
Abstract
For decades, no cancer therapy had been shown to improve average survival in metastatic melanoma. Two critical events have occurred, the discovery of melanoma driver mutation subsets and the discovery of immune checkpoint inhibitors, which have allowed for the development of modern, effective therapies. These findings have facilitated a rapid emergence of novel therapeutics for the disease with multiple FDA approvals in the last several years. The drugs vemurafenib, trametinib, and dabrafenib, which inhibit the commonly mutated BRAF pathway, have been approved based on improvements in survival outcomes. Agents that block immune checkpoints on lymphocytes allowing for immune cell activity against melanoma have also been approved based on improved survival outcomes such as ipilimumab and nivolumab. Pembrolizumab, another immune checkpoint inhibitor, has also been approved based on the response rate and duration of response in a phase 1 trial. Further agents and combinations of approved agents are positioned to possibly further increase this tally of approved drugs. This review will discuss recently approved novel agents and select drugs in development in advanced melanoma.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Melanoma has held a designation of ill repute among cancer subtypes, being a disease that has been resistant to modern chemotherapeutic approaches and results in the rapid demise of patients who are in the prime of their life. Up until a couple of years ago, typical treatments included chemotherapy drugs such as dacarbazine (DTIC) and temozolomide, which had low response rates and no improvement in overall survival [1]. Additionally, early attempts at immune therapy in the form of cytokines, such as interferon and interleukin-2 (IL-2), also had low response rates with high toxicity levels [2]. In the setting of these therapies, a metastatic melanoma patient’s average survival was 6–8 months, and no agent had been able to improve on this outcome in numerous randomized clinical trials [3].
Fortunately, a new generation of therapies for metastatic melanoma has arisen in the last few years resulting in improvements in response rates and overall survival outcomes. These advancements have taken place primarily on two separate fronts: (1) molecularly targeted inhibitors that work within the melanoma cell against abnormally activated protein kinases and (2) immune checkpoint inhibitors that work by enhancing T-lymphocyte function. Vemurafenib and ipilimumab represent the first in class of each of these approaches, respectively, and both agents have shown improved average overall survival outcomes for metastatic melanoma patients in randomized trials. Several other agents in both classes have subsequently emerged, continuing to improve clinical outcomes. This review will focus on the clinical development of modern novel melanoma molecularly targeted agents and immune checkpoint inhibitors, as well as combination approaches.
This article is based on previously conducted studies and does not involve any new studies of human or animal subjects performed by any of the authors.
Development of Therapies Targeting Driver Mutations in Melanoma
Targeting the BRAF Pathway in Advanced Melanoma
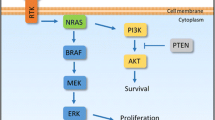
The molecular biology of melanoma is complicated with numerous mutations present and a variety of pathways impacted (Fig. 1). However, there are common driver mutations present at high frequencies that allow for a molecularly targeted approach. Critical to highlighting these common targetable mutation events was the work of Davies et al. in 2002 when they reported a high frequency of BRAF gene mutation (now known to be present in approximately 50% of melanomas), leading to the theory that the BRAF serine/threonine kinase could represent a commonly applicable therapeutic target [4]. In about 80–90% of mutated BRAF tumors, an activating V600E missense mutation is present, while about 10–20% of BRAF mutant tumors have V600K, V600D, and V600R mutations [5]. These activating mutations result in constitutive activation of the MAP (mitogen-activated protein) kinase pathway resulting in unchecked cell proliferation.

Schematic diagram showing select signaling pathways in melanoma. With activating BRAF mutations present in 50% of melanoma tumors and NRAS mutations present in approximately 20%, the MAP kinase pathway (RAS-RAF-MEK-ERK) plays an important role in the majority of melanomas. However, other pathways also can contribute to pathogenesis and resistance to BRAF and MEK inhibition, such as activation of the PI3K pathway and signaling through COT and CDK4
A landmark phase I trial of a potent BRAF V600 inhibitor PLX-4032 (now known as vemurafenib) demonstrated proof of principle that this targeted therapy approach could work [6]. This study demonstrated high response rates and acceptable tolerability, which led to further clinical development of the drug. A phase III study (BRIM-3) comparing vemurafenib with dacarbazine was performed with co-primary endpoints of overall survival (OS) and PFS [7]. This study included 675 patients with metastatic melanoma (95%) or unresectable stage IIIC (5%) with a BRAF V600E mutation who were previously untreated and showed a statistically significant improvement in survival for those treated with vemurafenib compared to dacarbazine [median OS 13.6 versus 9.7 months, respectively (HR 0.70, p = 0.0008)] [8]. The median PFS was also significantly prolonged with vemurafenib use at 6.9 months compared to 1.6 months for dacarbazine (HR 0.26, p < 0.001). Based on these results, the FDA approved vemurafenib for the treatment of BRAF V600E mutant metastatic melanoma in 2011. An increased incidence of cutaneous squamous cell carcinomas and keratoacanthomas has been noted with vemurafenib and other BRAF inhibitor therapies. In a study that analyzed three trials with a total of 520 patients who received vemurafenib, the incidence of squamous cell carcinoma was noted to be 19–26%, mostly keratoacanthomas [9]. The majority of patients continued therapy without dose reduction after resection. The SCC seems to be due to a paradoxical activation of the MAPK pathway that bypasses BRAF inhibition, as mentioned above [10].
Following on the footsteps of vemurafenib has been the development of other novel potent kinase inhibitors. Dabrafenib, similar to vemurafenib, is a potent inhibitor of mutant V600 BRAF kinase and has also been studied in a randomized phase III (BREAK-3) trial evaluating its activity against dacarbazine in patients with metastatic V600 mutated melanoma [11–13]. This trial showed significant improvement in the primary endpoint of PFS with dabrafenib (5.1 months) compared to dacarbazine (2.7 months) (HR = 0.33, p < 0.0001). Dabrafenib was FDA approved for single agent use for BRAF mutant metastatic melanoma based on these data. An update presented at ESMO 2014 showed a median OS of 20 and 15.6 months for the dabrafenib arm and dacarbazine arm, respectively, but did not reach statistical significance (HR 0.77 with 95% CI 0.52–1.13) [14]. More than half of the patients originally treated with dacarbazine crossed over to dabrafenib, potentially obscuring the OS benefit.
