lost to follow-up.

Patient disposition
Demographics and baseline clinical characteristics were broadly comparable between study groups (Table 1).
Efficacy
Primary Endpoints
A significantly larger proportion of patients in the risankizumab group achieved the primary study endpoint of PASI 90 at week 16 compared with the placebo group (NRI; Table 2). The difference in response rate for patients treated with risankizumab versus those treated with placebo was 38.8% (95% CI 7.8–69.7%; P = 0.035). Sensitivity analyses of the primary endpoint using LOCF and among the per protocol population were consistent with results from the primary analysis.
Secondary and Other Efficacy Endpoints
At week 16, 19 patients (46.3%) in the risankizumab-treated group achieved an sPGA score of 0 compared with 1 patient (11.1%) in the placebo-treated group (Fig. 2a). The adjusted between-group difference for risankizumab versus placebo treatment at week 16 was 35.2% (P = 0.051). During period B, 78% of patients (32/41) who continued risankizumab treatment (risankizumab/risankizumab) and 88.9% of patients (8/9) who switched from placebo to risankizumab (placebo/risankizumab) achieved an sPGA score of 0 at week 52 (Fig. 3a). An sPGA score of 0/1 was achieved by 73.2% of risankizumab-treated patients versus 22.2% of placebo-treated patients at week 16 (adjusted between-group difference, 50.9%; P = 0.004). At week 52, 90.2% of patients who received risankizumab/risankizumab achieved an sPGA score of 0/1 versus 100% of patients who received placebo/risankizumab (Fig. 3b).

Proportions of patients achieving a sPGA 0, b sPGA 0/1, c PASI 75, d PASI 90, e PASI 100, and f percentage improvement from baseline in PASI. NRI analysis for panels a–e and LOCF for panel f. LOCF last observation carried forward; NRI nonresponder imputation; PASI 75/90/100 ≥ 75%/≥ 90%/100% improvement in Psoriasis Area and Severity Index; PBO placebo; sPGA static Physician’s Global Assessment; RZB risankizumab. *P < 0.05; †P < 0.001; ‡P < 0.0001
The secondary endpoints of PASI 75 and PASI 100 at week 16 were achieved by 73.2% and 46.3% of the patients treated with risankizumab, respectively, versus 22.2% and 11.1% of the patients treated with placebo (Fig. 3c, e). Adjusted between-group differences for risankizumab versus placebo at week 16 were 50.9% (P = 0.004) for PASI 75 and 35.2% (P = 0.051) for PASI 100. By week 52, PASI 75 had been reached by 92.7% and 100% of patients treated with risankizumab/risankizumab and placebo/risankizumab, respectively; PASI 90 was achieved by 90.2% and 100% and PASI 100 by 78.0% and 88.9% (Fig. 3c–e).
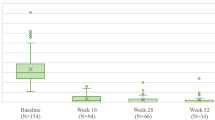
Percentage improvement from baseline in PASI at week 16 was 81.1% and 24.7% (LOCF) with risankizumab and placebo, respectively (least squares mean difference, 56.3%; nominal P < 0.001). By week 52, PASI had improved in both the risankizumab/risankizumab and placebo/risankizumab groups by almost 100% (97.6% and 99.4%, respectively; Fig. 3f).
Improvements in patient DLQI occurred over 16 weeks of risankizumab treatment, with 46.3% and 63.4% of patients treated with risankizumab achieving DLQI 0 or 0/1 (NRI), respectively, compared with 11.1% and 22.1% of patients treated with placebo (Fig. 4a). The adjusted between-group differences were 35.2% for DLQI 0 (nominal P = 0.051) and 52.3% for DLQI 0/1 (nominal P = 0.004). By the end of the study, ≥ 70% of patients in both treatment groups had achieved DLQI 0 or 0/1 (NRI), and there was an approximately 14-point improvement from baseline in DLQI (Fig. 4b, c).

Proportions of patients achieving a DLQI 0 and b DLQI 0/1 and c change from baseline in DLQI. NRI analysis used for panels a, b and LOCF for panel c. DLQI Dermatology Life Quality Index, LOCF last observation carried forward, NRI nonresponder imputation, PBO placebo, RZB risankizumab. *Nominal P < 0.05; †nominal P < 0.001; ‡nominal P < 0.0001
Safety
The evaluation of safety at each study visit showed that the AE profiles were generally similar between the treatment groups and that rates of SAEs, severe AEs, and AEs in ASI were low, with no meaningful differences between risankizumab and placebo during period A (Table 3). Rates of AEs with continued risankizumab treatment were similar through week 52.
