
Abstract
Introduction
Onychomycosis is a fungal nail infection, frequently caused by dermatophytes, which occurs in 2–14% of Western adults. The present study was set up to evaluate the efficacy and safety of a water-based, peelable nail polish (daily application), which acidifies the nail environment, versus a 5% amorolfine nail lacquer (weekly application) for topical treatment of mild-to-moderate onychomycosis.
Methods
One hundred two adults were randomized in this open, prospective, blinded trial. Clinical efficacy was evaluated at baseline and days 30, 60, 120, and 180, respectively. All patients underwent microbiological testing (at baseline and study end). The primary objective of this trial was the change in the percentage of healthy nail surface at day 180.
Results
The percentage of healthy surface between baseline and day 180 increased with 11.8% in the test product group and 13.2% in the amorolfine group, which were statistically comparable. Other onychomycosis-related parameters (dystrophy, discolouration, thickening, and healthy aspect, respectively) showed significant (p < 0.05) improvement after 180 days (versus baseline) with both treatments. Clinical performance was further confirmed by the frequency of patients showing onychomycosis improvement or success at the end of the study: 96.0% (test product) versus 79.6% (amorolfine). Microbiological results and improved quality of life confirmed clinical performance. Both treatments were well tolerated and appreciated for their properties and efficacy.
Conclusion
The present trial confirmed the clinical performance of daily acidification of the nail, as reflected by (1) a comparable increase of percentage of healthy nail surface following treatment with the test product versus amorolfine, (2) the overall improvement of other onychomycosis-related parameters, (3) user convenience, and (4) absence of side effects. These data indicate that daily application of an aqueous, acetic acid-based, peelable solution can be a convenient, safe, and equally effective alternative for the topical management of onychomycosis.
Trial Registration
ClinicalTrials.gov identifier; NCT03382717
Funding
Oystershell Laboratories.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Onychomycosis is a common nail infection with a worldwide prevalence of 5%, but this value may vary according to the studied area. Most common pathogens are dermatophytes, but also yeasts (e.g. Candida albicans) and non-dermatophyte moulds [1]. Depending on the location and the route of pathogen penetration, the following types of onychomycosis have been characterized: (1) disto-lateral onychomycosis, (2) white superficial onychomycosis, (3) proximal subungual onychomycosis, (4) total onychodystrophy, and (5) endonyx onychomycosis [2]. Disto-lateral subungual onychomycosis is the most common form and is usually caused by Trichophyton rubrum, which invades the nail bed and underside of the nail plate [3,4,5].
Onychomycosis gradually destroys the nail by affecting the nail plate, nail bed, and periungual tissue. Depending on the degree of infection, nail discolouration, thickening, dystrophy, brittleness, and loosening (onycholysis) are observed [6]. Although the disease is not life-threatening, its morbidity may negatively impact patients' quality of life [7].
Efficient treatment is challenging because of the inherent slow growth of the nail and its composition as well as patient compliance. Also, comorbidity in risk groups (e.g. elderly, diabetic, immunosuppressed, or psoriasis patients) will further hamper treatment [8].
Oral medication has been shown to be rather ineffective against specific forms of onychomycosis (e.g. endonyx forms). Manufacturers have focused on the development of topical products that affect dermatophytes through a physical, non-specific, or pharmacological mode of action, respectively [9]. Topical treatments are the best choice for the treatment of lateral onychomycosis. Most of the topical products are lacquers that need to be removed with solvents on a weekly basis, a fact that stands in the way of patient compliance.
A randomized, controlled, multicentre, open-label trial was performed to assess the clinical efficacy against onychomycosis of an aqueous, acetic acid-based, peelable nail polish (daily application) versus a medicated nail lacquer (5% amorolfine; Loceryl®; weekly application), serving as reference. After penetration, acetic acid acidifies the nail and consequently blocks fungal spreading, thereby allowing the infected part of the nail to grow out [10, 11]. The amorolfine nail lacquer elicits its action by destroying the fungal cell membrane [12].
The primary objective of the present study was to assess variation in the percentage of healthy surface of the great toenail after a treatment period of 180 days with both products in combination with changes in microbiological findings at baseline and at the end of the treatment (assessed via KOH staining and fungal culture). Clinical diagnosis was performed by blinded investigators using digital image analysis (contour tracing). Secondary objectives included evaluation of clinical efficacy against onychomycosis of the great toenail at distinct time points (days 30, 60, 120, and 180), microbiological efficacy of both products, product safety, impact on quality of life (QoL), and finally product efficacy, tolerance, and acceptability by subject’s self-assessment and medical examination.
