
Abstract
The therapy of psoriatic arthritis (PsA) has blossomed in the past decade. Inhibition of tumor necrosis factor (TNF) has been at the fore of this approach and has paved the way for the investigation of many other potential pro-inflammatory and signaling pathways. Most of the initial studies of TNF inhibitors in PsA have been conducted in specific populations, largely focusing on those with established, peripheral joint disease. That said, in excess of 10 years’ worth of real world clinical experience has led to increased confidence in the wider use of these agents. We are now faced with an exciting time of discovery of many new molecules; these not only include new, large protein biological agents, but also smaller synthetic chemical molecules, many of which can be administered orally. Those currently under development are discussed within this article. Whilst there is scarce data about their real world efficacy and safety profile, it is evident that the therapeutic armamentarium for treating PsA will greatly increase in the foreseeable future and this is anticipated to improve patient outcomes.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The optimal management of psoriatic arthritis (PsA) entails the adequate suppression of aberrant inflammatory processes that give rise to the clinical PsA phenotype of joint stiffness, pain, tenderness, and swelling. Categorized as one of the seronegative spondyloarthropathies, PsA can manifest as enthesitis, dactylitis, synovitis, arthritis, and/or axial inflammation, either in isolation or in any combination, although animal models and clinical studies suggest that enthesitis may be the primary lesion [1, 2]. The magnitude and chronicity of psoriatic joint disease and the consequential physical and financial burden, has, in recent years, prioritized PsA on the research agenda [3].
Compared to rheumatoid arthritis (RA), the optimal management of PsA still lags behind considerably. Prior to the advent of biological therapy for RA, disease-modifying anti-rheumatic drugs (DMARDs; methotrexate or sulfasalazine), were most well established option, initially as monotherapy and then in combination as required. The superior efficacy of the biological agents, often anchored to methotrexate therapy, are well established in preventing joint damage and minimizing long-term disability in RA [4–6].
However, for a number of reasons, making progress in the therapeutic field for PsA has been more complicated. Historically, PsA was viewed as a less prevalent or severe disease. In addition, it is considerably more heterogeneous in its evolution and manifestations compared to RA. To date, a reliable serum biomarker, such as the anti-citrullinated protein antibody in RA, does not exist for psoriatic joint disease detection, and this is likely to have negatively impacted on the generation of good-quality, robust clinical trials for PsA. Whilst conventional DMARDs have been employed as the mainstay of PsA therapy for decades, there is a surprising paucity of data to support their clinical efficacy, with clinical experience generally taking precedence over hard evidence. In addition, the majority of trials in PsA have focused on the treatment of peripheral arthritis in polyarticular disease [7], and have largely ignored those patients with primarily axial disease and other subgroups including oligoarthritis.
The current British Society of Rheumatology guidelines for the treatment of PsA were published in 2005, when anti-TNF therapy was not widely available [8]. Only one TNF inhibitor was licensed for active PsA in the UK at the time of publication (etanercept), and only one other anti-TNF therapy demonstrated evidence in PsA (infliximab). There are now four TNF inhibitors with proven efficacy in PsA, established by large, good-quality clinical trials, and a number of novel compounds in development which if prove to be safe, may translate into promising additions to the biological armamentarium against PsA [9–13]. This article assimilates all of the relevant data concerning both old and new biologic molecules, and provides an evidence-based review of the current and emerging therapeutic options for PsA. However, rather than systematically describe and appraise each clinical study in detail, the pertinent commonalities shall be summarized, and relevant differences highlighted. At the current time, patients within the UK must of course fulfill the criteria for biological therapy as stipulated by the National Institute for Health and Clinical Excellence (NICE); that is, they must have failed to improve on or tolerate first-line, disease-modifying agents including methotrexate, leflunomide, and sulfasalazine, either alone or in combination.
Anti-TNF Agents in Psoriatic Arthritis
Many pro-inflammatory cytokines have been identified in the pathogenesis of PsA, but amongst these, TNF-alpha exerts a key pro-inflammatory role [14]. Increased levels of TNF-alpha have been observed in skin, synovial fluid, and synovial tissue from patients with PsA [15] and allelic polymorphisms in the promoter region for TNF-alpha have been shown to correlate with certain aspects of the disease [16]. TNF inhibitors have demonstrated efficacy in both the skin and joint manifestations of psoriatic disease, as well as preventing radiographic damage [9–13, 17]. In the UK, four anti-TNF agents are licensed for the treatment of PsA—infliximab, etanercept, adalimumab, and golimumab. A fifth agent, certolizumab, has also shown promising efficacy in clinical trials [18, 19]. All trials encompass PsA as a single entity, with no primary endpoint data reporting results based on the PsA subtype.
Almost universally, PsA studies have sought to recruit patients with a predominantly peripheral distribution of joint disease. However, it is estimated that up to 40% of patients with PsA will develop some disease within the axial skeleton. Again, no specific data evaluating the effects of TNF inhibitors at this site has been forthcoming, and treatment guidance is based entirely upon data extrapolated from studies into other seronegative spondyloarthropathies, particularly ankylosing spondylitis (AS) [20, 21]. In AS, TNF inhibitors are effective at suppressing the clinical and imaging markers of inflammation [22] and it is this effect that confers the likelihood of benefit in axial PsA—the suppression of associated bone formation (ankylosis) in AS has yet to be determined [23].
