
Abstract
Importance
Type I interferon (IFN)-mediated monogenic autoinflammatory disorders (interferonopathies) are childhood-onset rare multisystemic diseases with limited treatment options. The Janus kinase (JAK) inhibitors are promising potential therapeutic candidates for immune-mediated chronic inflammatory skin diseases.
Objective
To review the use of JAK inhibitors to improve decision-making when treating interferonopathies with cutaneous manifestations.
Evidence Review
The MEDLINE, EMBASE, CINAHL, Scopus, and Web of Science databases were searched for studies that used JAK protein inhibitors to treat IFN-related monogenic diseases with cutaneous manifestations in humans. The search results are reported using the scoping review approach.
Findings
Seventeen open-label studies assessing the efficacy of ruxolitinib, baricitinib, or tofacitinib reported variable responses in patients with chronic atypical neutrophilic dermatosis with lipodystrophy and elevated temperature (CANDLE) and related syndromes, stimulator of IFN genes [STING]-associated vasculopathy with onset in infancy (SAVI), familial chilblain lupus (FCh-L), gain-of-function mutations of STAT1 (GOF-STAT1), or Aicardi-Goutiéres syndrome. JAK inhibitors improved clinical and analytical parameters and decreased flare numbers, plasma inflammatory markers, and expression of IFN-stimulated genes. BK viremia and upper respiratory infections were the most frequent and severe adverse events. Significant heterogeneity in efficacy assessment methods and poor reporting of safety events were detected.
Conclusions and Relevance
Evidence of the use of JAK inhibitors in patients with interpheronopathies is scarce and of low methodological quality. Future clinical trials should use validated scales and report drug safety in a more accurate way.
Similar content being viewed by others
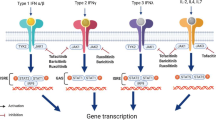

Avoid common mistakes on your manuscript.
Evidence on efficacy of Janus kinase inhibitors in patients with interpheronopathies is scarce and of low methodological quality. |
Future clinical trials should use validated scales and report drug safety in a more accurate way. |
Digital Features
This article is published with digital features, including a summary slide, to facilitate understanding of the article. To view digital features for this article go to: https://doi.org/10.6084/m9.figshare.14237444.
Introduction
Type I interferon (IFN)-mediated monogenic autoinflammatory disorders (type I interferonopathies) comprise a genetically and phenotypically heterogeneous group of autoinflammatory and autoimmune disorders characterized by constitutive activation of the antiviral type I IFN axis [1]. This activation is multifactorial, as in the case of lupus erythematosus, or is associated with monogenic, autosomal dominant, or recessive mutations [2].
There are several monogenic interferonopathies that cause significant morbidity and mortality from early stages of life, as well as skin involvement. Stimulator of interferon genes [STING]-associated vasculopathy with onset in infancy (SAVI, ORPHA:425120) is a monogenic interferonpathy due to a mutation in TMEM173. Patients present feet and hand involvement showing rash, blisters, pustules, or telangiectasia, in addition to fever flares, vasculitis, stunted growth, interstitial lung disease, or lymphadenopathy [2,3,4,5]. Chronic atypical neutrophilic dermatosis with lipodystrophy and elevated temperature (CANDLE, ORPHA:325004) is an interferonpathy related with proteasome dysfunction. Clinical features include cutaneous rash, acral perniosis, and annular and disseminated plaques or violaceus edema around the mouth and eyelids associated with fever flares. Joint swelling and arthralgia are also observed. A characteristic phenotype includes facial lipodystrophy, a protruding abdomen, and stunted growth [2, 6,7,8]. Familial chilblain lupus (FChL, ORPHA:481662) is an autosomal dominant condition due to a mutation in the three prime repair exonuclease 1 gene (TREX1) or in the SAM and HD domains containing deoxynucleoside triphosphate triphosphohydrolase 1 (SAMHD1) or STING1, respectively. Clinical features include chilblain acral lesions, fever, and interstitial lung disease [2, 9, 10]. Other important interferonopathies include Aicardi-Goutières syndrome (ORPHA:51), Singleton-Merten syndrome (ORPHA:85191), and gain-of-function mutations of STAT1 (GOF-STAT1) and SKIVL2, an unfolded protein response or spondyloenchondrodysplasia, where neurologic, immunologic, hepatologic, or rheumatologic conditions are associated with chilblain, panniculitis, psoriasis, oral and genital ulcers, hair disorders, and acrocyanosis, respectively [11, 12].