MEK inhibitors work by inhibiting MEK1/MEK2, a protein kinase that is just downstream of BRAF in the MAP kinase signaling pathway. Trametinib, which is an MEK1/MEK2 inhibitor, has been explored for its single-agent activity in the randomized phase 3 METRIC trial [15]. In this study, 322 patients with BRAF V600E/V600K mutant metastatic melanoma who had failed a prior chemotherapy were randomized (2:1) to receive trametinib or chemotherapy (choice of dacarbazine or paclitaxel). The primary endpoint of the study was PFS. The study was positive with the median PFS for trametinib being 4.8 versus 1.5 months for chemotherapy. An update presented at the Society for Melanoma Research 2013 Congress showed median OS of 15.6 vs. 11.5 months (HR 0.78; 95% CI 0.57–1.06), despite 65% crossover from the chemotherapy arm to trametinib arm [16]. Side effects of trametinib therapy included rash, diarrhea, fatigue, dermatitis, and edema. In May 2013, the FDA approved trametinib for treatment of patients with unresectable or metastatic melanoma with BRAF V600E/V600K mutation who have not received prior BRAF inhibitor therapy.
Overcoming Resistance to BRAF Inhibition with Combination Molecularly Targeted Approaches
The development of resistance to single BRAF and MEK inhibition therapy occurs in almost all cases of advanced melanoma with resistant tumor cells exhibiting a reactivation of the MAP kinase pathway. A variety of events occur triggering this resistance including the presence of NRAS mutations, COT-mediated MAPK activation, redirection of signaling through CRAF, PI3kinase pathway signaling via PTEN loss, PDGFR upregulation or AKT3 upregulation, secretion of hepatocyte growth factor, increased activation of FGFR3, and a variety of other events [17–22]. Given that MAP kinase pathway reactivation is a common cause of single-agent BRAF inhibitor resistance, the use of other MAP kinase inhibitors downstream of BRAF has been an area of interest.
The proof that BRAF and MEK coinhibition could bypass some of these resistance mechanisms and advance clinical outcomes was demonstrated in a phase 1/2 study evaluating the combination of dabrafenib and trametinib [23]. These data resulted in a series of pivotal phase 3 trials evaluating the BRAF/MEK inhibitor combinations. The COMBI-D trial was a randomized, double-blind, phase 3 study comparing the combination of dabrafenib and trametinib to dabrafenib plus placebo as first-line therapy in 423 patients with unresectable or metastatic BRAFV600E/K mutant melanoma [24]. Combination dabrafenib and trametinib resulted in a 25% reduction in risk of progression compared to dabrafenib alone (median PFS of 9.3 vs. 8.8 months, HR 0.75, p = 0.035). The median PFS decreased to 7.2 months when clinical progression and receipt of new anticancer therapy were included in the dabrafenib alone group. An updated OS analysis announcement in February 2015 showed a 29% risk reduction in death (HR = 0.71, p = 0.011) [25]. This study also demonstrated that combining an MEK inhibitor with BRAF inhibition can attenuate the increased risk of cutaneous malignancies with BRAF inhibitor therapy. Improvements in skin toxicities were noted in the combination group, such as squamous cell carcinoma (2% vs. 9%), hyperkeratosis (3% vs. 32%), alopecia (7% vs. 26%), and hand-foot syndrome (5% vs. 27%). This reduction in squamous cell carcinoma with BRAF/MEK combination therapy has been explained by the paradoxical activation of the MAP kinase pathway in squamous cell carcinoma lesions by a single-agent BRAF inhibitor because the malignant cells harbor RAS mutations [10]. Use of an MEK inhibitor in this situation nullifies this pathway activation by single-agent BRAF inhibition, thus the reduction in incidence of squamous cell lesions. It is important to note that single-agent BRAF inhibitor use does not cause squamous cell carcinomas, but results in acceleration in growth of pre-existing lesions. Other toxicities were seen at higher frequencies in the combination group, such as pyrexia (51% vs. 28%); hypertension (22% vs. 14%) and diarrhea (24% vs. 14%) were higher in the combination group.
The COMBI-V trial was an open-label phase 3 study that randomized patients with untreated advanced BRAF mutant melanoma (n = 704) to dabrafenib plus trametinib or vemurafenib alone [26]. The median PFS favored the combination group with a 44% risk reduction (median PFS of 11.4 vs. 7.3 months, HR = 0.56, p < 0.001). In addition, the overall response rate was 64% for the combination group compared to 51% for the vemurafenib group, which was statistically significant (p < 0.001). Median duration of response was also longer in the combination group (13.8 months compared to 7.5 months). The OS analysis at 12 months showed a 31% risk reduction (HR = 0.69, p = 0.005). Similar to COMBI-D, a significant reduction in hyperproliferative skin toxicity was seen in the COMBI-V trial. For example, the rate of cutaneous SCC and keratoacanthomas was only 1% in the combination group compared to 18% for the vemurafenib group. This has provided further evidence that combination BRAF and MEK inhibition is superior to single-agent BRAF inhibition. Based on the results of COMBI-D and COMBI-V, trametinib and dabrafenib combination therapy has become the standard approach to patients harboring a BRAF V600 mutation who are considered for molecularly targeted therapy.
Following the path of dabrafenib and trametinib, other BRAF/MEK combination studies have been performed or are in progress. The phase 3 coBRIM study evaluated the combination of the MEK inhibitor, cobimetinib, with vemurafenib. In this double-blind phase 3 trial, 495 patients with previously untreated unresectable stage 3 or metastatic V600 mutant melanoma were randomized to receive either vemurafenib plus cobimetinib or vemurafenib plus placebo [27]. The PFS was significantly prolonged with the combination represented by a 49% risk reduction of progression (median PFS of 9.9 vs. 6.2 months, HR = 0.51, p < 0.001). Interim OS analysis with a 9-month survival rate showed a 35% risk reduction (HR = 0.65, 95% CI 0.42–1.00), but it did not cross the pre-specified stopping boundary. Reductions in secondary cutaneous neoplasms, as noted in the other studies, were observed, and grade 3 or 4 adverse events with the combination arm were 65% as opposed to 59% in the single-agent arm. Cobimetinib is currently under priority review for FDA approval in combination with vemurafenib. Finally, the COLUMBUS study is an ongoing phase 3 randomized three-armed trial comparing LGX818 (BRAF inhibitor) plus MEK162 (MEK inhibitor) to LGX818 alone and vemurafenib alone. This study may present the therapeutic field with a third BRAF/MEK combination option.
Other trial approaches to overcoming BRAF pathway resistance beyond BRAF plus MEK combinations are also being studied. A trial evaluating an individualized molecular approach, named the LOGIC 2 trial, is underway that focuses on BRAFV600 mutant melanoma patients who have progressed on the LGX818/MEK162 combination and obtains a new tumor biopsy after progression. Based on the molecular findings of the progressing tumor, the patient continues the LGX818/MEK162 combination with an additional third agent, which would include BKM120 (PI3k inhibitor), LEE011 (CDK4/6 inhibitor), BGJ398 (FGFR inhibitor) or INC280 (cMET inhibitor). This study has the potential to show benefit of a more individualized approach in patients with emerging or pre-existing mutations that promote resistance to primary MAP kinase pathway inhibition. Studies evaluating combinations to overcome the PI3 kinase pathway resistance mechanism are also ongoing (NCT01512251, NCT01902173).