The licensed TNF inhibitors each have a distinct structure and target TNF-alpha in slightly different ways, but essentially potentiate the same overall anti-inflammatory effect. Infliximab is a chimeric (mouse/human) monoclonal IgG1 antibody, adalimumab and golimumab are recombinant fully human IgG1 monoclonal antibodies, and etanercept is a 75 kDa IgG1 fusion protein—the former three bind directly to both the circulating and membrane-bound TNF-alpha molecule, whereas etanercept reversibly binds soluble, circulating TNF-alpha [24]. Despite these structural differences, the different TNF inhibitors exhibit comparable efficacy on the joints [25, 26], although no direct head-to-head trials have been conducted. The four licensed agents have demonstrated efficacy at 12–16 weeks for PsA response criteria (PsARC) response, American College of Rheumatology (ACR) 20, 50, and 70 response, and Psoriasis Area Severity Index (PASI) response [9–13, 27–29]. Of note, the onset of clinical benefit of the TNF receptor construct on etanercept has been reported to be of slower onset in those with more extensive skin disease than for the monoclonal antibodies [9], although this was challenged by the outcomes of the more recent PRESTA trial [30]. Again, there are no direct head-to-head comparisons available. This slower onset of action is unlikely to pose a significant problem in the rheumatologic arena, where many PsA cases have a low PASI score (and therefore less cosmetic urgency to respond rapidly) and where current data leads us to believe that no association between PASI score and joint disease exists [31].
In addition to clinical features, subjective improvements in the Health Assessment Questionnaire (HAQ) are reported to be greatest with adalimumab [10, 27] and infliximab [11, 12] at 12 weeks, and with adalimumab [10], etanercept [28], golimumab [13] and infliximab [12] at 24 weeks, when compared to non-TNF-alpha biologic agents, such as ustekinumab (UST) [32] alefacept [33], and placebo. Specifically looking at the TNF-alpha inhibitors, a more recent indirect comparison meta-analysis measured the relative risks for the PsA response criteria (PsARC) and mean differences for improvements from baseline for the HAQ for PsARC responders and non-responders. Etanercept and infliximab yielded the largest mean difference in HAQ score among PsARC responders, while for non-responders, etanercept, infliximab and golimumab yielded similar mean differences and adalimumab a notably lower mean difference [34].
The key pathogenic importance of enthesitis and dactylitis in PsA has been established [2, 35, 36], and the original studies into TNF inhibitors suggested superiority of some agents (infliximab [11, 12]) over others. In total, seven randomized controlled trials have published data on enthesitis and dactylitis as secondary endpoints [10–13, 27, 32, 37]. In addition to infliximab [11, 12], golimumab [13] (a second generation agent licensed for PsA), has also shown significant benefits. Secondary endpoint data for etanercept (PRESTA trial) demonstrate a significant decrease in enthesitis (−66.0%, week 12; −75.0%, week 24) and dactylitis (−74.3%, week 12; −82.2%, week 24) [37]. However, it is worth noting that the PRESTA trial did not include a placebo arm.
The ADEPT trial reported efficacy of adalimumab on enthesitis and dactylitis as secondary endpoints [10]. Whilst the mean changes were greater in the treatment group over placebo [10] and these responses were maintained throughout 2 years of therapy [38], these changes were not statistically significant at any time point [10, 38].
These seven trials also include data for the anti-IL-12/23 agent ustekinumab [32], although this is only licensed at present for the treatment of psoriasis (PsO). Of these studies, they have all used different outcome measures, with only a proportion of patients having documented baseline dactylitis or enthesitis. However, research aside, it is seemingly evident from clinical experience that enthesitis and dactylitis respond well to TNF inhibitors, irrespective of the agent employed. Early intervention to prevent progressive joint destruction, pain and disability has been widely embraced in the treatment of RA [39, 40], and there is scope to adopt such an approach in early PsA, especially in patients who require a biologic agent for their skin disease.
The efficacy of TNF inhibitors in preventing bone destruction in PsA appears to be independent of the need for combination with DMARDs. This is not true for RA, where anti-TNF monotherapy does not provide the same benefit [40–46]. Often, a DMARD agent is required in addition to a TNF inhibitor if any attempt is to be made at ceasing joint erosion in RA. It is unclear whether the tendency to erosion repair seen in PsA treated with anti-TNF monotherapy is reflective of a distinct disease process that is intrinsically sensitive to the suppression of inflammation, or if it represents a disease-associated/phenotypic process of new bone formation, or both.