Methods
Trial Set-Up
This randomized controlled study, which was multicentre, comparative, investigator blinded, and open label, was approved by the ethics committee of the principal clinical trial centre (Military Hospital of Tunis, Tunisia) on 22 September 2016. The study was conducted in accordance with the principles of the Declaration of Helsinki 2013, Good Clinical Practice, and European Union Directive 2001/20/EC.
The entire study took place in two clinical trial facilities in Tunis (Tunisia), specialized in treatment of skin and nail disorders. Recruitment was performed by the Principal Investigator (dermatologist) of each trial centre and continued from 26 October 2016 (first patient, first visit) to 26 August 2017 (last patient, last visit). The trial was registered at ClinicalTrials.gov with the following code: NCT03382717.
Inclusion and Exclusion Criteria
Patients (> 18 years) were included after confirmed diagnosis of superficial or light-to-moderate disto-lateral onychomycosis (without matrix involvement, the infected area being smaller than 2/3 of the nail surface) on at least one great toe nail. Potassium hydroxide (KOH) staining was used to confirm the diagnosis [13]. Fungal culture was performed on samples of KOH-positive subjects to characterize dermatophyte infection. However, the outcome of these fungal cultures did not restrict subject inclusion since false-negative results regularly occur in clinically confirmed cases [14].
Beside positive diagnosis, patients must have stopped any systemic and/or topical antifungal treatment for at least 6 and 3 months, respectively, before inclusion. Finally, female subjects of childbearing potential needed to use an accepted contraceptive regimen at least 12 weeks prior to study start, during the study, and at least 1 month after study end.
Exclusion criteria were: non-compliance with the protocol, enrolment in another clinical trial during the test period, pregnant (or planning to be) or nursing women, known allergy to one of the ingredients of both products, patients suffering from serious or progressive diseases (uncontrolled diabetes, peripheral circulatory disease, HIV, psoriasis, lichen planus, immunosuppressive disorders), and patients with other skin diseases in the studied zone.
Sample Size, Informed Consent, Randomization, and Baseline Data
Based on previous available clinical data, a sample size of 100 subjects was calculated to allow correct statistical comparison between the test product and reference. Based on this information, 102 eligible subjects were recruited by the study staff and randomly allocated to two groups. Prior to the study, a randomization list was calculated by an external statistician using SAS software (version 9.4). For this purpose, block randomization with a block size of two was used.
Each subject received oral and written information concerning the studied product, its nature, and the duration and conditions of the study. Written consent was obtained before any study-specific procedures were performed in accordance with the Helsinki Declaration.
Following this informed consent, a patient screening number was assigned to each patient by the responsible investigator. A randomization list was provided prior to the start of the study. A unique randomization number attributed each included patient to one of the treatment groups. Baseline demographic data were collected on gender, age, height, weight, blood pressure parameters, and medication use.
Blinding
Discernible differences in the product properties (e.g. different bottle, odour) and in the administration process allowed patients to recognize both trial products. Therefore, blinding and unbiased evaluation was guaranteed by making digitalized macro-photographs of the toe nail, which were in turn analysed by two blinded evaluators. The detailed procedure is described below in “Evaluation of clinical efficacy”.
Study Medication, Dosage and Administration
The test product was supplied in glass bottles (with a brush applicator) by Oystershell Laboratories (Ghent, Belgium). This product consists of acetic acid (active ingredient), a peelable film-forming polymer (polyurethane), water, peppermint oil (penetration enhancer, solvent, perfume), an anti-oxidant (octyl gallate), preservatives, acetylated lanolin alcohols, and biotin.
The amorolfine nail lacquer reference (Loceryl®; available in a glass bottle) was provided by the Principal Investigator. This medicated nail lacquer contains 5% amorolfine (as amorolfine hydrochloride in ethanol, triacetin, butyl acetate, ethyl acetate, and ammonium methacrylate polymer).
The test product was applied once daily with the brush, covering the complete (cleaned) nail. After 24 h, the water-resistant film was removed by stripping and a new layer was applied. If new growth appeared, the nail was trimmed using a nail clipper.
