
Abstract
Introduction
Netakimab (NTK), an original humanized anti-interleukin-17 monoclonal antibody, showed therapeutic efficacy in moderate-to-severe plaque psoriasis in a phase 2 clinical study. Herein we report the results of 54 weeks of a phase 3 PLANETA trial aimed to evaluate the efficacy and safety of two NTK regimens vs. placebo.
Methods
Two hundred thirteen patients with moderate-to-severe plaque psoriasis were randomly assigned to receive NTK 120 mg once every 2 weeks (NTK Q2W), NTK 120 mg once every 4 weeks (NTK Q4W) or placebo. During the first 3 weeks, patients received subcutaneous injections of NTK or placebo (according to the allocation) once a week. Patients in the NTK Q2W group then received NTK at weeks 4, 6, 8 and 10. Subjects in the NTK Q4W group received NTK at weeks 6 and 10 and placebo at weeks 4 and 8. Patients in the placebo group received placebo injections at weeks 4, 6, 8 and 10. Treatment was unblinded at week 12. During the open-label phase, patients in both NTK groups continued to receive NTK Q4W. The primary efficacy endpoint was the proportion of patients in each group who achieved a ≥ 75% reduction from baseline in psoriasis area and severity index (PASI 75) at week 12.
Results
A total of 77.7%, 83.3% and 0% of patients had a PASI 75 response at week 12 in the NTK Q2W, NTK Q4W and placebo groups, respectively (P < 0.0001, Fisher’s exact test, ITT). The effect was maintained throughout the 1-year treatment. NTK showed a good safety profile and low immunogenicity.
Conclusion
Treatment with NTK results in high rates of sustained clinical response in patients with moderate-to-severe plaque psoriasis. The study is ongoing; thus, long-term use efficacy and safety data are forthcoming.
Clinical Trial Registration
The trial is registered at the US National Institutes of Health (ClinicalTrials.gov; NCT03390101).
Similar content being viewed by others


Avoid common mistakes on your manuscript.
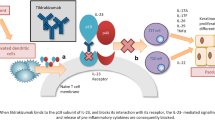
Netakimab is an original humanized anti-interleukin-17 monoclonal antibody which showed therapeutic efficacy in plaque psoriasis and ankylosing spondylitis in phase 2 clinical studies. The phase 3 PLANETA clinical trial was conducted to evaluate the efficacy and safety of Q4W and Q2W netakimab dosing regimens vs. placebo in patients with moderate-to-severe plaque psoriasis |
Netakimab demonstrated high rates of sustained clinical response as early as the first few weeks of treatment, a favourable safety profile and good tolerability in patients with moderate-to-severe plaque psoriasis and can be the treatment option for patients with moderate-to-severe plaque psoriasis |
In patients with moderate-to-severe plaque psoriasis NTK 120 mg should be administered once a week for the first 3 weeks followed by NTK 120 mg Q4W injections |
Digital Features
This article is published with digital features, including a summary slide, to facilitate understanding of the article. To view digital features for this article go to https://doi.org/10.6084/m9.figshare.14461808.
Introduction
Psoriasis is a chronic, systemic inflammatory disease with leading skin involvement that is associated with a range of comorbidities. Symptoms may vary from mild with a few local skin lesions to severe forms, affecting a significant proportion of the body surface. Psoriasis can seriously affect patient social activity and quality of life.
In 2012, the first published data established the pivotal role of interleukin-17 (IL-17) in the immunopathogenesis of psoriasis and suggested that blockade of IL-17 signalling can be one of the therapeutic options [1].
Netakimab (NTK) is a novel recombinant humanized IgG1 anti-IL-17 monoclonal antibody (mAb) with modified Fc-fragment (Fc-fragment crystallisable region) and CDR (complementarity-determining regions). The superiority of NTK at the dose of 120 mg over placebo was shown in phase 2 trials in moderate-to-severe psoriasis (NCT02762994) and in ankylosing spondylitis (NCT02763111) [2, 3].
The PLANETA study aimed to evaluate the efficacy and safety of NTK Q2W (once every 2 weeks) vs. NTK Q4W (once every 4 weeks) vs. placebo in patients with moderate-to-severe plaque psoriasis. In this article, we report the analysis of 1 year of the study.