Of note, while anti-TNF monotherapy may be efficacious in PsA, patients are still prone to a subsequent reduction in efficacy or frank treatment failure after a period of time [47]. From studies of TNF inhibitors in RA and inflammatory bowel disease (IBD), in addition to improving initial therapeutic efficacy, concurrent administration of methotrexate has additive benefits in that it may lessen the propensity for neutralizing or anti-drug antibody generation [48]. A reduction in such antibodies can retain the efficacy of an agent in the longer term, as a result of fewer side effects that may necessitate withdrawal (e.g. allergic reactions) or loss of effect through neutralization of the monoclonal antibody. Whether this is the case in PsA awaits further investigation: to date, there are no data showing superiority of TNF inhibitors in combination with DMARD versus anti-TNF-alpha monotherapy [10, 12, 17]. Of note, in all of the important trials of TNF inhibitors in PsA, methotrexate was allowed, but not required, and approximately half of these patients were treated with anti-TNF monotherapy. The data for patients receiving and not receiving concurrent methotrexate was comparable [49].
Of course, based on available evidence, there are certain specific circumstances that may lead the clinician to choose one TNF inhibitor over another. Where a patient has severe PsO, an agent with dual efficacy and availability should be selected. In theory, this should immediately exclude golimumab, which is not, as yet, licensed in the treatment of PsO. However, where it is used primarily for PsA, in our experience, a substantial and meaningful improvement in PASI is repeatedly seen, and it our experience that many rheumatologists are confident in prescribing golimumab where treatment for skin disease is a priority in addition to PsA.
It has repeatedly been shown that infliximab has superior outcomes over etanercept and adalimumab in terms of joint, functional status and rapid skin clearance [50] although for this very reason, it is often not chosen first line and is typically reserved for more recalcitrant and severe PsO (PASI >20). In addition, infliximab is often used second line as a consequence of its less convenient mode of administration (hospital-based infusion). Of adalimumab and etanercept, the data attempting to demonstrate which is superior for skin and joint disease is conflicting, and is rarely statistically significant [50]. As mentioned previously, there are data to suggest that etanercept may be slower to act [9], and thus adalimumab may be the most logical first-line choice of TNF-alpha inhibitor for patients requiring treatment for both PsO and PsA. However, over a prolonged treatment course (as the majority of patients will need), there are no robust data to refute that etanercept will reach equivalence with adalimumab in terms of achieving PASI 75, PASI 90 and ACR 20 [26]; therefore the urgency to achieve these responses should be tailored to the individual.
There is a greater body of evidence to show that etanercept is not efficacious in those with IBD, both in the induction and maintenance of remission [51, 52] and it no longer features in the most current Cochrane Systematic Review (2008), which does support the use of infliximab, adalimumab, and certolizumab [53]. As the latter is not currently licensed for use in PsA, either adalimumab or infliximab are recommended as treatment for patients with both IBD and PsA. From a practicality perspective, adalimumab may be more convenient, being self-administered at home.
What to Do When TNF-Inhibition Fails
The real world use of anti-TNF agents is associated with good drug retention in the short term. Large scale data from the Danish DANBIO registry showed that increased levels of C-reactive protein (CRP) at baseline were associated with both good treatment responses and can serve as a predictor of longer term drug retention [25]. This may be of clinical value in selecting cases with a greater burden of inflammation, which are most likely to benefit from treatment with TNF-alpha inhibitors.
TNF-inhibition irrevocably provides a powerful clinical benefit in terms of the signs and symptoms of PsA and can halt the progression of erosion at a population level in the small joints [54]. However, there is a paucity of evidence to confirm that these agents retard the progressive, structural changes seen in PsA and the subsequent joint fusion that occurs most perceptibly in the axial skeleton; this suggests that pathways involving cytokines other than TNF are crucial in new bone formation.
Whilst the investigation of many non-anti-TNF molecules often includes patients who have failed TNF-inhibition, it is worth noting that to date, there are no published, completed, randomized controlled trials in PsA that solely include these patients. As such, there is an unmet need to provide conclusive evidence for a clear management strategy for this patient group.
ANTI IL-12/IL-23 in Psoriatic Arthritis
Interleukin-23 (IL-23) receptor polymorphisms and IL-12B (p40 IL-12 and IL-23 subunit) polymorphisms have been reported in PsO and PsA [55]. This has been supported by the detection of elevated serum concentrations of IL-23 in spondyloarthropathy patients. A recent publication has shown that, in mice, IL-23 promotes a pathology that resembles spondyloarthritis (including new entheseal bone formation and aortic root inflammation) by acting on a previously unidentified subset of innate-like T cells that reside at the enthesis [1]. Collectively these findings provide robust genetic and molecular evidence for the key role of IL-23 in PsA based, entheseal driven pathology.
Ustekinumab, an anti-IL-12/IL-23 p40 subunit human monoclonal antibody, is licensed for the treatment of PsO, and exhibits impressive reduction in PASI scores in the vast majority of patients. Efficacy in PsA has been investigated in one active crossover, phase II placebo controlled trial of 146 patients, and demonstrated moderate results (42% achieved ACR20 at week 12, compared with 14% of those receiving placebo) [32]. Whilst it should be noted that response outcomes were lower than those reported for TNF inhibitors, it is also worth taking into account that many of these patients had previously failed on the latter. Extrapolating from the more substantial experience of using biologics in RA, prior anti-TNF failures are generally associated with poorer response rates to all forms of treatment, including different biologic agents, and thus negative selection bias may have played a role in this initial trial.