Different treatments, most of which involve immunosuppressants, have been employed with an irregular response. Corticosteroids have shown moderate or partial success in CANDLE [6] and SAVI [13], but have not been used in FCh-L [10]. Methotrexate has been reported to produce partial improvement in CANDLE cases [14] without affecting the evolution of FCh-L [10]. The use of cyclosporine and azathioprine is anecdotal, while isolated cases of CANDLE and SAVI treated with tumor necrosis factor alpha (TNFα) inhibitors have shown poor responses [13]. Immunosuppressants (biologic disease-modifying antirheumatic drugs) used to treat other interferonopathies have been shown to lack effectiveness or cause toxicity [11, 12].
Because the major signaling pathway activated by IFNs is the Janus kinase/signal transducer and activator of transcription (JAK/STAT) pathway [3], drugs inhibiting this pathway represent a therapeutic opportunity to these patients. In recent years, drugs inhibiting different proteins of the JAK/STAT pathway have been developed; some act specifically on a single component (e.g., filgotinib, JAK1; pacritinib, JAK2; decernotinib, JAK3) and others on several components (e.g., tofacitinib, JAK1 and JAK3; ruxolitinib and baricitinib, JAK1 and JAK2) [15].
The aim of this study was to report the current evidence of JAK inhibitor treatment in autoinflammatory IFN-related monogenic diseases with cutaneous manifestations using scoping review methodology [16].
Methods
Protocol Design
The protocol for the scoping review protocol used in this study has been previously published [17]. The study was conducted and reported using the methodology described in the Joanna Briggs Institute Reviewer’s Manual [18] and the PRISMA Extension for Scoping Reviews [19], respectively. When necessary, the authors of the included articles were contacted to obtain more data or clarify issues.
Eligibility Criteria
Studies that have used JAK protein inhibitors to treat IFN-related monogenic diseases with cutaneous manifestations in humans were included in the review. There were no restrictions regarding age, ethnicity, study design, or any other characteristics. IFN-related monogenic disease studies were included if written in English, involved human participants, and described the conditions formulated in the research question, regardless of publication date or format. Articles were excluded if they did not fit into the conceptual framework of the study. Non-scientific reviews and opinion articles were excluded.
Literature Search
Strategies for the literature search and eligibility criteria are described in detail in the Supplementary Methods of the Electronic Supplementary Material (ESM).
Data Charting
A data charting form was jointly developed by two authors to determine the variables to extract. These reviewers independently charted the data, discussed the results, and continuously updated the data charting form in an iterative process. Finally, variables related to patients (disease, drug, treatment response, and safety profile) and to the study design and metadata from primary sources were reported.
Collating, Summarizing and Reporting Results
The comprehensive search results are presented using a PRISMA flow diagram and references and primary studies are displayed by drug. After data extraction, a narrative and qualitative synthesis of references, studies, efficacy, and safety data was elaborated.
Compliance with Ethics Guidelines
This scoping review relates to previously conducted studies, and does not involve any new human or animal studies performed by the authors.
Protocol versus Overview
Our planned search strategy in the published protocol was compared to the final reported review methods.
Results
Search Results
We found 4897 articles on the use of drugs targeting the JAK/STAT pathway in the treatment of dermatological diseases. After filtering duplicates and selecting studies according to title, abstract, and keywords, 22 studies met the criteria for full-text review. Three more articles were included after reviewing the references of other studies and conducting manual searches (Fig. 1). After applying the inclusion criteria, only 17 studies were included in the scoping review (Fig. 1). Data related to the country, study and publication type, number of authors, and funding across all studies are presented in ESM Table S1. Lists of included and excluded articles and reasons for exclusion are given in ESM Tables S2 and S3. The JAK inhibitor drugs included in this review were baricitinib, ruxolitinib, and tofacitinib.

PRISMA flow diagram.JAK Janis kinase
Baricitinib
Six articles mentioning baricitinib were identified [11, 12, 20,21,22,23], and all were observational studies, including three case series and three case reports published in journals related to rheumatology, general medicine, immunology, and dermatology. The articles were written by an average of 26.56 (range 6–55) authors from the USA (n = 2), the Netherlands (n = 2), Bulgaria (n = 1), and Germany (n = 1). In one of six articles, the authors declared a conflict of interest. One article had private funding (Elli Lilly, Indianapolis, IN, USA) and one had public (US National Institutes of Health [NIH], Bethesda, MD, USA) funding (ESM Table S1).