Methods
PLANETA is the ongoing international, multicentre, phase 3, double-blind, placebo-controlled clinical study conducted in 24 investigational sites in the Russian Federation and the Republic of Belarus according to the Declaration of Helsinki following Good Clinical Practice guidelines. The study was approved by the Central Regulatory Authorities of the Russian Federation and the Republic of Belarus and Ethical Review Boards of each of the participating sites. The trial was registered at the US National Institutes of Health (ClinicalTrials.gov; NCT03390101).
Eligible patients were 18 years and older, had an established diagnosis of moderate-to-severe plaque psoriasis for at least 6 months, were candidates for or had received at least one course of phototherapy or systemic therapy, had at least a 10% body surface area (BSA) affected by psoriasis, a Psoriasis Area and Severity Index (PASI) score ≥ 10, and a static Physician’s Global Assessment (sPGA) score ≥ 3 at screening.
Patients were ineligible to participate in the study if they had other forms of psoriasis (e.g. guttate, erythrodermic or pustular) or any other skin diseases (e.g. eczema), previously used mAbs targeting IL-17 or its receptor, or received more than one drug containing mAbs or their fragments. Any mAb had to be withdrawn for at least 12 weeks; systemic therapy and phototherapy within 4 weeks; and the range of topical medications within 2 weeks before signing the informed consent (IC). All patients were given adequate time to reflect on the information, had their questions answered, gave free and voluntary consent to participate and signed an IC form.
All patients underwent chest x-ray and tuberculosis test (T-SPOT.TB test/QuantiFERON-TB Gold test/TB skin test) at screening. In case of ambiguous tuberculosis test results, patient underwent consultation with a specialized physician. If active/latent infection was excluded, the patient could be included in the study.
Eligible patients were stratified by body weight (< 100 kg/≥ 100 kg), previous treatment of psoriasis with mAb (previously treated/naive), PASI score (< 20/≥ 20) and presence of psoriatic arthritis (yes/no) and randomly assigned in a 2:2:1 ratio to receive NTK 120 mg Q2W (NTK Q2W group), NTK 120 mg Q4W (NTK Q4W group) or placebo. The randomization was performed with random sequence using an electronic centralized randomization system. The study investigators, trial team and patients were blinded to the treatment allocation during the first 12 weeks of the study.
The main treatment period included the blinded phase (weeks 0–12) followed by an open-label phase up to 54 weeks. The study was extended up to 3 years. During the first 3 weeks, all patients received subcutaneous injections of NTK or placebo (according to the allocation) once a week (induction phase). Patients in the NTK Q2W group then received the study drug at weeks 4, 6, 8 and 10. Subjects in the NTK Q4W group received NTK at weeks 6 and 10 and placebo at weeks 4 and 8 to preserve blinding. Patients in the placebo group received injections containing placebo at weeks 4, 6, 8 and 10.
The treatment was unblinded at week 12. During the open-label phase, patients in both NTK groups continued to receive the study drug Q4W (NTK Q2W/Q4W and NTK Q4W groups). Patients who received placebo were switched to the active treatment and were administered NTK at weeks 12, 13 and 14 (induction) and then Q4W through weeks 18–50 (placebo/NTK Q4W group). Patients were followed up for 4 weeks after the last injection of the study drug.
NTK was administered as two subcutaneous injections (1.0 ml containing NTK 60 mg); placebo was administered as two indistinguishable subcutaneous injections (1.0 ml each).
Study Outcomes
The primary efficacy endpoint was the proportion of patients who achieved a ≥ 75% reduction from baseline in PASI (PASI 75) at week 12. The secondary endpoints included the proportion of patients attaining PASI 75 at weeks 8, 16, 24, 42 and 52, PASI 90 and PASI 100 at weeks 8, 12, 16, 24, 42 and 52; the change from baseline in PASI, the proportion of patients with sPGA 0 or 1 and sPGA 0 at weeks 8, 12, 16, 24, 42 and 52; the proportion of patients with Dermatology Life Quality Index (DLQI) 0 or 1 at weeks 24, 42 and 52; the change from baseline in itch severity (measured by visual analogue scale [VAS], 0–100 mm) at weeks 1, 12, 24 and 52; Nail Psoriasis Severity Index (NAPSI) at weeks 12, 24 and 52; and DLQI at weeks 8, 12, 24, 42 and 52.