The results from the follow-on phase III (PSUMMIT1) study were reported at the 2012 European League Against Rheumatism (EULAR) Annual Congress, with similar results at a later end point of 24 weeks (Table 1) [56]. Of relevance, significant improvements in enthesitis and dactylitis were reported in the ustekinumab group. This was supported by week 52 data from PSUMMIT1, presented later in the year at the 2012 ACR Annual Conference (Table 1) [57]. Further detail was provided about the effects on enthesitis and dactylitis. At week 24, median changes in enthesitis and dactylitis scores were significantly higher than those patients seen for patients receiving placebo (Table 2; P < 0.001 for all comparisons). Improvements in enthesitis and dactylitis scores continue through to week 52 (Table 2).
The results of a second phase III RCT (PSUMMIT II) were also released at ACR 2012 [58]. PSUMMIT II recruited 312 patients, 180 of whom were TNF-alpha experienced. In this subgroup, significantly higher numbers of patients receiving ustekinumab achieved ACR20 responses at the primary endpoint (week 24) compared with placebo (Table 1). For all participants, including those who were biologic naive, significantly greater proportions achieved ACR50 at week 24 compared with placebo, although the improvement failed to reach significance for ACR70 (Table 1).
The positive experience with monoclonal antibody inhibition of the IL-12/IL-23 pathway raised the prospect of using small orally active molecules to attain the same feat. Successful trials with antibodies directed against IL-12/IL-23 in psoriatic disease, and the recent data published into the key role of IL-23 in driving enthesitis has supported the quest to find other molecules that can also inhibit IL-12/IL-23 pathways. Apilimod is an orally administrated small molecule that was developed from a novel triazine derivative identified through high-throughput IL-12 inhibitor screening [59]. It selectively and potently inhibits IL-12 and IL-23 production through the inhibition of transcription of both p35 and p40 genes and has shown clear dose–response reduction in the production/expression of IL-12/IL-23, the number of infiltrating immune cells and the clinical measures of PASI and PGA in phase I PsO studies. However, clinically optimal drug levels were associated with CNS-related adverse events, preventing the molecule from progressing to phase III trials. Similar problems were experienced in the phase-IIa randomized controlled trial of apilimod in patients with RA [60]. Whilst an orally available small molecule would provide superior convenience and cost effectiveness, to date, no apilimod derivatives with improved safety and pharmacokinetics have been forthcoming to the market.
Anti-IL-17 in Psoriatic Arthritis
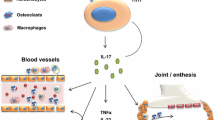
A novel cytokine target is IL-17, produced by both innate and adaptive immune cells including TH17 cells, which are induced by IL-23. Currently, there are three monoclonal antibodies that target IL-17 under investigation—ixekizumab (formerly LY2439821), secukinumab (formerly AIN457), and brodalumab (formerly AMG827). Initial investigation has focused primarily on the dermatological manifestations of psoriatic disease.
Of the many members of the IL-17 cytokine family, both IL-17A and IL-17F are expressed at elevated levels in psoriatic skin [61]. They bind as homodimers or heterodimers to the IL-17 receptor (IL-17R), a heterodimer of IL-17RA and IL-17RC subunits. IL-17RA is highly expressed on keratinocytes and in psoriatic skin—improvements induced by anti-IL-17A therapies further supports the hypothesis that IL-17 is a co-conspirator alongside many other cytokines in the pathogenesis of PsO. Brodalumab is a human IgG2 monoclonal antibody which prevents binding and biological activity of multiple members of the IL-17 cytokine family (IL-17A, IL-17C, IL-17E, IL-17A/F, IL-17F) though high affinity binding and antagonism of the IL-17RA receptor, and in both the phase I and II studies has demonstrated rapid, dose-dependent improvement in PASI scores. Further larger scale studies are required to confirm this effect.
Ixekizumab is a humanized IgG4-type monoclonal antibody that rapidly neutralizes IL-17, leading to improvements both in clinical measures of disease and histopathologic features in lesional skin (i.e. reduced acanthosis, hyperkeratosis and dermal lymphocytic infiltration). These changes are associated with a significant down-modulation of a broad array of genes in the skin from multiple inflammatory pathways. Similar changes were demonstrated in a trial exploring the efficacy of another monoclonal antibody—secukinumab—directed against IL-17, and reductions in PASI score were observed after just one dose [62]. Secukinumab differs structurally from ixekizumab in that it is a fully human monoclonal antibody of IgG1 kappa isotype.
In addition to efficacy in PsO, ixekizumab [63] and secukinumab [62] have demonstrated some worth in the management of RA. Efficacy in inflammatory disorders other than PsO is to be expected, as IL-17 acts as an effector cytokine much like TNF-alpha, and virtually all cell types have been demonstrated to have a biologic response to IL-17. IL-17 can synergize with other pro-inflammatory cytokines to stimulate release of additional pro-inflammatory cytokines and chemokines, nitric oxide, and matrix metalloproteinases. Larger scale clinical trials are required to ascertain the safety and efficacy profile not only in RA but also in many other inflammatory and autoimmune diseases.