Targeting Pathways in Other Melanoma Subsets
Evaluation of targeted treatments for melanoma patients whose tumors harbor mutations other than BRAF is also underway. The NRAS kinase is mutated in about 15–25% of untreated melanoma tumors. It is seen in about 20% of BRAF mutant patients who progressed on BRAF inhibitor therapy, but very rarely coexists with BRAF mutation prior to a BRAF inhibitor exposure [28]. Like BRAF, NRAS is a driver mutation, and it can activate multiple downstream pathways including the MAPK pathway through BRAF or CRAF and PI3K. NRAS mutations are usually seen in an older population and where chronic UV exposure has been present. Compared to BRAF mutant tumors, the NRAS mutation tends to be present in thicker tumors with a higher mitosis rate and is arguably more aggressive than BRAF mutated tumors [29, 30]. Targeting of RAS directly has proven challenging for drug developers. However, since NRAS mutations drive the MAPK pathway, downstream inhibition of MEK appears to be a rational approach. Trials exploring different MEK inhibitors as well as combination approaches with CDK 4/6 inhibitors or PI3K/AKT pathway inhibitors are being performed for patients who harbor NRAS mutations [31]. The results of a phase 2 trial evaluating binimetinib (MEK162) in patients with metastatic NRAS mutant melanoma (n = 117) were reported in September 2014 [32]. An ORR of 14.5% was observed including one person who achieved complete CR. Survival endpoints were also reported including a median PFS of 3.6 months (95% CI 2.6–3.8 months) and a median OS of 12.2 months (lower 95% CI of 7.9 months). Based on the efficacy noted in this study, a randomized phase 3 trial (NEMO) was launched and is currently ongoing in metastatic NRAS mutant melanoma patients comparing binimetinib to dacarbazine.
The use of MEK inhibitors may also play a role in a rarer melanoma subtype, ocular melanoma, which is also driven by MAP kinase pathway activation. Ocular melanoma is a type of melanoma typically excluded from melanoma trials because of its notoriously treatment-resistant nature, rapid rate of progression, and different genetics (high frequency of GNAQ and GNA11 mutations). In a recently reported study, the MEK inhibitor, selumetinib, was compared to temozolomide chemotherapy and showed a PFS of 15.9 weeks compared to 7 weeks, respectively (HR = 0.46, p < 0.001) [33]. Median OS was 9.1 vs. 11.8 months in the selumetinib arm (HR 0.66, p = 0.09). Although overall survival did not reach statistical significance, this was the first randomized trial to show a statistical improvement in PFS, which is a meaningful advance forward for this disease that lacks therapeutic options.
The growth factor receptor, c-kit, also plays an important role in a small subset of melanoma patients. Up to 20% of patients with mucosal, chronic sun-damaged and acral lentigeninous melanomas have mutations in c-kit, which drives pathogenesis in these tumors [34]. A variety of agents, including imatinib, dasatinib, sunitinib, and nilotinib potently, inhibits the ckit receptor and trials with the agents have either been performed or are currently ongoing. The most experience with targeted therapy in kit-mutant melanoma has been with imatinib, with several phase 2 trials being reported [35–39]. In non-enriched patients for kit-activating mutations, the efficacy of imatinib has been rather disappointing; however, this is not the case in patients who are selected by a specific activating mutation (e.g., exon 9, 11, 13 mutations). In a study by Hodi et al., imatinib was tested in advanced melanoma patients with kit mutations or amplification [38]. In this study, the ORR was 29%, which was significantly better than the null value of 5%. The response rate was highest in those with activating mutations with a 51% response rated. Median time to progression was relatively brief (3.9 months for the mutated group, 3.4 months for the amplified group), and median overall survival was 12.5 months. In a trial evaluating the kit inhibitor nilotinib, patients with advanced melanoma harboring kit alterations that were refractory to imatinib were studied [40]. The response rate for this approach was 18% with a median TTP of 3.3 months and OS of 9.1 months. Notably, no responses were seen in a separate cohort exploring those with brain metastases.
Targeting Advanced Melanoma with Novel Immune Checkpoint Inhibitors
Ipilimumab
Similar to the field of targeted mutation-based therapy, the field of immunotherapy in melanoma has also changed dramatically in the last few years. Activated CD8+ T-lymphocytes are an essential line of defense against tumor cells. These lymphocytes require interfacing with antigen-presenting cells via T cell receptors (TCR), and a number of proteins have been identified that influence the effect of this interaction (Fig. 2) [41]. After the TCR interaction and a secondary signal via costimulatory molecules have activated the lymphocyte, inhibitory molecules may be expressed in order to limit or restrain T-cell function (an immune checkpoint). This process serves to protect the host from over activation of the immune system and autoimmunity. An important inhibitory immune checkpoint signal involves the interaction of CD28 and CTLA-4 (CD152) on T-cells, which act as costimulatory and coinhibitory signals respectively, with either CD80 (B7-1) or CD86 (B7-2) on the antigen-presenting cells. Inhibition of a negative immune checkpoint, such as CTLA-4, can release the negative impact on lymphocytes, allowing them to remain active against the tumor.
Ipilimumab, a monoclonal antibody against the CTLA-4 protein, has been evaluated in two landmark phase 3 clinical studies, showing improved survival compared to vaccine therapy in one trial and improved survival compared to DTIC in another. In the first study, 676 previously treated metastatic melanoma patients were randomized 3:1:1 to receive ipilimumab plus GP100 vaccine (melanoma peptide vaccine), ipilimumab alone, or GP100 vaccine alone [42]. The primary endpoint of overall survival was successful with the median OS being 10.0, 10.1, and 6.4 months for each arm, respectively. Interestingly, patients who respond to ipilimumab may develop initial progression followed by the observed response. This observation has led to the creation of a set of immune response criteria that can capture these data, unlike the traditional RECIST criteria. In clinical practice, this potential for delayed response requires the clinician to often consider continuation of therapy in the setting of asymptomatic radiographic or clinical progression to allow for this delayed response. However, in cases of symptomatic progression, waiting for delayed response can be challenging and in certain situations impractical.
Ipilimumab therapy has also been associated with distinctive immune-related adverse events (IRAEs). These IRAEs can include colitis, dermatitis, hepatitis, endocrinopathies and neuropathies, all of which are related to an inflammatory effect of lymphocyte activation. While most individuals had tolerable side effects, severe immune-related adverse events (grade 3 or higher) were noted in 10–15% of patients treated with ipilimumab.