The safety analysis included all data obtained after the first injection of NTK/placebo. Adverse events (AEs) were reported according to Common Terminology Criteria for Adverse Events (CTCAE) v. 4.03 [4].
Blood samples for the immunogenicity analysis were obtained before the first dose of NTK/placebo, at weeks 12, 24, and 54. The samples were analyzed using a validated enzyme-linked immunosorbent assay (ELISA; drug-tolerant method). In case of detection of NTK-binding antibodies, we performed the analysis for the antibodies with neutralizing activity. The immunogenicity endpoint was the proportion of patients with binding/neutralizing anti-drug antibodies at weeks 12, 24 and 54.
Sample Size
This trial tested three hypotheses. The main study hypothesis stated the non-inferiority of NTK Q4W vs. Q2W at a significance level of 0.05. The second and the third hypotheses stated the superiority of NTK over placebo for each of the dosing regimens, Q4W and Q2W, respectively, each at a significance level of 0.05.
To calculate the sample size, we used literature data about the efficacy of ixekizumab in patients with moderate-to-severe plaque psoriasis [5]. Assuming 89.1% and 82.6% PASI 75 response rate at week 12 in the Q2W and Q4W groups, respectively, a sample size of 154 patients (77 in each NTK group) would provide approximately 80% power to demonstrate non-inferiority with a 20.4% margin and 5% type I error rate. Non-inferiority margin was calculated as 0.25 of the lower boundary 95% confidence interval (CI) (81.5%; 88.9%) for the difference between assumed PASI 75 response rate at week 12 in the Q2W and placebo groups, assuming the response rate of 3.9% in the placebo group. Also, a sample size of 193 patients (77 in each NTK group and 39 in the placebo group) would provide > 80% power to demonstrate the superiority of NTK over placebo for each group with a 0% margin and 5% type I error rate. With a potential withdrawal rate of 10%, the study should involve 213 patients.
Statistical Analysis
The statistical analysis was performed using two-sided hypothesis tests. The statistical significance level was set as 0.05.
Quantitative data were tested for normality using the Shapiro-Wilk test. Normally distributed quantitative data were analysed using the two-sample Student’s t-test and analysis of variance. Non-normally distributed data were analysed using the Mann-Whitney, Wilcoxon, Kruskal-Wallis and Friedman tests. Benjamini-Yekutieli adjustment was used for multiple comparisons. The categorical data were processed using frequency tables, Fisher’s exact test and Pearson’s chi-squared test.
Statistical hypotheses were tested by two steps:
-
1.
Superiority hypotheses of NTK Q2W and NTK Q4W groups over placebo group;
-
2.
Non-inferiority hypothesis of NTK Q4W and NTK Q2W.
A statistical comparison of all secondary efficacy endpoints was performed between the NTK Q2W/Q4W and NTK Q4W groups. Secondary efficacy endpoints in the placebo/NTK Q4W group were assessed descriptively because comparison with the NTK Q2W/Q4W and NTK Q4W groups was not feasible because of the difference in NTK treatment durations.
The efficacy and safety analyses were performed according to the intention-to-treat (ITT) principle and included all patients randomized in the study (n = 213). For dichotomous responder-type endpoints, missing responses at a post-baseline visit were imputed as a non-responder. For continuous endpoints, no missing data imputation rules were applied.
Results
From December 2017 (date of the first visit) to June 2019 (date of completion of the analysed period by the last participant), a total of 213 patients were randomized: NTK Q2W group (n = 85), NTK Q4W group (n = 84) and placebo group (n = 44); 201 patients completed the 54 weeks of the trial, and 12 subjects discontinued treatment. The reasons for early withdrawal were AEs, pregnancy, IC withdrawal and loss to follow-up (Fig. 1).

Patient disposition flow chart. IC informed consent, NTK netakimab, Q2W every 2 weeks, Q4W every 4 weeks, AEs adverse events
The baseline demographics and clinical characteristics were consistent across the three groups. The median patient age was 42, 41.5 and 39 years in the NTK Q2W/Q4W, NTK Q4W and placebo/NTK Q4W groups, respectively. All patients were Caucasian. The median disease duration was 100–120 months. Previous therapy included phototherapy, systemic/topical corticosteroids and immunosuppressants. There were differences in the frequency of systemic therapy (more frequent use of methotrexate in the placebo/NTK Q4W group) that were considered insignificant because all eligible patients did not use systemic therapy for at least 4 weeks before signing the IC (Table 1).