Mapping Studies
Two clinical trials on the use of baricitinib to treat patients with interferonopathies were identified that were registered in November 2012 and November 2016 (NCT01724580 and NCT02974595, respectively).
Efficacy and Safety
Twenty-four patients with diagnoses of CANDLE (n = 11), SAVI (n = 4), CANDLE-like syndrome (n = 3), FCh-L due to TREX1 deficiency (n = 3), GOF mutations of STAT1' (n = 1), and Aicardi-Goutiéres syndrome (n = 2) were included. Of 11 patients with CANDLE syndrome, nine showed improvement in the daily diary score (DDS) and/or reduction in the use of prednisone. Four of those 11 patients with CANDLE syndrome had increased remission time. Baricitinib was used for an average of 39.5 (18–58.8) months at doses of 0.10–10 mg/24 h. In terms of safety, infections (BK viremia/respiratory tract infections) were frequently found.
On average, patients with SAVI received treatment with baricitinib for 29.6 (range 18.2–31.9) months at doses of 2–10 mg/24 h. One patient experienced a decrease in the DDS. Decreased prednisone use and remission were not reported in this case. Respiratory tract infections were the most frequent adverse events. The treatment was administered to four patients presenting CANDLE-related conditions for 17.5 (range 2–37) months at doses of 0.5–9 mg/24 h; one, two, and zero patients presented improved DDS, decreased use of corticosteroids, and remission, respectively. These patients experienced more adverse effects, including infectious and cerebrovascular or coagulation issues. Patients with FCh-L were treated on average for 3 months at doses of 4 mg/24 h. All patients achieved marked improvement in cutaneous involvement and relief of joint pain. The most common adverse effect was repeated mild respiratory infections.
Patients suffering from Aicardi-Goutières syndrome and GOF mutations of STAT1′ syndrome were treated at doses of 2 mg for 8 and 18 months, respectively. In both cases, skin improvement was observed. No adverse events were described in these patients (Table 1).
Ruxolitinib
Mapping Studies
Two full texts, three letters to the editor, and one congress abstract of a case series or case reports using ruxolitinib were published in immunology (n = 3) and two in rheumatology journals (n = 2) [9, 10, 13, 24,25,26]. On average, 14 (range 1–27) authors located in Australia, France, Italy, and Greece contributed to each of these studies. None of the studies were registered. In one of the studies, one of the authors declared a conflict of interest. Four studies had a public funding source and one study included both public and private funding. One study did not provide funding information (ESM Table S1).
Efficacy and Safety
Nine patients were included in these studies (eight with SAVI and one with FCh-L due to TREX1 deficiency). The mean time of treatment was 13 (6–32) months. The ruxolitinib dosage range was 0.25–1.3 (range 2.5–10 mg/12 h) mg/kg/day. All patients showed improvement in clinical and respiratory symptoms. However, respiratory symptoms worsened after treatment in three cases and one patient died. Six adverse effects were reported with ruxolitinib, including respiratory infections, BK viremia, and hypercapnia-induced cerebral edema in three patients, respectively (Table 2).
Tofacitinib
Mapping Studies
Five case series or case report articles (2 full text articles, 2 congress abstracts, and one 1 letter to the editor) were published in rheumatology (n = 3) and allergy–immunology (n = 2) journals [27,28,29,30,31]. On average, 10.4 (range 5–18) authors located in Germany, Russia, Italy, China, and South Korea contributed to each study. None of the studies were registered. Two authors declared a conflict of interest for receiving public funding. Two of the studies received public funding, while a third received private and public funding (ESM Table S1).
Efficacy and Safety
Seven patients were included in these studies (4 with SAVI, 2 with FCh-L with gain-of-function in STING, and 1 with CANDLE). Tofacitinib dosage range was 5–10 mg/day. All patients showed cutaneous improvement, reducing pain and discomfort. Lipodystrophy reduction and hair regrowth were reported in the patient with CANDLE. One of four patients with SAVI showed pulmonary symptom relief and two showed rash improvement with tofacitinib treatment. Adverse effects were reported in only two patients. An increase in creatine kinase and low-density lipoprotein cholesterol plasma levels was found in one case (Table 3).
Standardized Type-I INF-Response Gene Score
An INF signature (the INF-Response Gene Score [IRG-S]), using nanostring technology based on the expression of 28 target genes, was developed and validated by Kim et al. [32] to have a reliable and standardized readout to evaluate the severity and treatment response for type I interferonopathies. Nine of the 17 studies assessed the expression of INF target genes as a tool to evaluate treatment response. Overall, a maximum of seven genes were tested in these studies, with different genes tested in each study, with the exception of one study in which 25 of the 28 genes included in the IRG-S were tested (Fig. 2) [22].