Based on the results of these data, ipilimumab was FDA approved in March 2011. A subsequent phase 3 placebo-controlled trial evaluated 502 previously untreated patients with metastatic melanoma using a higher dose of ipilimumab (10 mg/kg as compared to 3 mg/kg) in combination with dacarbazine vs. dacarbazine alone [43]. This trial also showed a significant improvement in its primary endpoint of OS, with those receiving the ipilimumab/dacarbazine combination having a median OS of 11.2 months compared to 9.2 months for dacarbazine alone (HR 0.72, p < 0.001).
PD-1 Inhibition
Like CTLA-4, the programmed death-1 (PD-1) receptor is an important negative regulator of T-lymphocyte activity (inhibitory checkpoint) [44]. PD-1 interacts with the PD-1 ligand (PD-L1), which can be expressed on a variety of tissue surfaces including the tumor cell surface or tumor-infiltrating macrophages, resulting in suppression of T-lymphocyte function and tumor evasion of immune regulation. Multiple studies have shown an inverse correlation between PD-L1 expression in tumor cells and poor prognosis in multiple tumor types [41].
Pembrolizumab is a humanized monoclonal IgG4 antibody that binds to PD-1, blocks the negative interaction with PD-L1, and ultimately results in a rejuvenation and potentiation of T-lymphocytes, which can then have antitumor activity. The FDA granted accelerated approval in September 2014 for patients with unresectable or metastatic melanoma following treatment with ipilimumab or a BRAF inhibitor (in V600E/K mutant melanoma), based on the phase 1 Keynote-001 trial [45]. This trial explored pembrolizumab therapy in 173 advanced melanoma patients who met the criteria of progression on ipilimumab and a BRAF inhibitor (if applicable). Pembrolizumab demonstrated an ORR of 24% with response duration ranging from 1.4 months to over 8.5 months. In a follow-up phase 3 trial (Keynote-002), 540 ipilimumab refractory patients were randomized to pembrolizumab 2 mg/kg every 3 weeks, 10 mg/kg every 3 weeks, or chemotherapy. Results presented at the 2014 Society of Melanoma Research Congress showed 6-month PFS rates of 34, 38, and 16 percent for the 2 and 10 mg/kg pembrolizumab arms and the chemotherapy arm, respectively [46]. Treatment-related grade 3 or higher adverse events occurred in 11% of patients in the 2 mg/kg arm, 14% in the 10 mg/kg arm, and 26% with chemotherapy. In the phase 3 Keynote-006 trial, 834 patients with unresectable or metastatic melanoma who had received no more than one prior systemic therapy (required to be CTLA-4 and PD-1 inhibitor naïve) were randomized to receive pembrolizumab at 10 mg/kg every 3 weeks, 10 mg/kg every 2 weeks, or 4 cycles of ipilimumab 3 mg/kg every 3 weeks [47]. In this study, both pembrolizumab arms appeared to have similar efficacy outcomes, and both were superior to ipilimumab in the studies primary endpoints of PFS and OS. The median PFS for the q2 week pembrolizumab regimen was 5.5 months compared to 4.1 months for the q3 week regimen and 2.8 months for ipilimumab (HR 0.58, p < 0.001 for q2 week regimen versus ipilimumab). The median OS has not been reached for any of the groups at the time of publication; however, the 12-month survival landmark demonstrated a survival of 74.1% (q2 week pembrolizumab), 68.4% (q3 week), and 58.2% (ipilimumab). The difference between the pembrolizumab arms was statistically superior to ipilimumab for the 12-month OS analysis (HR 0.63).
Nivolumab is a fully human monoclonal PD-1 blocking antibody and has also been extensively evaluated in a series of phase 3 trials in advanced melanoma patients. The FDA approved the use of nivolumab for patients with unresectable or metastatic melanoma following treatment with ipilimumab or a BRAF inhibitor in December 2014 based on the phase 3 CheckMate-037 study [48]. In this trial, 405 patients were assigned to either nivolumab or chemotherapy in a 2:1 ratio, and the first interim analysis reported objective responses of 31.7% (95% CI 23.5–40.8) vs. 10.6% (95% CI 3.5–23.1) favoring nivolumab. Another phase 3 trial (CheckMate-066) enrolled 418 patients with previously untreated metastatic melanoma without a BRAF mutation to receive nivolumab or dacarbazine [49]. The 1-year landmark survival rate was 72.9% for nivolumab compared to 42.1% for dacarbazine (HR for death 0.42; 95% CI 0.25–0.73; p < 0.001). Median OS had not been reached for the nivolumab group at the time of publication, while the median OS was 10.8 months for dacarbazine. The median PFS for nivolumab was 5.1 months compared to 2.2 months for dacarbazine (HR 0.43, p < 0.001). Additionally, an improved ORR was noted for nivolumab of 40% compared to only 13.9% for those treated with dacarbazine chemotherapy. The improvements in PFS, response, and OS were seen regardless of PD-1 ligand expression in patient tumors. Interestingly, as with pembrolizumab, these studies have demonstrated responses in both ipilimumab-pretreated and ipilimumab-naïve patients, suggesting that a lack of benefit from ipilimumab does not preclude benefit from anti-PD-1 therapy.
Other approaches to interfering with the PD-1/PD-L1 interaction have been made with the development of anti-PD-L1 antibodies. A phase 1 trial of BMS-936559, a monoclonal antibody that targets PD-L1, has been reported showing activity among patients with metastatic melanoma. In a dose escalation trial, 55 patients with metastatic melanoma were included with nine objective responses (17% ORR) [50]. Among these responders were three who had a complete response. An additional 14 (27%) patients had stable disease lasting more than 24 weeks. Common side effects included infusion-related reactions (10%), diarrhea (9%), and rash (7%). Grade 3 or higher adverse events were uncommon (9%). Another anti-PD-L1 antibody (MPDL3280A) is being explored in a phase 1 trial in patients with melanoma, renal cancer, non-small-cell lung cancer, and other tumor types [51]. Results from the melanoma cohort (n = 44) were reported, showing a 29% ORR in 38 evaluable patients. The responses were durable with 10 of 11 patients continuing with therapy at the time of study report. In this group of patients, no dose-limiting adverse events were noted with common adverse events including fatigue, headache, diarrhea, and pruritus. MPDL3280A is currently being combined with vemurafenib in a phase 1 trial for patients with BRAF V600 mutant melanoma.