Efficacy Analysis
A total of 77.7% of patients in the NTK Q2W group and 83.3% of patients in the NTK Q4W group achieved PASI 75 at week 12 compared to 0% of patients in the placebo group; a 95% CI of 67.1–88.2% (P < 0.0001) for the NTK Q2W vs. placebo and 95% CI of 73.6–93% (P < 0.0001) for the NTK Q4W vs. placebo. The lower bound of the 95% CI for the NTK Q4W and NTK Q2W groups (73.6% and 67.1%, respectively) exceeds the superiority margin (0%). Thus, the efficacies of NTK in both dosing regimens were superior to that of placebo.
The difference in PASI 75 rate at week 12 between the NTK Q4W and NTK Q2W groups was 5.6% with a 95% CI of − 7.4 to 18.8% (P = 0.4603). The lower bound of the 95% CI (− 7.4%) did not exceed the pre-specified margin of − 20.4%. Thus, the efficacy of NTK Q4W was not inferior to that of NTK Q2W.
During the first 12 weeks of treatment and afterward (when NTK was administered Q4W in both groups), the proportion of patients attaining PASI 75/90/100 and relative PASI change vs. baseline did not differ between the NTK Q2W and NTK Q4W groups. Most patients achieved PASI 75 after as early as 8 weeks of therapy (58.8% and 59.5% in the NTK Q2W and NTK Q4W groups, respectively). By week 52, 94.1% and 92.9% of patients achieved PASI 75, 78.8% and 77.4% of patients achieved PASI 90, and 61.2% and 56% of patients achieved PASI 100 in the NTK Q2W/Q4W and NTK Q4W groups, respectively. There were no statistically significant differences between the two groups throughout the analysis period (P > 0.05) (Fig. 2).

Response to netakimab treatment (ITT analysis). ITT intention-to-treat principle, NTK netakimab, PASI Psoriasis Area and Severity Index, Q2W every 2 weeks, Q4W every 4 weeks. a PASI 75 responders; b PASI 90 responders; c PASI 100 responders
At the first assessment time point (week 8), 56.5% and 60.7% of patients had “clean or almost clean skin” (sPGA 0 or 1) and 48.2% and 45.2% (week 16) had “clean skin” (sPGA 0) in the NTK Q2W and NTK Q4W groups, respectively (P > 0.05). At week 52, 84.7% and 85.7% of patients achieved the sPGA score of 0 or 1, and 61.2% and 58.3% of patients had the sPGA score of 0 in the NTK Q2W/Q4W and NTK Q4W groups, respectively (P > 0.05).
As early as the first week of treatment, patients in both NTK groups reported a reduction in itch severity; the effect was maintained throughout the 1-year treatment period. The use of NTK resulted in a significant improvement in psoriatic nail damage assessed by NAPSI regardless of the dosing regimen. Patient-reported outcomes as measured by the DLQI showed that the impact of psoriasis on quality of life significantly decreased during therapy. By week 52, 69.4% of patients in the NTK Q2W/Q4W group and 69.1% of patients in the NTK Q4W group had a DLQI score of 0 or 1 (no impact on quality of life) (Table 2).
The patients in the placebo/NTK Q4W group had a reduction of clinical signs of psoriasis after as early as 4 weeks of therapy with NTK with the effect increasing through week 52: 38.6%, 77.3%, 86.4% and 95.5% of patients achieved PASI 75 at weeks 16, 24, 42 and 52, respectively. By week 52, 81.8% and 65.9% of patients achieved PASI 90/100, respectively, and 70.5% of patients achieved a sPGA score of 0 (Fig. 2). The same significant improvement was observed in itch severity (median change from baseline was − 10, − 37.5 and − 37 at weeks 12, 24 and 52, respectively, P < 0.0001), nail damage (median change from baseline in NAPSI was 0, − 11 and − 19 at weeks 12, 24 and 52, respectively, P < 0.0001) and quality of life (50%, 50% and 68.2% of patients had DLQI 0 or 1 at weeks 24, 42 and 52, respectively).