Amorolfine was applied once a week with a reusable spatula (supplied with the product). Prior to use, the nail was filed and cleaned with isopropanol wipes.
Evaluation of Clinical Efficacy
Patients were treated with the test product or reference, respectively, for a period of 180 days. Onychomycosis evolution was evaluated at distinct time points, day (D) 30, D60, D120, and D180, and compared with D0 (baseline). The primary objectives were to assess variation in the % of healthy surface of the great toenail at the end of the study (D180) compared with baseline in both treatment groups. Diagnosis was performed using digital image analysis [15]. For each photograph, a blinded dermatologist traced the healthy surface. Next, a second evaluator, also blinded, determined the percentage of healthy surface and assigned the following scores: 100% healthy surface, > 66.6% healthy surface, 33.3–66% healthy surface, and < 33.3% healthy surface.
Secondary objectives implied evaluation of the following parameters:
-
(1)
Clinical efficacy against onychomycosis of the great toenail at D30, D60, and D120.
-
(2)
Microbiological efficacy of the product (KOH staining method).
-
(3)
Product safety.
-
(4)
Impact on the quality of life (QoL) of the subjects using the NailQoL questionnaire, with assessment on D0, D60, and D180 [16].
-
(5)
Product efficacy, tolerance, and acceptability by subject’s self-assessment.
-
(6)
Impact on onycholysis, nail dystrophy, nail discolouration, nail thickening, and onychomycosis evolution at D30, D60, D120, and D180.
Safety Evaluation
At each visit, the local and global tolerance (collection of all adverse events and subjective signs) were evaluated. In addition, all patients were asked to report adverse events in a log book. Study staff investigated all adverse events and determined the relationship to the treatment.
Subjective Questionnaire
A subjective questionnaire was included to evaluate product usability and functionality of the test product (completion on days 30, 60, 120, and 180). Questions were related to packaging, cosmetic effects, overall satisfaction, tolerance, product application, and intention to purchase.
Statistical Analyses
Clinical efficacy and safety were evaluated in the intent-to-treat (ITT) population, including all subjects eligible for the study and who received at least one application of the test product and had at least one post-dose efficacy evaluation. Briefly, continuous data were summarized by their mean, standard deviation (SD), median, and minimum and maximum. Categorical data were summarized by frequencies and percentages.
Mean absolute changes in % healthy surface from baseline (D0) at day 180 between the test product and reference were compared with an independent t test after having verified the assumptions of normality (QQ-plot) of the differences. Additionally, changes in mean % healthy surface from baseline in function of visit for subjects treated with test product versus reference product were studied in more detail using generalized linear mixed effects models.
Mean absolute changes in NailQol global score from baseline (D0) at day 180 between test product and reference were compared with an independent t-test after having verified the assumptions of normality (QQ-plot) of the differences.
To compare changes in nail dystrophy, discolouration, and nail thickening between baseline and day 180, five categories were reduced to two (none to slight versus moderate to severe). The same was done for healthy aspect of the nail. The McNemar test for paired data was used to test if there was a change in nail dystrophy, discolouration, and nail thickening between baseline and day 180.
All descriptive and statistical analyses were performed in R version 3.3.1. (R development core team, 2017). p < 0.05 was considered statistically significant. No imputation of missing data was performed. The amount of missing data is presented in the tables wherever appropriate.
Baseline Data
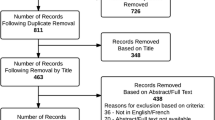
Study data were collected between October 2016 and August 2017. In total, 102 subjects were randomized into the study (n = 52, test product group; n = 50, reference group). For two patients in the test product group and one patient in the reference group, post-dose efficacy and safety data were not available (lost to follow-up). In total, 99 subjects (n = 50, test product group; n = 49, reference group) were included in the intention-to-treat (ITT) population. A CONSORT flow chart is shown in Fig. 1. A summary of demographic characteristics is presented in Table 1.

CONSORT Flow chart (ITT population)
Prior to product application (D0), no significant differences were found between the two treatment groups for average healthy surface, secondary clinical parameters, and average NailQoL score.