Adjuvant Approaches with Immune Checkpoint Inhibitors
In the adjuvant setting, the EORTC 18071 trial explored ipilimumab compared to placebo in 951 patients with stage III melanoma after wide local excision and lymph node resection [52]. Median recurrence-free survival was 26.1 vs. 17.1 months, favoring the ipilimumab group (hazard ratio 0.75; 95% CI 0.64–0.90; p = 0.0013). Overall survival results remain pending at this time; however, ipilimumab has been submitted for FDA review for use in the adjuvant setting. Ipilimumab is also being explored in two other randomized phase 3 trials. The cooperative group study E1609 is evaluating two different doses of ipilimumab, 10 mg/kg and 3 mg/kg, compared to interferon therapy in patients with resected stage IIIB, IIIC, and IV (M1a, b) melanoma (NCT01274338). This study completed accrual in 2014 and is yet to be reported. Another actively accruing trial, CheckMate-238, compares ipilimumab (at 10 mg/kg dosing) to nivolumab in patients with resected stage IIIB, IIIC, and IV melanoma (NCT02388906). Finally, two separate randomized trials evaluating pembrolizumab in the adjuvant setting are planned to be conducted through cooperative group mechanisms beginning in 2015 (EORTC, SWOG).
Combination Immune Checkpoint Inhibitors
Single-agent PD-1 inhibition with nivolumab and pembrolizumab has demonstrated higher rates of activity and tolerability compared to what has previously been seen with ipilimumab. However, the potential for the combination of a PD-1 inhibitor and ipilimumab has been highlighted in a series of phase 1, 2, and 3 trials [53–55]. In a striking phase 1 trial, the combination of ipilimumab and nivolumab in previously treated metastatic melanoma patients resulted in surprising response rates (range 21–53%) with both rapid and deep responses noted. An update on 1- and 2-year OS rates for this trial has been presented, and they are 82% and 75%, respectively, with a median OS of about 40 months [56]. Additionally, of those patients obtaining a response, the bulk of these patients is seeing depths of tumor shrinkage of >80%, which is durable. Grade 3 or 4 adverse events were noted in 53% of patients, and the most common ones were elevated lipase and AST. A phase 2 trial confirmed the efficacy of the combination compared to ipilimumab and has now been published [54]. A three-arm placebo-controlled, blinded phase 3 trial (CheckMate-067) has now been reported, further describing the efficacy and adverse event profile of the nivolumab/ipilimumab combination [55]. This study explored the front-line use of nivolumab and ipilimumab compared to nivolumab alone and ipilimumab alone in metastatic melanoma patients with a primary endpoint of PFS. The median PFS for the nivolumab/ipilimumab combination group was significantly improved compared to ipilimumab (11.5 versus 2.9 months, HR 0.42, p < 0.001) and numerically improved compared to nivolumab (6.9 months). It should be noted that the trial was not designed to evaluate statistical difference between nivolumab single arm and the combination arm. The status of PD-L1 expression on tumors was evaluated, and patients were stratified based on presence (>5% expression) or absence (<5%). There was no difference between the combination arm and nivolumab arms for patients expressing PD-L1 (median PFS of 14 months for both groups); however, in those with negative PD-L1 expression a significant difference was noted between the two groups (median PFS 11.2 months for combination and 5.3 months for nivolumab alone). As expected, there was a higher frequency of grade 3/4 events in the combination arm (55%) compared to either single-treatment arm (nivolumab: 16.3%; ipilimumab: 27.3%). It is important to note that patient toxicities in the combination arm were could be managed safely with established algorithms and that no patients died from treatment-related toxicity in the combination arm. The trial has not yet matured enough for overall survival data.
Multiple preclinical studies and a few clinical studies showed a synergistic benefit of CTLA-4 blockade and GM-CSF-secreting tumor cell vaccines [57–59]. A phase 2 randomized trial enrolled 245 previously treated unresectable stage 3 or metastatic melanoma patients to ipilimumab plus sargramostim vs. ipilimumab alone [60]. At median follow-up duration of 13 months, there was no difference in PFS; however, the median OS significantly favored the sargramostim group at 17.5 versus 12.7 months. The 1-year OS rate for the combination was 68.9% compared to 52.9% for ipilimumab alone (HR 0.64, p = 0.01). Interestingly, there was a reduction of toxicity in the combination group, particularly gastrointestinal and pulmonary adverse events. The proposed mechanism for improved efficacy that resulted in increased OS is theorized to be an improved antigen presentation with GM-CSF via recruitment of dendritic cells and macrophages. However, it is also possible that the improved toxicity profile also may have impacted outcomes.
One unanswered question in the BRAF V600 mutated subset of melanoma patients is whether to use initial therapy focused on the BRAF pathway, target immune checkpoints or utilize a combination of these approaches. There are a variety of trials seeking to answer these questions, including trials comparing upfront ipilimumab and nivolumab versus dabrafenib and trametinib followed by cross-over on progression (NCT02224781), combining nivolumab with dabrafenib and/or trametinib (NCT02357732), and combining pembrolizumab with dabrafenib and trametinib (NCT02130466).
Other Agents in Development
A variety of other approaches are being pursued for melanoma therapy that can activate the immune system in other ways. One agent that has been tested in a phase 3 trial is talimogene laherparepvec, also known as TVEC. This agent is a modified herpes simplex virus that has been designed to replicate in tumor cells and transcribe granulocyte macrophage colony-stimulating factor (GM-CSF) [61]. The virus must be directly injected into tumors where it can multiply and cause cell lysis and release of GM-CSF into the tumor microenvironment. The GM-CSF can then attract immune cells such as dendritic cells that can stimulate an anticancer immune response. TVEC has been studied in a phase 3 trial of 436 metastatic or unresectable melanoma patients who were randomized to receive either T-VEC intratumoral injections or subcutaneous GM-CSF [62]. The primary endpoint was a durable response rate with secondary endpoints of the overall response rate and overall survival. The durable response rate for TVEC was noted to be 16.3% compared to 2.1% for GM-CSF (p < 0.01). The response rate was also higher at 26.4% compared to 5.7%. A trend toward improved overall survival was noted but not statistically significant (p = 0.051). Common side effects with T-VEC included fatigue, chills, and pyrexia. TVEC has been submitted to the FDA for approval; however, final FDA approval is pending. Criticisms of the study include the GM-CSF control arm, which was given the treatment subcutaneously rather than intra-tumorally. Additionally, GM-CSF is known to have no significant activity in melanoma as a single agent. Finally, the clinical significance of these trial results compared to study results of the previously immune checkpoint agents and targeted therapies, which have shown more impressive survival outcomes, is unclear. TVEC is currently being studied in combination with pembrolizumab in advanced melanoma patients.
In addition to CTLA-4 and PD-1, there are numerous other immune checkpoints. These include activating checkpoints such as OX-40, GITR, and CD137 as well as inhibiting checkpoints such as TIM-3 and LAG-3. Antibodies to these immune checkpoint receptors have been created and are in early development in clinical trials. Positive clinical activity from these trials would certainly make combination approaches with currently available drugs an exciting challenge in the future. Novel cytokine therapy development has also been recently undertaken. Interleukin-15 (IL-15) is a cytokine that activates T-cell lymphocytes and natural killer (NK) cells. The cytokine IL-15 has been studied in a phase 1 trial in advanced melanoma and renal cell carcinoma patients [63]. Dose-limiting toxicities that were seen included hypotension, thrombocytopenia, and liver transaminase elevation. Activity was seen in two patiets with lung metastases. ALT-803 is a novel engineered compound in development that activates the IL-15 receptor, but has been shown to have improved biologic activity and longer half-life than IL-15. It is currently in a phase I dose escalation trial in patients with advanced melanoma.