Safety
Throughout the placebo-controlled period, treatment-emergent AEs (TEAEs) were reported in 20%, 16.7% and 20.5% of patients receiving NTK Q2W, NTK Q4W and placebo, respectively (P > 0.05). Treatment-related AEs were registered in 9.4%, 10.7% and 6.8% of patients receiving NTK Q2W, NTK Q4W and placebo, respectively (P > 0.05).
Safety Results of NTK Q2W/Q4W and NTK Q4W Groups
During the 54 weeks, 40% and 40.5% of patients had TEAEs and 4.7% and 2.4% of patients had grade 3/4 TEAEs in the NTK Q2W/Q4W and NTK Q4W groups, respectively (P > 0.05). Two patients (2.4%) in the NTK Q2W/Q4W group and one patient (1.2%) in the NTK Q4W group were withdrawn from the trial because of the AE “positive test for tuberculosis”, which was considered not related to the study treatment; subsequently, the diagnosis of active tuberculosis was not confirmed.
A total of 5% or more of patients had hypercholesterolaemia, neutropaenia, hyperbilirubinaemia and upper respiratory tract infections. The most common treatment-related AEs were represented by neutropaenia. The AEs were mostly mild to moderate (grade 1/2). Severe treatment-related AEs were (one case each): grade 3 neutropaenia, grade 4 neutropaenia and grade 3 lymphopaenia. The only treatment-related serious AE (SAE) was one case of grade 3 pneumonia in the NTK Q4W group, which led to hospitalization. The patient presented with low-grade fever and mild cough 2 days after the third NTK injection and was admitted to the hospital. Based on the results of physical examination and chest x-ray, lower lobe pneumonia was diagnosed. After 1 week of treatment with ceftriaxone, the patient completely recovered and continued the participation in the trial.
Safety Results of the Placebo/NTK Q4W Group
From week 13 to week 54, TEAEs were recorded in 50% of patients; 15.9% of patients had grade 3/4 TEAEs. The most frequent TEAEs were hypercholesterolaemia (4.6%), neutropaenia (6.8%) and upper respiratory tract infections (4.6%). Two patients had serious TEAEs not related to NTK. One patient with arterial hypertension, type II diabetes and obesity had worsening of the arterial hypertension, which led to hospitalization. The second patient discontinued treatment because of the development of type II diabetes and collapse.
Treatment-related AEs were represented by neutropaenia, infections, hyperbilirubinaemia, elevated aspartate aminotransferase (AST), elevated alanine aminotransferase (ALT), hypercholesterolaemia and dermatitis. Severe therapy related AEs were represented by single cases of grade 3 neutropaenia and elevated ALT (Table 3).
Immunogenicity
The immunogenicity analysis included 210 patients. One patient (NTK Q2W/Q4W group) had binding anti-drug antibodies at week 12; at this time, he had 52.9% PASI improvement. He achieved PASI 100 at week 24 and maintained 94.1% PASI improvement at week 52. The second patient (NTK Q4W group) had binding anti-drug antibodies at week 24; at this time, he achieved PASI 100 and maintained the response throughout the analysis period. Neutralizing antibodies were not detected.
Discussion
The aim of this phase 3 clinical trial was to evaluate and compare the efficacy and safety of NTK Q2W and Q4W dosing regimens versus placebo during the first 12 weeks of treatment with further assessment of the efficacy and safety of 1-year treatment with NTK in patients with moderate-to-severe plaque psoriasis.
PASI 75 is considered to be a reliable and objective tool to assess the efficacy of biologics and is commonly used in the clinical trials of IL-17 inhibitors in patients with psoriasis [6, 7]. In the PLANETA study, the blinding was maintained throughout the double-blind period; well trained and experienced dermatologists assessed the treatment efficacy. Thus, the risk of bias in the measurement of the outcomes is low.
The assessment of the primary endpoint demonstrated that both NTK dosing regimens were significantly more effective than placebo and that the Q4W dosing regimen was as effective as Q2W during the 12 weeks of treatment. The analysis of all secondary efficacy endpoints showed no significant differences between the NTK Q2W/Q4W and NTK Q4W treatment groups throughout the 1-year treatment period.