Studies that have assessed the expression of interferon target genes as a tool to evaluate treatment response. See footnote to Table 1 for definitions of SAVI, CANDLE, FCh-L and GOF-STAT1
Discussion
This is the first scoping review on the use of JAK inhibitors in monogenic type I interferonopathies, and it provides an overview of currently available research evidence based on observational studies, most of which are cases or case series. The majority of evidence is derived from non-registered studies conducted in the USA and European countries and supported by private–public collaborative funding. In terms of treatment efficacy, baricitinib, ruxolitinib, and tofacitinib showed variable clinical and analytical improvement in most patients with these monogenic diseases, with cases of patients with CANDLE or SAVI being the most commonly reported. With the exception of studies that were registered, a low systematization was identified in both the conduct and reporting of these studies, making it difficult to synthesize their results. Specifically, the reporting of adverse events was poor. These deficiencies were more evident when the cases were reported in abstract form.
Summary of Findings
First, related to diagnosis, Sanchez-Montalegre et al. [22] establish criteria to define a group of patients with CANDLE-related conditions that may classify patients with undetermined interpheronopaty. This may be very helpful in cases where patients present incomplete forms of the disease or to guide diagnoses in this type of rare disease. Second, related to severity measurement for these diseases, a DDS, a daily specific scale, was used to assess patients with CANDLE and SAVI in above-mentioned study. The authors of a study that explored its validity came to the conclusion that the outcome measures in SAVI should be further refined and validated across other outcome measures in unrelated pediatric inflammatory diseases [33]. Widespread use of the standardized type I IRG-S would make it possible to measure severity and treatment response for these diseases [32, 34], improving replicability and making comparison across studies feasible. Third, related to risk/benefit, important conclusions could not be drawn given the small patient numbers and follow-up times due to illness. Generally, most patients experienced improvement with these drugs, except for a group treated with baricitinib and defined as having CANDLE-like syndrome. However, as previously mentioned, an efficacy measurement was not performed in most cases with a scale that allowed a comparison of results. Thus, although a possible publication bias has not been studied, there is evidence of the preferential publication of favorable treatment results, and this must be taken into account when evaluating that the efficacy of the treatment [35]. Fourth, regarding safety, mild-moderate respiratory infections and BK viremia were the most frequent adverse events, without any apparent differences across drugs and diseases. Finally, related to the study methodology, to our knowledge baricitinib was the only drug tested in registered clinical trials.
Strengths and Limitations
This scope review was conducted and reported based on an a priori protocol, using the latest methodology for conducting and reporting this type of study. Two authors selected the articles for inclusion and extracted the data. The number of databases consulted was wide and authors were contacted when necessary, thus ensuring more reliable research results and increasing the internal validity. However, due to time and funding, the study was limited to articles published in English. Since this study was a substudy, the three-step search strategy was not research-specific, so some studies might have been missed. Furthermore, despite best efforts, information on all consulted cases was not obtained. Finally, the risk of bias of the included studies was not evaluated, nor was a quantitative synthesis performed on the evidence found.
Research Gaps
Evidence on the use of JAK-STAT pathway inhibitors in interferonopathies was scarce, but is increasing. To date, the methodology used in these studies was of a low quality; most studies were observational. Although specific efficacy scales have been developed and validated, they were not applied in most studies. Collection of adverse events should be systematized. Overall, clinical improvement was observed in patients, regardless of disease type and treatment used, and with a safety profile in which mild-moderate respiratory infections and BK viremia were notable. However, these findings should be interpreted with caution.
Conclusions
Studies with a more robust methodology, such as clinical trials, are necessary to evaluate efficacy and safety of JAK inhibitors in patients with interpheronopathies. It is advisable to standardize outcomes evaluated and time-points for evaluating efficacy and to report drug safety in a more systematic and accurate way. Finally, efforts should be made to make these treatments accessible to developing countries.
References
Lee-Kirsch MA. The type i interferonopathies. Annu Rev Med. 2017;14(68):297–315.
Frémond M-L, Crow YJ. Mendelian disorders of immunity related to an upregulation of type I interferon. In: Sullivan K, Stiehm ER, editors. Stiehm’s immune deficiencies. Amsterdam: Elsevier; 2020. p. 751–72.
Nan Y, Wu C, Zhang YJ. Interplay between Janus kinase/signal transducer and activator of transcription signaling activated by type I interferons and viral antagonism. Front Immunol. 2017;11(8):1758.