Schematic diagram showing select immune checkpoint interactions between T-cells and tumor or antigen-presenting cells. There are several important receptors that are expressed on the T-lymphocyte that can either up- or downregulate T-cell activity. Depicted are select important immune checkpoint receptors and their corresponding ligands for which drug development is ongoing. Examples of immune checkpoint receptors that downregulate T-cell function are CTLA-4, PD-1, and TIM-3. Immune checkpoint receptors that upregulate T-cell function are OX-40, GITR, and CD-40. Blockade of CTLA-4 (ipilimumab) or PD-1 (nivolumab or pembrolizumab) results in the sustained activity of T-lymphocyte function and enhanced tumor cell destruction
Finally, adoptive T-cell therapy is an investigational approach to immunotherapy, which has been under study for over several decades [64]. The adoptive T-cell approach has traditionally required resection of tumor tissues and isolation of tumor-infiltrating lymphocytes (TILs). The TILs are then grown in culture in the laboratory, activated or enhanced in vitro, and given back to the patient by infusion often along with interleukin-2 therapy. While this approach has shown response rates between approximately 30–50% in small trials in melanoma patients, it has remained a very laborious treatment that can be done at only a few select centers in the world. New approaches including instilling chimeric antigen receptors on the surface of peripherally obtained lymphocytes (CAR-T cells) that target certain cancers may be a more practical method of introducing this novel cellular therapy into the clinic. These chimeric antigen receptors can be designed to recognize common proteins on the malignant cell, which thereby can activate an immune response in vivo. Studies evaluating CAR-T cells are underway in a variety of different cancer types.
Conclusion
It is currently an exciting time for metastatic melanoma clinical research, with six new drugs FDA approved in the last 4 years (Table 1). Each of these has shown single-agent activity in improving overall survival for patients with this devastating cancer. These agents are additionally being explored as adjuvant therapies for patients with high-risk resected stage III patients. For BRAF mutant melanomas, the combination of BRAF inhibitor and MEK inhibitor has become the standard approach because of the superior survival outcomes compared to single-agent BRAF inhibitors. In addition, evaluation of unique combinations of agents such as PI3K, AKT, CDK4, and ERK inhibitors may prove valuable for patients who develop resistance to BRAF/MEK inhibitors based on early work evaluating the molecular mechanisms of BRAF inhibitor resistance. Targeted approaches for non-BRAF mutant melanoma patients are also ongoing, including MEK inhibitors for patients with NRAS mutations and c-kit inhibitors for patients with these c-kit mutations. Certainly critical to the success of the targeted therapy tactic is an improved understanding of the drivers of resistance to targeted drugs, which would allow for development of further individualized molecular approaches in later lines of therapy. Immune checkpoint inhibition has been explored in parallel with molecularly targeted drugs and has shown significant improvement in patients with advanced melanoma. Ipilimumab, nivolumab, and pembrolizumab are currently approved as single agents for advanced melanoma because of their ability to extend survival outcomes. However, combinational approaches such as combining immune checkpoint inhibitors with each other, with other immunotherapies, and with molecularly targeted drugs may expand on these outcomes. Based on recent positive clinical studies, it is anticipated that nivolumab and pembrolizumab will become FDA approved in the front-line setting, and potentially the nivolumab and ipilimumab combination will also become approved. The optimal sequence and combination of targeted drugs and immunotherapies are still unknown and remain an active area of clinical research. One area of concern is the potential cost burden that these new agents may introduce for patients and society. For example, a course of ipilimumab has the potential to cost over $100,000. The other immune checkpoint inhibitors and oral targeted agents also have a similar degree of cost, particularly if patients remain on them for many months. For patients receiving no benefit high costs add an extra burden; however, for patients that achieve long-term survival these costs may be viewed through a different lens, and a positive return for the high cost is more tangible. Although many factors go into the cost of drugs, including the price of development of new agents, this issue drives the need for improved biomarkers to help select patients who would benefit from a particular agent so that patients who will not benefit can avoid both the toxicity of the drug as well as the cost.
With that said, it is difficult to deny that the advances that have been made in therapeutic development for metastatic melanoma over the last few years are quite astonishing. The sharp rise in response rates from 10% with the older drugs to >50% with newer agents has given the treating physician agents which are much more likely to palliate cancer-related symptoms and thus improve quality of life. Additionally, these new drugs have extended survival of the average patient, which over the last several decades has never been done before. Not only is the average survival of the metastatic melanoma patient improving for the first time in history, but there is a large, currently undefined subset of patients who have the potential to experience a long term survival advantage measured in years, not months. Of all things, this gives us hope, that as we progress towards an improved understanding of how to use these currently approved and upcoming drugs, we will see the elimination of this once dismal disease in our lifetime.
References
Anderson CM, Buzaid AC, Legha SS. Systemic treatments for advanced cutaneous melanoma. Oncology (Williston Park). 1995;9(11):1149–1158 (discussion 1163–1144, 1167–1148).
Atkins MB, Lotze MT, Dutcher JP, et al. High-dose recombinant interleukin 2 therapy for patients with metastatic melanoma: analysis of 270 patients treated between 1985 and 1993. J Clin Oncol. 1999;17(7):2105–16.
Manola J, Atkins M, Ibrahim J, Kirkwood J. Prognostic factors in metastatic melanoma: a pooled analysis of Eastern Cooperative Oncology Group trials. J Clin Oncol. 2000;18(22):3782–93.
Davies H, Bignell GR, Cox C, et al. Mutations of the BRAF gene in human cancer. Nature. 2002;417(6892):949–54.
Long GV, Menzies AM, Nagrial AM, et al. Prognostic and clinicopathologic associations of oncogenic BRAF in metastatic melanoma. J Clin Oncol. 2011;29(10):1239–46.
Flaherty KT, Puzanov I, Kim KB, et al. Inhibition of mutated, activated BRAF in metastatic melanoma. N Engl J Med. 2010;363(9):809–19.
Chapman PB, Hauschild A, Robert C, et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med. 2011;364(26):2507–16.
Chapman PB, Hauschild A, Robert C, et al. Updated overall survival (OS) results for BRIM-3, a phase III randomized, open-label, multicenter trial comparing BRAF inhibitor vemurafenib (vem) with dacarbazine (DTIC) in previously untreated patients with BRAFV600E-mutated melanoma. ASCO Meeting Abstracts. 2012;30(15_Suppl):8502.