To date, only one small proof-of-concept study has investigated IL-17A as a target for the treatment of PsA. McInnes et al. [64] randomized 42 patients to two injections of secukinumab (given at 3-week intervals) or placebo (2:1 randomization). The trial failed to meet the primary endpoint of ACR20 at week 6 (39% active group vs. 23% placebo). At week 28, the ACR20 response rate in the active treatment arm was 43%, suggesting that the week 6 response rate was maintained at 28 weeks. Again, it is worth noting that many of the participants had previously failed TNF-inhibition therapy, which may prejudice the outcome. Coupled with the brief treatment course, no firm conclusions can be made from these data about the true efficacy of secukinumab in PsA. Interestingly, post hoc analysis of data from a more recent phase II study of secukinumab in PsA by the same authors demonstrated a week 6 ACR20 response of 10% in those previously exposed to TNF inhibitors, compared to 62% in those who were biologic naïve [65]. Whilst the numbers in the latter study were small, it will be interesting to see if this observation is repeated in larger cohorts. Overall, the proportion of patients in these studies demonstrating sustained improvements in clinical scores are encouraging and these support the rationale for larger clinical trials of IL-17A monoclonal antibodies in the spondyloarthropathies.
Other Biologic Agents
Leucocyte-Function-Associated Antigen 3 (LFA-3)/CD2 Blocker
Alefacept was the first US Food and Drug Administration (FDA) sanctioned biologic agent for PsO in 2003 [66]. Alefacept is a human fusion protein with a dual mechanism of action—firstly, it inhibits T-cell activation by binding CD2 on T cells and thus inhibiting leukocyte function antigen-3/CD2 interaction with antigen-presenting cells [67]. Secondly, alefacept also bridges between CD2 on target lymphocytes and immunoglobulin Fc receptors on natural killer cells. CD2 expression is higher on memory effector than naive T cells. By binding CD2 on memory T cells and interacting with CD16 receptors on natural killer cells and macrophages, alefacept induces selective apoptosis of CD4 memory effector cells, whilst largely sparing naive T cells [67]. However, the former action necessitates close monitoring of the CD4 count to ensure that it does not drop below 250 cells/mm3.
Whilst demonstrating efficacy in moderate-to-severe PsO, alefacept was rapidly superseded by the TNF inhibitors. However, as for most biologics, no randomized controlled trials have directly compared the efficacy of alefacept with the other agents. In 2008, Brimhall et al. [68] performed a quantitative meta-analysis of available randomized controlled trials of four biologic agents: alefacept, efalizumab (now withdrawn), etanercept, and infliximab. Across all trials, efficacy was measured by achievement of PASI 75 after 10–14 weeks of treatment—the study showed that all agents were efficacious for improving PsO, though alefacept was the least effective of the agents studied [68]. Pooled relative risk of achieving PASI 75 was 4, 7, 12, and 19 for alefacept, efalizumab, etanercept, and infliximab, respectively, compared with placebo. The corresponding numbers needed to treat were 8, 4, 3, and 2. Alefacept is also slower to act than TNF inhibitors for most PsO patients [68].
Two studies have assessed the role of alefacept in PsA [33, 69]. In the first, 185 patients were randomized 2:1 to receive either 15 mg intramuscular (IM) alefacept with methotrexate (MTX) or placebo and MTX weekly for 12 weeks. ACR20 response at week 24 was 54% for alefacept + MTX versus 23% for placebo + MTX [33]. Response rates did not differ according to the duration of prior treatment with MTX.
In the second study, 185 patients with active PsA despite at least 3 months treatment with MTX were similarly randomized to concurrently receive at least eight once-weekly injections of 15 mg IM alefacept or placebo (double-blinded), followed by a 12-week observational period; 54% achieved ACR20 with alefacept + MTX at week 24, versus 23% of placebo + MTX [69]. All eligible participants (160 in total) were then entered into an extension phase and received open-label treatment with alefacept (15 mg IM weekly) + MTX for 12 weeks. At week 24 of the extension phase, ACR20 reached 55% and 51% for the two initial groups, respectively. Amongst patients who received alefacept plus MTX in both phases of the study, the proportion achieving ACR50 increased with the additional course of alefacept from 17% to 34%, and achieving ACR70 from 7% to 12% [69].
Whilst these trials have demonstrated some efficacy in PsA and a favorable safety profile, the emergence of more efficacious molecules means that alefacept is now unlikely to gain a dominant status in the hierarchical management of PsA.
CD28 Receptor Inhibitors
Full antigen-induced activation of naïve T cells requires two discrete signals from the antigen-presenting cell. Antigen is presented to the T-cell receptor in the context of a major histocompatibility complex molecule, but full activation occurs only when the binding of CD80 or CD86 to the CD28 molecule on the T cell produces a secondary co-stimulatory signal. After activation, the T cell expresses CTLA-4, which competes with CD28 for binding to CD80 or CD86, leading to homeostatic down-modulation of activated T cells.