We observed very low efficacy in the placebo group—the PASI 75 response rate was 0% at week 12. In phase 3 clinical trials of ixekizumab the PASI 75 response rate in the placebo group varied from 2.4 to 7.3% [6]. In core clinical trials of secukinumab the placebo effect according to PASI 75 at week 12 varied from 0 to 4.5% [7]. We found the systematic review of 27 studies aimed to determine the outcome in the placebo groups in randomized controlled trials of systemic drugs for chronic plaque-type psoriasis. Included trials used different outcome measures (BSA, mean percentage of PASI score reduction, decrease in the average global score, investigator’s overall assessment and investigator’s final judgment). The efficacy outcomes in placebo groups were low in most studies. The authors conclude that the effect of treatment in placebo groups varied widely across studies in an unpredictable way [8]. Thus, the efficacy results obtained in the PLANETA study are consistent with the literature data.
The patients who received placebo during the first 12 weeks of the study and then were switched to NTK showed the improvement in clinical signs of psoriasis after as early as 4 weeks of therapy with the active drug, with the effect increasing through week 52; 95.5%, 81.8% and 65.9% of patients achieved PASI 75, 90 and 100, respectively; 70.5% of patients achieved sPGA score 0 after 40 weeks of treatment with NTK. Significant improvement was observed in itch severity, nail damage and quality of life. Thus, the observed dynamics confirm the validity of the results obtained in the NTK Q2W/Q4W and NTK Q4W treatment groups.
NTK was well tolerated. The safety profile was comparable in both NTK dosing regimens and placebo during the placebo-controlled period and throughout the open-label NTK treatment period. All treatment-related AEs were expected and common for all IL-17 inhibitors: neutropaenia, lymphopaenia and upper respiratory tract infections. None of them led to treatment discontinuation.
The following limitations of the trial could be considered: patients included in this trial had rather short duration of the disease (approximately 10 years) and < 10% of patients in each group were exposed to biologics prior to the inclusion in the study.
Thus, in patients with moderate-to-severe plaque psoriasis NTK 120 mg should be administered once a week for the first 3 weeks followed by NTK 120 mg Q4W injections.
Conclusion
NTK demonstrated high rates of sustained clinical response as early as the first few weeks of treatment, a favourable safety profile and good tolerability in patients with moderate-to-severe plaque psoriasis. However, as the study is ongoing, more efficacy and safety data of long-term NTK use will be obtained over the coming years.
References
Zhu S, Qian Y. IL-17/IL-17 receptor system in autoimmune disease: mechanisms and therapeutic potential. Clin Sci (Lond). 2012;122(11):487–511.
Erdes S, Nasonov E, Kunder E, et al. Primary efficacy of netakimab, a novel interleukin-17 inhibitor, in the treatment of active ankylosing spondylitis in adults. Clin Exp Rheumatol. 2020;38(1):27–34.
Bakulev AL, Samtsov AV, Kubanov AA, Khairutdinov VR, Kokhan MM, Artemyeva AV, Derbin SI, Chernyaeva EV, Ivanov RA. Long-term efficacy and safety of netakimab in patients with moderate-to-severe psoriasis. Results of phase II open-label extension clinical study BCD-085–2-ext. Vestnik dermatologii i venerologii. 2019;95(3):54–64 (In Russ.).
Common Terminology Criteria for Adverse Events (CTCAE). https://www.eortc.be/services/doc/ctc/ctcae_4.03_2010-06-14_quickreference_5x7.pdf. Accessed 13 Jan 2021.
Farahnik B, Beroukhim K, Zhu TH, et al. Ixekizumab for the treatment of psoriasis: a review of phase III trials. Dermatol Ther (Heidelb). 2016;6(1):25–37.
European medical Agency. Assessment Report EMA/CHMP/389874/2014. https://www.ema.europa.eu/en/documents/assessment-report/cosentyx-epar-public-assessment-report_en.pdf. Accessed 25 Jan 2021.
Gordon K, Blauvelt A, Papp K, et al. Phase 3 trials of Ixekizumab in moderate-to-severe plaque psoriasis. N Engl J Med. 2016;375:345–56.
Spuls P, Witkamp L, Bossuyt PM, Bos JD. The course of chronic plaque-type psoriasis in placebo groups of randomized controlled studies. Arch Dermatol. 2004;140(3):338–44.
Acknowledgements
We acknowledge the collaboration and commitment of all investigators and staff.