Lacouture ME, Duvic M, Hauschild A, et al. Analysis of dermatologic events in vemurafenib-treated patients with melanoma. Oncologist. 2013;18(3):314–22.
Su F, Viros A, Milagre C, et al. RAS mutations in cutaneous squamous-cell carcinomas in patients treated with BRAF inhibitors. N Engl J Med. 2012;366(3):207–15.
Kefford R, Arkenau H, Brown MP, et al. Phase I/II study of GSK2118436, a selective inhibitor of oncogenic mutant BRAF kinase, in patients with metastatic melanoma and other solid tumors. ASCO Meeting Abstracts. 2010;28(15_Suppl):8503.
Trefzer U, Minor D, Ribas A, et al. BREAK-2: a phase IIA trial of the selective BRAF kinase inhibitor GSK2118436 in patients with BRAF mutation-positive (V600E/K) metastatic melanoma. Pigment Cell Melanoma Res. 2011;24(5):1020.
Hauschild A, Grob J, Demidov L, et al. Dabrafenib in BRAF-mutated metastatic melanoma: a multicentre, open-label, phase 3 randomised controlled trial. Lancet. 2012;380(9839):358–65.
Hauschild A, Grobb J, Demidov L, et al. 1092PDAN update on overall survival (OS) and follow-on therapies in break-3, a phase III, randomized trial: dabrafenib (D) vs. dacarbazine (DTIC) in patients (pts) with BRAF V600E mutation-positive metastatic melanoma (MM). Ann Oncol. 2014;25(Suppl 4):iv378.
Flaherty K, Robert C, Hersey P, et al. Improved survival with MEK inhibition in BRAF-mutated melanoma. N Engl J Med. 2012;367(2):107–14.
Schadendorf D, Flaherty KT, Hersey P. Overall survival update on METRIC, a randomized phase 3 study to assess efficacy of trametinib compared with chemotherapy in patients with BRAFV600E/K mutation-positive advanced or metastatic melanoma. Pigment Cell Melanoma Res. 2013;26(6):997.
Chapman P. Mechanisms of resistance to RAF inhibition in melanomas harboring a BRAF mutation. Am Soc Clin Oncol Educ Book. 2013;33:e80–2.
Nazarian R, Shi H, Wang Q, et al. Melanomas acquire resistance to B-RAF(V600E) inhibition by RTK or N-RAS upregulation. Nature. 2010;468(7326):973–7.
Das Thakur M, Salangsang F, Landman AS, et al. Modelling vemurafenib resistance in melanoma reveals a strategy to forestall drug resistance. Nature. 2013;494(7436):251–255.
Atefi M, von Euw E, Attar N, et al. Reversing melanoma cross-resistance to BRAF and MEK inhibitors by co-targeting the AKT/mTOR pathway. PLoS One. 2011;6(12):e28973.
Johannessen CM, Boehm JS, Kim SY, et al. COT drives resistance to RAF inhibition through MAP kinase pathway reactivation. Nature. 2010;468(7326):968–72.
Montagut C, Sharma SV, Shioda T, et al. Elevated CRAF as a potential mechanism of acquired resistance to BRAF inhibition in melanoma. Cancer Res. 2008;68(12):4853–61.
Flaherty KT, Infante JR, Daud A, et al. Combined BRAF and MEK inhibition in melanoma with BRAF V600 mutations. N Engl J Med. 2012;367(18):1694–703.
Long GV, Stroyakovskiy D, Gogas H, et al. Combined BRAF and MEK inhibition versus BRAF inhibition alone in melanoma. N Engl J Med. 2014;371(20):1877–88.
Robert C, Karaszewska B, Schachter J, et al. Improved overall survival in melanoma with combined dabrafenib and trametinib. N Engl J Med. 2015;372(1):30–9.
Ascierto PA, McArthur GA, Dreno B, et al. coBRIM: a phase 3, double-blind, placebo-controlled study of vemurafenib versus vemurafenib + cobimetinib in previously untreated BRAF (V600) mutation-positive patients with unresectable locally advanced or metastatic melanoma (NCT01689519). J Transl Med. 2015;13:2061.
Goel VK, Lazar AJ, Warneke CL, Redston MS, Haluska FG. Examination of mutations in BRAF, NRAS, and PTEN in primary cutaneous melanoma. J Invest Dermatol. 2006;126(1):154–60.
Ekedahl H, Cirenajwis H, Harbst K, et al. The clinical significance of BRAF and NRAS mutations in a clinic-based metastatic melanoma cohort. Br J Dermatol. 2013;169(5):1049–55.
Devitt B, Liu W, Salemi R, et al. Clinical outcome and pathological features associated with NRAS mutation in cutaneous melanoma. Pigment Cell Melanoma Res. 2011;24(4):666–72.
Sosman JA, Kittaneh M, Lolkema MPJK, et al. A phase 1b/2 study of LEE011 in combination with binimetinib (MEK162) in patients with NRAS-mutant melanoma: early encouraging clinical activity. ASCO Meeting Abstracts. 2014;32(15_Suppl):9009.
van Herpen C, Agarwala SS, Hauschild A, et al. LBA35overall survival and biomarker results from a phase 2 study of MEK1/2 inhibitor binimetinib (MEK162) in patients with advanced NRAS-mutant melanoma. Ann Oncol. 2014;25(Suppl 4). doi:10.1093/annonc/mdu438.43.
Carvajal RD, Sosman JA, Quevedo F, et al. Phase II study of selumetinib (sel) versus temozolomide (TMZ) in gnaq/Gna11 (Gq/11) mutant (mut) uveal melanoma (UM). ASCO Meeting Abstracts. 2013;31(15_Suppl):CRA9003.
Postow MA, Carvajal RD. Therapeutic implications of KIT in melanoma. Cancer J. 2012;18(2):137–41.
Carvajal RD, Antonescu CR, Wolchok JD, et al. KIT as a therapeutic target in metastatic melanoma. JAMA. 2011;305(22):2327–34.
Wyman K, Atkins MB, Prieto V, et al. Multicenter phase II trial of high-dose imatinib mesylate in metastatic melanoma: significant toxicity with no clinical efficacy. Cancer. 2006;106(9):2005–11.
Guo J, Si L, Kong Y, et al. Phase II, open-label, single-arm trial of imatinib mesylate in patients with metastatic melanoma harboring c-Kit mutation or amplification. J Clin Oncol. 2011;29(21):2904–9.
Hodi FS, Corless CL, Giobbie-Hurder A, et al. Imatinib for melanomas harboring mutationally activated or amplified KIT arising on mucosal, acral, and chronically sun-damaged skin. J Clin Oncol. 2013;31(26):3182–90.