Through week 52, and considering all patients exposed to risankizumab, 20 patients (40.0%) experienced AEs and 2 (4.0%) experienced a total of 3 SAEs, all of which were considered by the investigator to be unrelated to treatment; the study drug was not discontinued. One patient experienced an exacerbation of chronic obstructive pulmonary disease of moderate severity during period A and then a worsening of open-angle glaucoma (mild severity) during period B. A second patient experienced community-acquired pneumonia of moderate severity during period B that resolved with treatment.
No increase was observed in the rates of AEs overall (events/100 patient-years) in patients who received risankizumab during both study periods compared with the rates observed through week 16, and there was no notable difference in the rates of AEs in the patients who switched from placebo to risankizumab versus those who continued risankizumab through week 52.
Upper respiratory infections (nasopharyngitis, respiratory tract infection) were the most common AEs in patients who received risankizumab through week 52. The only occurrence of an SAE in an ASI was the case of moderate community-acquired pneumonia described above. Few AEs in ASI were reported through week 52; no patient experienced a serious hypersensitivity reaction, malignancy, or adjudicated cardiovascular event. There were no deaths or discontinuations of the study drug due to AEs in the study (Table 3).
The incidence of anti-risankizumab antibodies and risankizumab-neutralizing antibodies was monitored throughout the study. Among all patients who received risankizumab and who had ≥ 1 assessment postbaseline, 7 (14.6%) had treatment-emergent anti-risankizumab antibodies and 1 (2.1%) had risankizumab-neutralizing antibodies.
Discussion
The findings of this phase 3, randomized, double-blind, placebo-controlled, clinical study demonstrate that risankizumab was highly effective in the treatment of patients with moderate to severe plaque psoriasis in the Russian Federation.
Treatment with risankizumab resulted in a rapid onset of clinical response (within 4 weeks of the first dose), which was maintained through week 52. At week 16, 61.0% of patients receiving risankizumab had achieved PASI 90 [compared with 22.2% of patients treated with placebo during period A; the difference in response rate was 38.8% (95% CI 7.8–69.7%; P = 0.035)]. This was maintained through week 52, at which time 90.2% of the patients treated with risankizumab throughout the study and 100% of the patients who had received placebo/risankizumab had achieved PASI 90. Commensurate improvements in PASI 100 and sPGA were observed. These results are consistent with previous multinational phase 3 risankizumab clinical studies in which a PASI 90 response rate of 73–75% with risankizumab versus 2–5% with placebo was achieved at week 16 [13, 16]. The response rate at week 52 was similarly maintained in these studies with continuous risankizumab (81–86%), whereas patients who switched from placebo to risankizumab achieved PASI 90 at a response rate of 78–85% [13, 16].
Clearance of psoriatic skin lesions following treatment with risankizumab was associated with improvements in patient-reported quality of life (QoL), with 72% of all patients achieving DLQI 0 by week 52, indicating a minimal effect of psoriasis on QoL at this time.
The improvements in clinical and QoL outcomes achieved by 16 weeks of risankizumab treatment were sustained through 52 weeks with continued risankizumab treatment; patients who were allocated to placebo treatment during period A and then switched to risankizumab during period B achieved similar improvements in clinical and QoL parameters.
Risankizumab was well tolerated in the IMMpress study (rate of AEs/SAEs was 24%/2% over 16 weeks and 40%/4% over 52 weeks). The safety profile was consistent with other phase 3 studies in patients with plaque psoriasis carried out in other countries (rate of AEs/SAEs over 16 weeks: 50%/2% in UltIMMa-1, 46%/2% in UltIMMa-2, 46%/2% in IMMhance, 56%/3% in IMMvent, and 71%/5% in IMMerge) [13,14,15,16]. In the IMMpress study, there was a single SAE in the ASI, a serious infection (pneumonia) during period B. There were no reports of malignancy, no instances of serious hypersensitivity reactions, and no adjudicated cardiovascular events.