Abatacept is a recombinant, fully human fusion protein that consists of the extracellular domain of CTLA-4, linked to a modified Fc potion of human IgG1. It selectively binds to the CD80 or CD86 receptor on the antigen-presenting cell, blocking the second signal T-cell activation of the CD28 receptor and thus decreases serum levels of cytokines and proteins implicated in the pathogenesis of psoriatic and other inflammatory diseases [70]. Administered by monthly infusion, abatacept is licensed in Europe and the United States for the treatment of RA in adult patients with an inadequate response to DMARDs or TNF inhibitors. In 2010, it was also approved in Europe for moderate-to-severe juvenile idiopathic arthritis in children aged 6 and over.
A phase I trial has shown good clinical efficacy for abatacept in PsO, with a reduction in intralesional T-cell populations [71]. These observations of abatacept in PsO and other inflammatory arthritides spurred the development of trials in PsA. Initial case reports in PsA of two patients who previously failed TNF inhibitors described significant improvement in clinical signs and symptoms of PsA after regular treatment with abatacept [72, 73]. Formal investigation by Mease et al. [70, 74] confirmed a significant ACR20 response in patients randomized to 10 mg/kg abatacept compared to placebo (48% vs. 19%, P = 0.006), but no significant difference in those treated with 3 mg/kg abatacept. Magnetic resonance image assessment of synovitis and psoriatic target lesion scores also improved with abatacept. Unfortunately, TNF failures did not respond as well as others in the trial (ACR20 31% vs. 56%), as seen in many studies of alternative biologics following TNF failure.
Because abatacept was the first therapy targeting the inhibition of co-stimulatory signals to prevent T-cell activation, its use in early disease [75] and in biologic-naïve patients with active RA [76, 77] has generated particular interest and investigation [78–81]. These data may support the investigation of abatacept in biologic-naïve patients with early inflammatory joint disease who have had an inadequate response to MTX. However, it is worth noting that patients included in these studies were exposed to concomitant corticosteroids [76, 77].
Small Molecules in Psoriatic Arthritis
In contrast to biologics, which target soluble extracellular cytokine or cellular receptors, small molecule inhibitors target intracellular enzymes within a signaling pathway, e.g., tyrosine kinases. In essence, this can make it more difficult to anticipate their complete biological effects and may cause potential for unintended side effects, as these molecules will often interact with the same enzymes but in non-targeted cells involved other biological processes. However, if this can be overcome, small molecule inhibitors have several advantages over biologic agents in that they can be administered orally, and as synthetic compounds, they are comparatively inexpensive to manufacture.
Protein Kinase C Inhibitors
The protein kinase C (PKC) family consists of 10 isoenzymes and each play key roles in cellular signaling, migration, survival and death [82]. Most isoforms are ubiquitously expressed, except PKCγ and PKCθ. PKCγ is exclusively found within the brain, whilst high protein levels of PKCθ are seen mostly in hematopoietic cells and skeletal muscle [83].
PKCθ (along with PKCβ and PKCδ) are functionally important for T and B cell signaling [83, 84]. PKCθ is central to T-cell activation as it is the only isoenzyme that is selectively translocated to the T-cell/antigen-presenting cell contact site immediately after cell-to-cell interaction [85]. Furthermore, PKCθ is essential for IL-2 production, which is required for T-cell proliferation. PKCα in T cells is required for proliferation [86], in addition to IFNγ and IL-17 production. PKCβ is a prerequisite for B cell antigen receptor function, and PKCδ for the induction of tolerance [87]. Identification of specific PKC isoenzymes in T and B cells has promoted their attractiveness as therapeutic targets for inflammatory and autoimmune diseases [88].
To date, only one PKC inhibitor—sotrastaurin (AEB071)—is in the exploratory phase of drug development for autoimmune diseases [89]. Sotrastaurin has a strong inhibitory activity on three PKC isoforms—PKCθ, PKCα and PKCβ, and weaker activity on PKCδ, PKCε, and PKCη, meaning that in addition to T-cell activity, it has inhibitory effects on several other cells [82]. It affects more than 200 kinases, including those important for early T-cell activation such as Lck and ZAP-70. Phase II trial data have shown promising efficacy in PsO and transplant rejection [90]. The proven potent inhibition of IL-17 by sotrastaurin makes this molecule a potential future therapeutic target in PsA [91].
Janus Kinase Inhibitors
Another family of kinases commanding interest in PsO are the Janus kinases (JAK), of which there are four members—JAK1, JAK2, JAK3, and TYK2 [92]. These enzymes, which are expressed primarily in immune and hematopoietic cell lineages, form part of the signaling apparatus used by receptors for various cytokines and growth factors. When such receptors are engaged by their specific ligands, JAKs phosphorylate and thus activate members of the signal transducer and activator of transcription (STAT) family [93]. There are seven STATs (STAT 1, 2, 3, 4, 5a, 5b, and 6), each with a variety of distinct effects on gene transcription in cells of the immune system that are critical in processes such as lymphocyte differentiation, immune regulation and inflammation [94].