Kim KB, Eton O, Davis DW, et al. Phase II trial of imatinib mesylate in patients with metastatic melanoma. Br J Cancer. 2008;99(5):734–40.
Carvajal RD, Lawrence DP, Weber JS, et al. Phase II study of nilotinib in melanoma harboring KIT alterations following progression to prior KIT inhibition. Clin Cancer Res. 2015;21(10):2289–2296.
Zang X, Allison JP. The B7 family and cancer therapy: costimulation and coinhibition. Clin Cancer Res. 2007;13(18 Pt 1):5271–9.
Hodi FS, O’Day SJ, McDermott DF, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. 2010;363(8):711–23.
Robert C, Thomas L, Bondarenko I, et al. Ipilimumab plus dacarbazine for previously untreated metastatic melanoma. N Engl J Med. 2011;364(26):2517–26.
Ribas A. Tumor immunotherapy directed at PD-1. N Engl J Med. 2012;366(26):2517–9.
Robert C, Ribas A, Wolchok JD, et al. Anti-programmed-death-receptor-1 treatment with pembrolizumab in ipilimumab-refractory advanced melanoma: a randomised dose-comparison cohort of a phase 1 trial. Lancet. 2014;384(9948):1109–17.
Ribas A, Puzanov I, Dummer R. A randomized controlled comparison of pembrolizumab and chemotherapy in patients with ipilimumab-refractory melanoma. Pigment Cell Melanoma Res. 2014;27(6):1169–241.
Robert C, Schachter J, Long G, et al. Pembrolizumab versus ipilimumab in advanced melanoma. N Engl J Med. 2015;372(26):2521–32.
Weber JS, D’Angelo SP, Minor D, et al. Nivolumab versus chemotherapy in patients with advanced melanoma who progressed after anti-CTLA-4 treatment (CheckMate 037): a randomised, controlled, open-label, phase 3 trial. Lancet Oncol. 2015;16(4):375–84.
Robert C, Long GV, Brady B, et al. Nivolumab in previously untreated melanoma without BRAF mutation. N Engl J Med. 2015;372(4):320–30.
Brahmer JR, Tykodi SS, Chow LQ, et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N Engl J Med. 2012;366(26):2455–65.
Hamid O, Sosman JA, Lawrence DP, et al. Clinical activity, safety, and biomarkers of MPDL3280A, an engineered PD-L1 antibody in patients with locally advanced or metastatic melanoma (mM). ASCO Meeting Abstracts. 2013;31(15_Suppl):9010.
Eggermont AM, Chiarion-Sileni V, Grob JJ, et al. Adjuvant ipilimumab versus placebo after complete resection of high-risk stage III melanoma (EORTC 18071): a randomised, double-blind, phase 3 trial. Lancet Oncol. 2015;16(5):522–30.
Wolchok J, Kluger H, Callahan M, et al. Nivolumab plus ipilimumab in advanced melanoma. N Engl J Med. 2013;369(2):122–33.
Postow MA, Chesney J, Pavlick AC, et al. Nivolumab and ipilimumab versus ipilimumab in untreated melanoma. N Engl J Med. 2015;372(21):2006–17.
Larkin J, Chiarion-Sileni V, Gonzalez R, et al. Combined nivolumab and ipilimumab or monotherapy in untreated melanoma. N Engl J Med. 2015;373(1):23–34.
Sznol M, Kluger HM, Callahan MK, et al. Survival, response duration, and activity by BRAF mutation (MT) status of nivolumab (NIVO, anti-PD-1, BMS-936558, ONO-4538) and ipilimumab (IPI) concurrent therapy in advanced melanoma (MEL). ASCO Meeting Abstracts. 2014;32(15_Suppl):LBA9003.
Hodi FS, Mihm MC, Soiffer RJ, et al. Biologic activity of cytotoxic T lymphocyte-associated antigen 4 antibody blockade in previously vaccinated metastatic melanoma and ovarian carcinoma patients. Proc Natl Acad Sci. 2003;100(8):4712–7.
van den Eertwegh AJ, Versluis J, van den Berg HP, et al. Combined immunotherapy with granulocyte-macrophage colony-stimulating factor-transduced allogeneic prostate cancer cells and ipilimumab in patients with metastatic castration-resistant prostate cancer: a phase 1 dose-escalation trial. Lancet Oncol. 2012;13(5):509–17.
Hodi FS, Butler M, Oble DA, et al. Immunologic and clinical effects of antibody blockade of cytotoxic T lymphocyte-associated antigen 4 in previously vaccinated cancer patients. Proc Natl Acad Sci. 2008;105(8):3005–10.
Hodi FS, Lee S, McDermott DF, et al. Ipilimumab plus sargramostim vs ipilimumab alone for treatment of metastatic melanoma: a randomized clinical trial. JAMA. 2014;312(17):1744–53.
Johnson D, Puzanov I, Kelley M. Talimogene laherparepvec (T-VEC) for the treatment of advanced melanoma. Immunotherapy. 2015;7(6):611–9.
Andtbacka R, Kaufman H, Collichio F, Amatruda T, Senzer N, Chesney J, et al. Talimogene laherparepvec improves durable response rate in patients with advanced melanoma. J Clin Oncol. 2015;33(25):2780–8. doi:10.1200/JCO.2014.58.3377.
Conlon KC, Lugli E, Welles HC, et al. Redistribution, hyperproliferation, activation of natural killer cells and CD8 T cells, and cytokine production during first-in-human clinical trial of recombinant human interleukin-15 in patients with cancer. J Clin Oncol. 2015;33(1):74–82.
Rosenberg SA, Restifo NP. Adoptive cell transfer as personalized immunotherapy for human cancer. Science. 2015;348(6230):62–8.
Acknowledgments
No funding or sponsorship was received for this study or publication of this article.
All named authors meet the International Committee of Medical Journal Editors (ICMJE) criteria for authorship for this manuscript, take responsibility for the integrity of the work as a whole, and have given final approval for the version to be published.
Conflict of interest
Dr. Lijo has no conflicts of interest. Dr. Cowey has served as a consultant, speaker and received research funding from Bristol-Myers Squibb and Genentech. He has also served as a consultant and received research funding from Merck, and Glaxosmith Kline.
Compliance with ethics guidelines
This article is based on previously conducted studies and does not involve any new studies of human or animal subjects performed by any of the authors.
Open Access
This article is distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License (http://creativecommons.org/licenses/by-nc/4.0/), which permits any noncommercial use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0), which permits use, duplication, adaptation, distribution, and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
John, L., Cowey, C.L. The Rapid Emergence of Novel Therapeutics in Advanced Malignant Melanoma. Dermatol Ther (Heidelb) 5, 151–169 (2015). https://doi.org/10.1007/s13555-015-0080-7
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13555-015-0080-7