Direct detection of fungal infection with the KOH staining method was positive for all recruited subjects, except for one patient in the test product group, showing a false-negative result. Indeed, fungal culture was positive for this patient, and for this reason, the Principal Investigator decided to include this patient. Consequent fungal culture was positive for a majority of the subjects (67% test product group, 52% reference group), with Trichophyton (T.) rubrum being the most common pathogen (88.6% and 80.8% for test product and reference, respectively). Other dermatophytic fungi were also detected: T. mentagrophytes (5.7%, test product group; 3.9%, reference group) and Microsporum canis (3.9%, reference group). A few non-dermatophytic fungi were also detected: Aspergillus niger (7.7%, reference group), Aspergillus flavus (2.9%, test product group), and Candida albicans (2.9%, test product group; 3.9%, reference group), respectively.
Results
Efficacy Evaluation
Primary efficacy of both treatments was compared in terms of percentage of healthy surface. As shown in Table 2, the percentage of healthy surface between baseline and D180 increased by 11.8% (SD = 13.9; n = 48) in the test product group and 13.2% (SD = 15.0; n = 46) in the reference group, respectively. The observed difference was comparable between both treatment groups. Furthermore, the percentage of healthy surface after 30, 60, 120, and 180 days of treatment showed a significant increase in % of healthy surface in function of treatment duration (p < 0.001). This improvement was similar for both treatments over the whole treatment period (p = 0.889). An example of nail improvement following treatment with both products is shown in Fig. 2.

Onychomycosis improvement in function of time following treatment with test product and reference, respectively
The effect of both treatments on other onychomycosis-related parameters is summarized in Table 3. Significant differences for each treatment (D180 versus baseline) and between treatments, respectively, are indicated in the last column.
After 180 days of treatment, a reduction of 46.0% (test product) and 41.0% (reference), respectively, of KOH-positive subjects was observed. Similar results were observed for the fungal culture: the % of subjects with positive culture decreased from 67.0% to 23.0% (test product) and from 52.0% to 10.0% (reference) after 180 days of use.
Finally, both treatments resulted in improvement of subjects' quality of life, as reflected by the reduction of the NailQoL global score in function of treatment duration. After 180 days, the mean NailQoL score decreased by 27.9 units (test product) and 26.7 units (reference) compared with baseline. This improvement was similar for both treatment groups [mean difference 1.2 units; 95% confidence interval (CI): − 3.8 to 5.9, p = 0.628].
Safety Evaluation
Local tolerance of the treatment was assessed by the investigator via clinical evaluation and the patient's response at each visit during the trial. Overall, both treatments were very well tolerated with a score = 3 during each visit for almost all subjects. A score of 2 was given once by a subject from the test product group (D180) and two times by different subjects from the reference group (day 120 and day 180).
Subjective Questionnaire
The subjective questionnaire data clearly confirmed product safety and usability. All subjects confirmed that the product was: (1) well tolerated, (2) able to cover the nail infection, (3) highly resistant to water and physical stress, (4) easy to apply (visibility, mode of application) and to remove, and (5) allowed application of a top coat. This was further reflected by the high product satisfaction and intention to buy.
Discussion
Fungal infections are responsible for 23% of foot diseases and 50% of nail conditions in patients seen by dermatologists. However, they are less common in the general population (3% to 5%) [17]. Prevalence varies among populations, which may be related to different screening techniques. In one large European project, including 13,695 people with a range of foot conditions, a fungal infection (diagnosed by microscopy and culture) was present in 35% of the patients [18]. One prospective study in Spain (1000 adults, aged > 20 years) reported a prevalence of fungal toenail infection of 2.7% (infection defined as clinically abnormal nails with positive microscopy and culture) [19]. In Denmark, a study (5755 adults, aged > 18 years) reported a prevalence of fungal toenail infection of 4.0% (determined by positive fungal cultures) [20]. The incidence of mycotic nail infections may have increased over the past few years. This may be explained by the increasing use of systemic antibiotics, immunosuppressive treatment, more advanced surgical techniques, and the increasing incidence of HIV infection [21].
During recent years, different topical products have been put on the market for the treatment of onychomycosis. They are used either alone or in combination with systemic treatments, resulting in higher cure rates. For topical treatment, both medicated nail solutions and medical devices with a physical mode of action are commercially available. In the present study, an aqueous, acetic acid-based, peelable nail polish, which inhibits fungal growth by acidification of the nail environment, was compared with Loceryl®, a nail lacquer containing 5% amorolfine [22, 23]. Amorolfine is a morpholine antifungal drug that disrupts the fungal cell membrane [12].