A limitation of this study was the relatively small sample size (n = 50) as compared with the earlier phase 3 studies (n = 300–600 patients), which can lead to higher variation in the results, limit the power to detect infrequent AEs, and make direct comparisons to other risankizumab studies difficult. Despite this limitation, however, the results of this study suggest a robust efficacy response and high tolerability of risankizumab in the treatment of moderate to severe psoriasis in the Russian Federation. Overall, the results of IMMpress are consistent with the benefit/risk profile established for risankizumab in other populations.
Conclusion
In conclusion, risankizumab was highly effective and well tolerated in this study of patients with chronic plaque psoriasis in the Russian Federation.
References
Griffiths CE, Barker JN. Pathogenesis and clinical features of psoriasis. Lancet. 2007;370(9583):263–71.
Takeshita J, Grewal S, Langan SM, et al. Psoriasis and comorbid diseases: epidemiology. J Am Acad Dermatol. 2017;76(3):377–90.
Parisi R, Symmons DP, Griffiths CE, Ashcroft DM, Identification and Management of Psoriasis Associated Comorbidity (IMPACT) Project Team. Global epidemiology of psoriasis: a systematic review of incidence and prevalence. J Investig Dermatol. 2013;133(2):377–85.
World Health Organization. Global report on psoriasis. 2016. https://apps.who.int/iris/handle/10665/204417. Accessed 10 Feb 2021.
Gelfand JM, Weinstein R, Porter SB, Neimann AL, Berlin JA, Margolis DJ. Prevalence and treatment of psoriasis in the United Kingdom: a population-based study. Arch Dermatol. 2005;141(12):1537–41.
Kubanov AA, Bakulev AL, Fitileva TV, et al. Disease burden and treatment patterns of psoriasis in Russia: a real-world patient and dermatologist survey. Dermatol Ther (Heidelb). 2018;8(4):581–92.
Parisi R, Iskandar IYK, Kontopantelis E, et al. National, regional, and worldwide epidemiology of psoriasis: systematic analysis and modelling study. BMJ. 2020;28(369): m1590.
Iskandar IYK, Parisi R, Griffiths CEM, Ashcroft DM, Global Psoriasis Atlas. Systematic review examining changes over time and variation in the incidence and prevalence of psoriasis by age and gender. Br J Dermatol. 2021;184(2):243–58.
Chan TC, Hawkes JE, Krueger JG. Interleukin 23 in the skin: role in psoriasis pathogenesis and selective interleukin 23 blockade as treatment. Ther Adv Chronic Dis. 2018;9(5):111–9.
Hawkes JE, Yan BY, Chan TC, Krueger JG. Discovery of the IL-23/IL-17 signaling pathway and the treatment of psoriasis. J Immunol. 2018;201(6):1605–13.
Singh S, Kroe-Barrett RR, Canada KA, et al. Selective targeting of the IL23 pathway: generation and characterization of a novel high-affinity humanized anti-IL23A antibody. MAbs. 2015;7(4):778–91.
Papp KA, Blauvelt A, Bukhalo M, et al. Risankizumab versus ustekinumab for moderate-to-severe plaque psoriasis. N Engl J Med. 2017;376(16):1551–60.
Gordon KB, Strober B, Lebwohl M, et al. Efficacy and safety of risankizumab in moderate-to-severe plaque psoriasis (UltIMMa-1 and UltIMMa-2): results from two double-blind, randomised, placebo-controlled and ustekinumab-controlled phase 3 trials. Lancet. 2018;392(10148):650–61.
Reich K, Gooderham M, Thaci D, et al. Risankizumab compared with adalimumab in patients with moderate-to-severe plaque psoriasis (IMMvent): a randomised, double-blind, active-comparator-controlled phase 3 trial. Lancet (London, England). 2019;394(10198):576–86.
Warren RB, Blauvelt A, Poulin Y, et al. Efficacy and safety of risankizumab vs. secukinumab in patients with moderate-to-severe plaque psoriasis (IMMerge): results from a phase III, randomized, open-label, efficacy-assessor-blinded clinical trial. Br J Dermatol. 2021;184(1):50–9.