Members of the JAK family can combine to form homodimers or heterodimers; these unique pairings give rise to the signaling of specific cytokines. For example, IL-12 and IL-23 have been reported to signal through JAK2–TYK2 heterodimers [92], thus targeting of this pathway would be expected to produce similar therapeutic efficacy to ustekinumab [95]. JAK inhibitors also inhibit signaling from many other cytokines, including IL-17 (which activates multiple JAKs via IL-17R) [96], IL-20 [97], IL-22 (JAK1), and IFNγ (JAK1/2 heterodimers) [92]. JAK3 specifically transduces signals from IL-2, IL-7, IL-15, and IL-21, which are integral to lymphocyte function and survival, and inhibition of their signaling may result in modulation of multiple aspects of the immune response [98]. JAK inhibition therefore has the potential to positively impact on many inflammatory disorders.
Data from phase I and II studies has established that cytokine signaling through the JAK pathway is an important component of the pathogenesis of both PsO and RA, and promising efficacy has been achieved with a compound now known as tofacitinib (CP-690550) in both disorders [99–101]. First described in 2003, it was initially described as a JAK3 inhibitor that could prevent allograft rejection [102]. However, it is now considered to powerfully inhibit both JAK1 and JAK3 (which can function as signaling heterodimers), and to a much lesser extent, JAK2 [103]. The reported success in phase III trials means that it has recently been licensed for treatment of RA in the US [100]. The response to tofacitinib of coincidental PsO in patients with inflammatory RA provides a logical argument for investigation of efficacy in PsA, and is likely that trial data will soon become available in this domain.
Therapeutic inhibition of more selective JAK inhibitors is under investigation. The most extensively studied is ruxolitinib (INCB28050), a selective JAK1/JAK2 inhibitor that has been investigated primarily in myelofibrosis. Latterly, a topical preparation of ruxolitinib has been developed for use in the treatment of PsO, and in a phase-IIb proof-of-concept study of 29 patients, has been reported to improve mean total lesion scores (approximately 53% reduction) and PGA at the 1 and 1.5% concentrations after 28 days of continuous use [93, 104]. However, no firm conclusions can be drawn from this small study, and the high placebo response rate (32% reduction in mean total lesion scores) may simply reflect improved compliance with a topical regimen during the trial.
In terms of inflammatory arthritis, significant efficacy (as assessed by improvements in clinical, histologic, and radiographic signs of disease), has been achieved in the rat adjuvant arthritis model, with doses of ruxolitinib providing partial and/or periodic inhibition of JAK1/JAK2 and no inhibition of JAK3 [105]. Diminution of inflammatory Th1 and Th17 associated cytokine mRNA levels were observed in the draining lymph nodes of treated rats. Ruxolitinib was also effective in multiple murine models of arthritis, with no evidence of suppression of humoral immunity or adverse hematological effects [105]. As a result, clinical evaluation of ruxolitinib for RA is currently underway in humans.
Another orally active small molecule undergoing extensive investigation in a number of inflammatory disorders is ASP015K. This molecule selectively targets JAK1/JAK3 and in a 6-week phase-IIa POC study of patients with PsO, ASP015K was well tolerated and demonstrated dose-dependent improvements in PASI change from baseline [106]. ASP015K is currently being investigated in three phase-IIb studies in patients with RA in the United States, Europe and Japan, with no efficacy data released to date.
A more selective small molecule—CEP33779—which selectively targets only JAK2, has shown efficacy in two mouse models of RA by inhibiting the transduction of signals for several essential pro-inflammatory cytokines, including IL-6, IFNγ, and IL-12 [107]. It has been proposed that use of a selective, rather than pan-JAK inhibitor, avoids the potential complications of immunosuppression, whilst targeting critical signaling pathways involved in autoimmune disease progression. Further safety data are needed on all agents before this conclusion can be definitively drawn.
Phosphodiesterase Inhibitors
Phosphodiesterase-4 plays a key role in degrading cyclic-AMP in cells—inhibition leads to diminished T-cell secretion of pro-inflammatory cytokines, including TNF-alpha, IFN-γ, PDE-4, nitric oxide synthase, IL-2, IL-17, and IL-23 and blocks the degradation of cAMP. Apremilast, a phosphodiesterase-4 inhibitor, is currently in phase III trials in PsO, AS, and PsA. Positive results have been reported in PsO; in one phase-IIb, double-blind, four-arm, randomized controlled study, 352 patients received either 30 mg, 20 mg, or 10 mg oral apremilast twice daily or placebo with 41% of the active treatment group achieving PASI 75 compared to just 6% in the placebo arm [108].
In a similar phase II study of 204 patients with PsA, a modest but significant improvement in ACR20 was achieved at both 20 mg twice daily and 40 mg once daily doses. A significant difference was not seen in ACR70, with very few patients achieving such a marked improvement in disease activity [109]. Preliminary phase III data for apremilast from the PALACE 1 (Psoriatic Arthritis Long-Term Assessment of Clinical Efficacy) study in active PsA was recently presented at the 2012 ACR annual meeting. A primary endpoint of ACR20 at week 16 was achieved in 31.3% (P = 0.0140) of patients receiving apremilast 20 mg twice daily and 41.0% (P < 0.0001) receiving apremilast 30 mg twice daily compared with 19.4% patients receiving placebo. Adverse events included gastrointestinal upset, headache, and upper respiratory tract infections. Most were mild or moderate in severity and necessitated study discontinuation in up to 7%.