All subjects were diagnosed with either superficial onychomycosis or light-to-moderate disto-lateral onychomycosis on at least one great toenail. Fungal infection was further confirmed using the KOH staining method [13] and fungal culture, respectively.
Both females and males were included, with a higher proportion of females. Average age was 54.3 ± 11.8 years and 52.3 ± 14.2 years in the test product and reference group, respectively. At baseline (D0), both treatment arms were homogeneous for all studied parameters.
At the end of the study (D180), three subjects (n = 2, test product; n = 1, reference) did not complete the trial (lost to follow-up). For this reason, these subjects were excluded from the data analyses, resulting in a final number of 99 patients.
The primary objective of this study implied the evolution in % healthy surface between baseline and D180 following treatment with both products. For the test product, an increase of 11.8% (n = 48) was observed, whereas treatment with the reference product resulted in an increase of 13.2% (n = 46). The difference between the two treatments was not significantly different. At the end of the study, application of both products resulted in a significant (p < 0.001) improvement compared with baseline.
With both treatments, other onychomycosis-related parameters showed a significant improvement compared with baseline, being more pronounced (but not significantly different) for nail dystrophy, discolouration, nail thickening, and healthy aspect of the nail, respectively. The more pronounced effects may be explained by differences in nail penetration (polarity of the active substance), different modes of action, the condition of the nail, inter- and intra-individual variations, and so on. All these factors may contribute to a different but not necessarily significant outcome.
Clinical efficacy was further reflected by the improvement in patient’s quality of life, as evaluated using the NailQoL questionnaire [16]. This was observed in both treatment arms. Finally, both treatments were well tolerated, thereby confirming product safety.
The mode of action of the test product, which contains acetic acid, relies on acidification of the nail. Following application, acid penetration and the consequent pH decrease of the nail environment will inhibit acid-sensitive keratolytic enzymes, which are essential for dermatophyte nail penetration [10, 24,25,26]. In turn, fungal growth inhibition allows the infected part to grow out in vivo, without further fungal spreading.
Susceptibility of dermatophytes to acids has been demonstrated in independent experiments and literature reports. Results of a “minimum inhibitory concentration” (MIC) assay confirmed fungal growth inhibition following exposure to different organic acids, including acetic acid. We established MIC values varying between 0.0976% W/W (T. rubrum) and 0.195% (T. mentagrophytes), respectively (unpublished data). For amorolfine, MIC values varying between 4 and 16 µg/ml were reported, depending on the tested dermatophyte species [27]. Although the higher MIC value was for acetic acid, Trichophyton mentagrophytes growth was clearly inhibited following nail penetration, as shown in a validated bovine hoof assay [10]. As the nail behaves like a hydrogel [28], the low molecular weight and polarity of acetic acid allow quick penetration (within 20 min, in-house results). These data clearly contradict the results reported by Ghannoum et al. [29]. In their set-up, no activity was observed with three acid-based medical devices. However, these observations must be put in perspective. First, only a very small volume of product was applied once, which does not correspond with the instructions for use (twice daily application for the acetic acid-based product). Furthermore, the authors do not provide information regarding the hydration status of the nail, type of agar (buffered or not), or condition of the nails. For this reason, their results are inconclusive. Indeed, in a validated bovine hoof assay, both an acetic acid-based solution and amorolfine were able to penetrate the nail and inhibit Trichophyton mentagrophytes growth [10]. These in vitro data are further confirmed by the results of the present clinical study and previous clinical data with an equivalent medical device, showing a comparable outcome for product performance and safety [30]. The latter product also relies on acidification of the nail environment via penetration of acetic acid but must be applied twice-daily compared with the investigational product (once daily). Besides being water-based, the test product offers additional advantages because the film can be easily removed after 24 h.
Finally, product usability and safety were clearly confirmed by the subjective questionnaire data. Indeed, product application and removal were highly appreciated by the patients. Furthermore, the water- and physical-stress-resistant film can cover the affected nail, which is clearly a cosmetic advantage.
Limitations of the present trial include a relatively low number of patients. However, sample size was calculated using data of earlier clinical trials with acetic acid-based and medicated topical treatments, confirming a comparable outcome in view of product performance and safety. Another limitation implies a treatment period of 180 days. Depending on the degree of infection, a treatment period of at least 6 months may be required for full recovery of the infected nail.