Funding
Sponsorship for this study and the Rapid Service Fee were funded by JSC BIOCAD, Ul. Italianskaya 17, St Petersburg, Russia, 191186.
Authorship
All named authors meet the International Committee of Medical Journal Editors (ICMJE) criteria for authorship for this article, take responsibility for the integrity of the work as a whole, and have given their approval for this version to be published.
Authors’ Contributions
Conceptualization, methology and design: Antonina V. Artemeva, Arina V. Zinkina-Orikhan, Nikita A. Zolkin, Ivan V. Kuryshev; Data acquisition, data analysis and interpretation: Andrey L. Bakulev, Muza M. Kokhan, Alexey V. Samtsov, Vladislav R. Khairutdinov; Nikita A. Zolkin, Ivan V. Kuryshev, Alexey N. Petrov, Antonina V. Artemeva, Arina V. Zinkina-Orikhan, Maria A. Morozova; Writing—original draft preparation: Maria A. Morozova, Antonina V. Artemeva, Arina V. Zinkina-Orikhan; Writing—review and editing: Luís Puig, Andrey L. Bakulev, Muza M. Kokhan, Alexey V. Samtsov, Vladislav R. Khairutdinov, Nikita A. Zolkin, Ivan V. Kuryshev.
Disclosures
Luís. Puig has received consultancy/speaker’s honoraria from and/or participated in clinical trials sponsored by Abbvie, Almirall, Amgen, Baxalta, Biogen, Boehringer Ingelheim, Celgene, Gebro, Janssen, JSC BIOCAD, Leo-Pharma, Lilly, Merck-Serono, MSD, Mylan, Novartis, Pfizer, Regeneron, Roche, Sandoz, Samsung-Bioepis, Sanofi and UCB. Andrey L. Bakulev has received consultancy/speaker’s honoraria from and/or participated in clinical trials sponsored by Abbvie, Amgen, Boehringer Ingelheim, Celgene, Janssen, JSC BIOCAD, Leo-Pharma, Lilly, MSD, Novartis, Pfizer, Sanofi, UCB and Zeldis Pfarma. Muza M. Kokhan has received consultancy/speaker’s honoraria from and/or participated in clinical trials sponsored by Celgene, Janssen, JSC BIOCAD, Leo-Pharma, Lilly, Novartis, Pfizer and Sanofi. Alexey V. Samtsov has received consultancy/speaker’s honoraria from and/or participated in clinical trials sponsored by Celgene, Glenmark, Jadran and JSC BIOCAD. Vladislav R. Khairutdinov has received consultancy/speaker’s honoraria from and/or participated in clinical trials sponsored by Abbvie, Belupo, Bosnalec, Celgene, Glenmark, Jadran, Janssen, JSC BIOCAD, Leo-Pharma, Lilly, MSD, Novartis, Pfizer, Sanofi and Sun Pharma. Maria A. Morozova, Antonina V. Artemeva, Arina V. Zinkina-Orikhan, Nikita A. Zolkin, Ivan V. Kuryshev and Alexey N. Petrov are JSC BIOCAD employees.
Compliance with Ethics Guidelines
The study was approved by the Central Regulatory Authorities of the Russian Federation (Authorization by the Ministry of Healthcare of the Russian Federation No. 616 of November 28, 2017) and the Republic of Belarus (Authorization by the Ministry of Healthcare of the Republic of Belarus No. 01-11-14/1368 of February 05, 2018) and Ethical Review Boards of each of the participating sites. A list of the Ethics Committee is provided as a Supplement. The study was performed in accordance with the Helsinki Declaration of 1964 and its later amendments. All subjects provided informed consent to participate in the study. There was no identifiable information in our paper.
Data availability
The datasets generated during and/or analysed during the current study are available from the corresponding author on reasonable request.
Author information
Authors and Affiliations
Corresponding author
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Puig, L., Bakulev, A.L., Kokhan, M.M. et al. Efficacy and Safety of Netakimab, A Novel Anti-IL-17 Monoclonal Antibody, in Patients with Moderate to Severe Plaque Psoriasis. Results of A 54-Week Randomized Double-Blind Placebo-Controlled PLANETA Clinical Trial. Dermatol Ther (Heidelb) 11, 1319–1332 (2021). https://doi.org/10.1007/s13555-021-00554-4
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13555-021-00554-4