Blauvelt A, Leonardi CL, Gooderham M, et al. Efficacy and safety of continuous risankizumab therapy vs treatment withdrawal in patients with moderate to severe plaque psoriasis: a phase 3 randomized clinical trial. JAMA Dermatol. 2020;156(6):649–58.
AbbVie Inc. Skyrizi™ (risankizumab-rzaa). Full prescribing information. North Chicago, IL, USA: AbbVie Inc.; 2019.
AbbVie S.r.l. Skyrizi™ (risankizumab). Summary of product characteristics, Campoverde di Aprilia, Italy: AbbVie S.r.l.; 2019.
Acknowledgements
Funding
AbbVie funded this study and participated in the study design, research, analysis, data collection, and interpretation of data and in the writing, reviewing, and approval of the publication. All authors had access to relevant data and participated in the drafting, review, and approval of this publication. No honoraria or payments were made for authorship. AbbVie funded the journal’s Rapid Service Fee.
Medical Writing and Editorial Assistance
Medical writing support was provided by Moira A. Hudson, PhD, and Janet E. Matsuura, PhD, of ICON (Blue Bell, PA, USA) and was funded by AbbVie Inc.
Authorship
All named authors meet the International Committee of Medical Journal Editors (ICMJE) criteria for authorship for this article, take responsibility for the integrity of the work as a whole, and have given their approval for this version to be published.
Author Contributions
Study concept and design: TW, MK, KA. Acquisition of data: LO, AE, AS, AK. Analysis and interpretation of data: all authors. Writing of the manuscript: all authors, MH. Critical revision of the manuscript for important intellectual content: all authors. Statistical analysis: TW. All authors had access to relevant data and participated in the drafting, review, and approval of this publication.
Disclosures
Liudmila Odnopozova has no conflict of interest to disclose. Anton Edin has received grant/research support as a principal investigator in clinical trials from AbbVie, Eli Lilly, LEO Pharma, GSK, Bayer, Novartis. Alexey Sukharev has received grant/research support as a principal investigator in clinical trials from AbbVie, Eli Lilly, Novartis, and Pfizer. Tianshuang Wu and Kerstin Aydin are full-time salaried employees of AbbVie and may own stock/stock options. Maureen Kelly was a full-time salaried employee of AbbVie at the time of this research and may own stock/stock options. She has since retired from AbbVie. Alkes Khotko has worked as principal investigator in clinical research for AbbVie, Bristol Myers Squibb, Eli Lilly, Galderma, Janssen, Leo, Novartis, Sanofi, and Amgen over the last 3 years.
Compliance with Ethics Guidelines
The study was conducted in accordance with the International Council for Harmonisation guidelines, the applicable regulations and guidelines governing clinical study conduct, and the Declaration of Helsinki. The protocol was approved by a local independent ethics committee at each location as well as a central ethics committee (Ethics Counsel at the Ministry of Health of the Russian Federation, Moscow, Russian Federation; Supplementary Table S1), and all patients provided written informed consent before the initiation of any screening or study-specific procedures.
Data Availability
AbbVie is committed to responsible data sharing regarding the clinical trials we sponsor. This includes access to anonymized, individual and trial-level data (analysis data sets), as well as other information (e.g., protocols and clinical study reports), as long as the trials are not part of an ongoing or planned regulatory submission. This includes requests for clinical trial data for unlicensed products and indications. These clinical trial data can be requested by any qualified researchers who engage in rigorous, independent scientific research, and will be provided following review and approval of a research proposal and Statistical Analysis Plan (SAP) and execution of a Data Sharing Agreement (DSA). Data requests can be submitted at any time and the data will be accessible for 12 months, with possible extensions considered. For more information on the process, or to submit a request, visit the following link: https://www.abbvie.com/our-science/clinical-trials/clinical-trials-data-and-information-sharing/data-and-information-sharing-with-qualified-researchers.html.
Author information
Authors and Affiliations
Corresponding author
Additional information
The affiliation shown for Maureen Kelly was her affiliation at the time of this research.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Odnopozova, L., Edin, A., Sukharev, A. et al. Risankizumab for the Treatment of Moderate to Severe Plaque Psoriasis in the Russian Federation. Dermatol Ther (Heidelb) 12, 2063–2075 (2022). https://doi.org/10.1007/s13555-022-00776-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13555-022-00776-0