Conclusions
In conclusion, the test product is an efficient and safe treatment for mild-to-moderate cases of onychomycosis. At study end, the increase in % of healthy surface of the nail was comparable to the medicated nail lacquer Loceryl®. Clinical performance of the test product was further confirmed by: (1) the high number of patients with onychomycosis improvement or success (96.0% vs. 79.6% for reference), (2) the more pronounced positive evolution of different onychomycosis-related parameters in function of time, (3) the positive impact on quality of life of the patients, and (4) confirmed safety.
References
Chabasse D, Pihet M. Mycological diagnosis of onychomycosis. J Mycol Med. 2014;24:269–78.
Piraccini BM, Alessandrini A. Onychomycosis: a review. J Fungi. 2015;1:30–43.
Elewski BE. Onychomycosis: pathogenesis, diagnosis and management. Clin Microbiol Rev. 1998;11:415–29.
Ghannoum M, Isham N. Fungal nail infections (Onychomycosis): a never-ending story? PLoS Pathog. 2014;10:e1004105.
Baran R, Hay RJ. New clinical classification for onychomycoses. J Mycol Med. 2014;24:247–60.
Shirwaikar AA, Thomas T, Shirwaikar A, Lobo R, Prabhu KS. Treatment of onychomycosis: an update. Ind J Pharm Sci. 2008;70:710–4.
Bunyaratavej S, Leeyaphan C, Chayangsu O, Bunyaratavej S, Kulthanan K. Onychomycosis: a study or self-recognition by patients and quality of life. Indian J Dermatol Venerol Leprol. 2015;81:270–4.
Elewski BE, Tosti A. Risk factors and comorbidities for onychomycosis. J Clin Aesthet Dermatol. 2015;8:38–42.
Del Rosso JQ. The role of topical antifungal therapy for onychomycosis and the emergence of newer agents. J Clin Aesthet Dermatol. 2014;7:10–8.
Sleven R, Lanckacker E, Delputte P, Maes L, Cos P. Evaluation of topical antifungal products in an in vitro onychomycosis model. Mycoses. 2016;59:327–30.
Kunert J. Physiology of keratinophilic fungi. In: Biology of dermatophytes and other keratinophilic fungi. In: Kushwaha, RKS, Guarro (eds.) Revista Iberoamericana de Micología, Bilbao. 2000;77–85.
Polak A. Preclinical data and mode of action of amorolfine. Dermatology. 1992;184:3–7.
Lim CS, Lim SL. Practical tip: Chicago Sky Blue (CSB) stain can be added to the routine potassium hydroxide (KOH) wet-mount to provide a color contrast and facilitate the diagnosis of dermatomycoses. Dermatol Online J. 2011;17:11.
Gupta AK, Jain HC, Lynde CW, Watteel GN, Summerbell RC. Prevalence and epidemiology of unsuspected onychomycosis in patients visiting dermatologists’ offices in Ontario, Canada—a multicenter survey of 2001 patients. Int J Dermatol. 1997;36:783–7.
Kaliyadan F, Manoj J, Venkitakrishnan S, Dharmaratnam AD. Basic digital photography in dermatology. Indian J Dermatol Venerol Leprol. 2008;74:532–6.
Warshaw EM, Foster JK, Cham PM, Grill JP, Chen SC. NailQoL: a quality-of-life instrument for onychomycosis. Int J Dermatol. 2007;46:1279–86.
Evans EGV. The rationale for combination therapy. Br J Dermatol. 2001;145(suppl 60):9–13.
Roseeuw D. Achilles foot screening project: preliminary results of patients screened by dermatologists. J Eur Acad Dermatol Venereol. 1999;12:S6–9.
Del Palacio A, Cuetara MS, Garau M, Perea S. Onychomycosis: a prospective survey of prevalence and etiology in Madrid. Int J Dermatol. 2006;45:874–6.
Svejgaard EL, Nilsson J. Onychomycosis in Denmark: prevalence of fungal nail infection in general practice. Mycoses. 2004;47:131–5.
Trepanier EF, Amsden GW. Current issues in onychomycosis. Ann Pharmacother. 1998;32:204–14.
Iorizzo M, Harmane I, Derveniece A, Mikazans I. Ciclopirox 8% HPCH nail lacquer in the treatment of Mild-to-moderate onychomycosis: a randomized, double-blind amorolfine controlled study using a blinded evaluator. Skin Appendage Disord. 2016;1:134–40.