Rodero MP, Crow YJ. Type I interferon-mediated monogenic autoinflammation: the type I interferonopathies, a conceptual overview. J Exp Med. 2016;213(12):2527–38.
Liu Y, Jesus AA, Marrero B Y, et al. Activated STING in a vascular and pulmonary syndrome. N Engl J Med. 2014;371(6):507–18.
Torrelo A. CANDLE syndrome as a paradigm of proteasome-related autoinflammation. Front Immunol. 2017;8:927.
Liu Y, Ramot Y, Torrelo A, et al. Mutations in proteasome subunit β type 8 cause chronic atypical neutrophilic dermatosis with lipodystrophy and elevated temperature with evidence of genetic and phenotypic heterogeneity. Arthritis Rheum. 2012;64(3):895–907.
Torrelo A, Colmenero I, Requena L, et al. Histologic and immunohistochemical features of the skin lesions in CANDLE syndrome. Am J Dermatopathol. 2015;37(7):517–22.
Saldanha RG, Balka KR, Davidson S, et al. A mutation outside the dimerization domain causing atypical STING-associated vasculopathy with onset in infancy. Front Immunol. 2018;9:1535.
Briand C, Frémond ML, Bessis D, et al. Efficacy of JAK1/2 inhibition in the treatment of chilblain lupus due to TREX1 deficiency. Ann Rheum Dis. 2019;78(3):431–3.
Meesilpavikkai K, Dik WA, Schrijver B, et al. Baricitinib treatment in a patient with a gain-of-function mutation in signal transducer and activator of transcription 1 (STAT1). J Allergy Clin Immunol. 2018;142(1):328-330.e2.
Meesilpavikkai K, Dik WA, Schrijver B, et al. Efficacy of baricitinib in the treatment of chilblains associated with aicardi-goutières syndrome, a type I interferonopathy. Arthritis Rheumatol. 2019;71(5):829–31.
Volpi S, Insalaco A, Caorsi R, et al. Efficacy and adverse events during janus kinase inhibitor treatment of SAVI syndrome. J Clin Immunol. 2019;39(5):476–85.
Cavalcante MP, Brunelli JB, Miranda CC, et al. CANDLE syndrome: chronic atypical neutrophilic dermatosis with lipodystrophy and elevated temperature-a rare case with a novel mutation. Eur J Pediatr. 2016;175(5):735–40.
Schwartz DM, Bonelli M, Gadina M, O’Shea JJ. Type I/II cytokines, JAKs, and new strategies for treating autoimmune diseases. Nat Rev Rheumatol. 2016;12(1):25–36.
Pham MT, Rajić A, Greig JD, Sargeant JM, Papadopoulos A, Mcewen SA. A scoping review of scoping reviews: advancing the approach and enhancing the consistency. Res Synth Methods. 2014;5(4):371–85.
Gómez-García F, Gómez-Arias PJ, Hernandez J, et al. Drugs targeting the JAK/STAT pathway for the treatment of immune-mediated inflammatory skin diseases: protocol for a scoping review. BMJ Open. 2019;9(5):e028303.
Joanna Briggs Institute. The Joanna Briggs Institute reviewers’ manual 2015: Methodology for JBI scoping reviews. 2015. http://joannabriggs.org/assets/docs/sumari/ReviewersManual_Mixed-Methods-Review-Methods-2014-ch1.pdf.
Tricco AC, Lillie E, Zarin W, et al. PRISMA extension for scoping reviews (PRISMA-ScR): checklist and explanation. Ann Intern Med. 2018;169(7):467–73.
Boyadzhiev M, Marinov L, Boyadzhiev V, Iotova V, Aksentijevich I, Hambleton S. Disease course and treatment effects of a JAK inhibitor in a patient with CANDLE syndrome. Pediatr Rheumatol. 2019;17(1):1–7.
Sanchez GAM, Reinhardt A, Ramsey S, et al. Response to JAK1/2 inhibition with baricitinib in “CANDLE”, “SAVI” and “CANDLE-like” diseases. A new therapeutic approach for type I IFN-mediated autoinflammatory diseases. Arthritis Rheumatol. 2017;69(Suppl. 10):3208.
Sanchez-Montalegre GA, Reinhardt A, Ramsey S, et al. JAK1/2 inhibition with baricitinib in the treatment of autoinflammatory interferonopathies. J Clin Invest. 2018;128(7):3041–52.