Auvinen T, Tiihonen R, Soini M, Wangel M, Sipponen A, Jokinen JJ. Efficacy of a topical resin lacquer, amorolfine and oral terbinafine for treating toenail onychomycosis: a prospective, randomized, controlled, investigator-blinded, parallel-group clinical trial. Br J Dermatol. 2015;173:940–8.
Guo J, Brosnan B, Furey A, Arendt E, Murphy P, Coffey A. Antifungal activity of Lactobacillus against Microsporum canis, Microsporum gypseum and Epidermophyton floccosum. Bioeng Bugs. 2012;3:102–11.
Lastauskiene E, Zinkeviciene A, Girkontaite I, Kaunietis A, Kvedariene V. Formic acid and acetic acid induce a programmed cell death in pathogenic Candida species. Curr Microbiol. 2014;69:303–10.
Lind H, Jonsson H, Schnurer J. Antifungal effect of dairy propionibacteria— contribution of organic acids. Int J Food Microbiol. 2005;98:157–65.
Adimi P, Hashemi SJ, Mahmoudi M, Mirhendi H, Shidfar MR, Emmami M, et al. In-vitro activity of 10 antifungal agents against 320 dermatophyte strains using microdilution method in tehran. IJPR. 2013;12:537–45.
Smith KA, Hao J, Kevin L. Effects of organic solvents on the barrier properties of human nail. J Pharm Sci. 2011;100:4244–57.
Ghannoum M, Sevin K, Sarkany M. Amorolfine 5% nail lacquer exhibits potent antifungal activity compared to three acid-based devices indicated for the treatment of onychomycosis: an in vitro nail penetration assay. Dermatol Ther (Heidelb). 2016;6:451.
Eertmans F, Doss N, Rossel B, Regidor PA. Nail acidification versus amorolfine in the local management of onychomycosis. A comparative, prospective, randomized, blinded trial. Int Educ Appl Sci Res. 2017;2:3–6.
Acknowledgements
Funding
Sponsorship for this study and article processing charges were funded by Oystershell Laboratories, which is also the manufacturer of the investigational product and employer of the co-authors Frank Eertmans and Bart Rossel. The sponsor was involved in the study planning as well as in the decision to publish, but was not involved in data collection, data analysis, and data interpretation. All authors had full access to all data in this study and take complete responsibility for the integrity of the data and accuracy of the data analysis.
Medical Writing and/or Other Assistance
Oystershell Laboratories engaged the CRO Dermscan (Tunis, Tunisia) to independently design and perform the study. Co-author Nejib Doss, who performed the clinical trial, was responsible for patient investigation/recruitment/treatment, and data collection. Statistical analyses were performed by an independent, external consultant, Els Adriaens (Adriaens Consulting, Bellegem, Belgium).
Authorship
All named authors meet the International Committee of Medical Journal Editors (ICMJE) criteria for authorship for this manuscript, take responsibility for the integrity of the work as a whole, and have given final approval for the version to be published.
Disclosures
Frank Eertmans is an employee of the Oystershell company, which is the manufacturer of the test product and funded this study. Bart Rossel is an employee of the Oystershell company, which is the manufacturer of the test product and funded this study. Nejib Doss is a clinical investigator, associated with Dermscan Tunisia, who performed the study. Els Adriaens has nothing to disclose.
Compliance with Ethics Guidelines
All procedures followed were in accordance with the ethical standards of the responsible committee on human experimentation (institutional and national) and with the Helsinki Declaration of 1964, as revised in 2013. Informed consent was obtained from all patients for being included in the study.
Open Access
This article is distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License (http://creativecommons.org/licenses/by-nc/4.0/), which permits any noncommercial use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
Author information
Authors and Affiliations
Corresponding author
Additional information
Enhanced digital features
To view enhanced digital features for this article go to https://doi.org/10.6084/m9.figshare.6840176.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

Cite this article
Eertmans, F., Doss, N., Rossel, B. et al. Daily Application of an Aqueous, Acidifying, Peelable Nail Polish versus Weekly Amorolfine for Topical Onychomycosis Treatment: A Prospective, Randomized, Blinded Trial. Dermatol Ther (Heidelb) 8, 463–473 (2018). https://doi.org/10.1007/s13555-018-0254-1
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13555-018-0254-1