Zimmermann N, Wolf C, Schwenke R, et al. Assessment of clinical response to janus kinase inhibition in patients with familial chilblain lupus and TREX1 mutation. JAMA Dermatol. 2019;155(3):342–6.
Frémond ML, Rodero MP, Jeremiah N,et al. Efficacy of the Janus kinase 1/2 inhibitor ruxolitinib in the treatment of vasculopathy associated with TMEM173-activating mutations in 3 children. J Allergy Clin Immunol. 2016;138(6):1752–5.
Manoussakis MN, Mavragani CP, Nezos A, Zampeli E, Germenis A, Moutsopoulos HM. Type I interferonopathy in a young adult. Rheumatology (Oxford). 2017;56(12):2241–3.
Volpi S. Efficacy of the JAK inhibitor ruxolitinib in two patients with SAVI syndrome [abstract]. Pediatr Rheumatol Online J. 2017;15(Suppl 2):65.
Galimberti AM, Wiel LC, Gortani G, et al. Response to tofacitinib in a case with familiar CANDLE-like disease. Pediatr Rheumatol. 2018;16(Suppl 2):52.
König N, Fiehn C, Wolf C, et al. Familial chilblain lupus due to a gain-of-function mutation in STING. Ann Rheum Dis. 2017;76(2):468–72.
Rodionovskaya S, Zaytseva S, Salugina S, Fedorov E, Tsymbal I. Successful use of tofacitinib in a 6-year-old patient with SAVI syndrome. Pediatr Rheumatol. 2017;15(Suppl 2):109–10.
Seo J, Kang JA, Suh DI, et al. Tofacitinib relieves symptoms of stimulator of interferon genes (STING)–associated vasculopathy with onset in infancy caused by 2 de novo variants in TMEM173. J Allergy Clin Immunol. 2017;139(4):1396-1399.e12.
Tang X, Xu H, Zhou C, et al. STING-Associated vasculopathy with onset in infancy in three children with new clinical aspect and unsatisfactory therapeutic responses to tofacitinib. J Clin Immunol. 2020;40(1):114–22.
Kim H, De Jesus AA, Brooks SR, et al. Development of a validated interferon score using nanostring technology. J Interf Cytokine Res. 2018;38(4):171–85.
Sanchez GA, de Jesus AA, Goldbach-Mansky R. Monogenic autoinflammatory diseases: disorders of amplified danger sensing and cytokine dysregulation. Rheum Dis Clin North Am. 2013;39(4):701–34.
de Jesus AA, Hou Y, Brooks S, et al. Distinct interferon signatures and cytokine patterns define additional systemic autoinflammatory diseases. J Clin Invest. 2020;130(4):1669–82.
Canestaro WJ, Hendrix N, Bansal A, Sullivan SD, Devine EB, Carlson JJ. Favorable and publicly funded studies are more likely to be published: a systematic review and meta-analysis. J Clin Epidemiol. 2017;92:58–68.
Acknowledgements
The authors would like to thank Stefano Volpi for his contribution to this review.
Funding
No Rapid Service Fee was received by the journal for the publication of this article. This work was supported, in part, by project PIN-0316-2017 of the Consejería de Salud, Junta de Andalucía (Spain) to JR. No funding was received from any pharmaceutical company. The funders had no role in the study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Editorial Assistance
The authors would like to thank Editage (http://www.editage.com) for English-language editing.
Authorship
All named authors meet the International Committee of Medical Journal Editors (ICMJE) criteria for authorship for this article, take responsibility for the integrity of the work as a whole, and have given their approval for this version to be published.
Disclosures
Juan Ruano is a member of the journal’s Editorial Board. Pedro J. Gómez-Arias, Francisco Gómez-García, Jorge Hernández-Parada, Ana M. Montilla-López and Esmeralda Parra-Peralbo have nothing to disclose.
Compliance with Ethics Guidelines
This article is based on previously conducted studies and does not contain any studies with human participants or animals performed by any of the authors.
Data Availability
All data generated or analyzed during this study are included in this published article/as supplementary information files. All data generated or analyzed during this study are included in this published article/as supplementary information files.
Author information
Authors and Affiliations
Corresponding author
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Gómez-Arias, P.J., Gómez-García, F., Hernández-Parada, J. et al. Efficacy and Safety of Janus Kinase Inhibitors in Type I Interferon-Mediated Monogenic Autoinflammatory Disorders: A Scoping Review. Dermatol Ther (Heidelb) 11, 733–750 (2021). https://doi.org/10.1007/s13555-021-00517-9
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13555-021-00517-9