TNF Antagonist Gene Therapy
A recombinant adeno-associated virus (rAAV) serotype 2-based vector containing human TNF-immunoglobulin (IgG1) Fc fusion gene (rAAV-TNFR:Fc) has been developed for clinical use [110]. Once injected into the affected joint(s), the DNA coding for a therapeutic protein is incorporated into native tissue cells by the process of gene transfer. The use of a vector is required to facilitate uptake of the single-stranded DNA by cells within the joint and incorporate it into the genome. Subsequent transcription/translocation should then provide sustained concentrations of the therapeutic protein within the joint [110]. rAAV-TNFR:Fc is based on an adeno-associated virus—a naturally occurring, non-pathogenic, non-integrating and non-replicating virus that depends on a helper virus for replication [111].
To date, one phase I/II study of patients with inflammatory arthritis (including PsA) treated with rAAV-TNFR:Fc has been published [112]—127 patients were randomized to receive intra-articular injections of escalating dose concentrations of the gene, or placebo (3:1 randomization) into a single target joint. Injection site reactions occurred in 20% and were dose-related. Septic arthritis developed in one patient 15 weeks after administration of rAAV-TNFR:Fc, which was deemed ‘probably related’ to the gene therapy, due to the increased risk of infection caused by expression of TNFR:Fc protein in the joint. In terms of efficacy, patient reported outcome measures (global visual analogue scores) yielded a greater difference between drug and placebo than clinical examination for the target joint, but this was not statistically significant [112].
Other Molecules in Phase I/II Studies
Several novel agents are in phase I and II trials for a number of inflammatory/immune cell driven disorders, including PsO and RA. Based on experience, for some, it is likely that the natural evolution of investigation for many of these agents will extend to PsA, whilst safety concerns and disappointing efficacy data may halt the progression of others into the clinical domain. Table 3 lists the new compounds currently in early clinical trials in psoriatic disease and other inflammatory arthritides that have not been discussed elsewhere in this paper.
Conclusion
The advent of biologic therapies has revolutionized the treatment of PsA and facilitated a real, meaningful, and measurable reduction in both disease progression and symptomatology. With more than a decade of safety data for TNF-alpha inhibitors, confidence in the use of biologics is increasing, and the net is being cast ever wider in the search for new biomarkers, molecular pathways, and therapeutic targets.
The impressive efficacy of TNF inhibitors in inflammatory disease has led to a significantly greater understanding of the inflammatory cascade and allowed for the identification of more direct molecular targets. Numerous agents, both biological and non-biological are in development which can precisely modulate or inhibit key molecules in the pathogenesis of inflammatory arthritis, and are showing promising results in phase II/III trials. The relative efficacy of these agents remains to be established, and, in time, head-to-head trials will be required to determine the best treatment options for patients.
The prospect of preventing radiographic damage in RA and PsA has led to a re-evaluation of how patients with inflammatory arthritides are managed—attempts are being made to identify specific phenotypic subgroups of patients who are more likely to derive benefit from selected treatments. Not only will this hasten the attainment of symptomatic relief, but could potentially reduce the economic burden imposed by ‘trial and error’ therapeutics and significantly lessen the physical and psychosocial morbidity of chronic disease.
The exciting search for the ultimate inhibitor of musculoskeletal inflammation continues, in terms of superior efficacy, safety, tolerability, mode of administration, and the ability to specifically target aberrant, pathogenic inflammatory pathways in multiple organ systems, without causing damage to healthy structures. Psoriatic disease is an ideal disease model, where aberrations in common inflammatory pathways give rise to the musculoskeletal, cutaneous and/or systemic phenotype, and is anticipated that in future, treatment options may become tailored to an individual’s clinical phenotype with the aid of imaging, serological and genetic biomarkers. The key challenge facing rheumatologists will then be how best to integrate all of the new, targeted molecules into daily practice, although the increasing armamentarium at their disposal will allow the provision of a significantly improved quality of life for many more patients.
References
Sherlock JP, Joyce-Shaikh B, Turner SP, et al. IL-23 induces spondyloarthropathy by acting on ROR-γt+ CD3+ CD4− CD8− entheseal resident T cells. Nat Med. 2012;18:1069–76.
McGonagle D, Khan MA, Marzo-Ortega H, O’Connor P, Gibbon W, Emery P. Enthesitis in spondyloarthropathy. Curr Opin Rheumatol. 1999;11:244–50.
Cortesi PA, Scalone L, D’Angiolella L, et al. Systematic literature review on economic implications and pharmacoeconomic issues of psoriatic arthritis. Clin Exp Rheumatol. 2012;30:S126–31.
Upchurch KS, Kay J. Evolution of treatment for rheumatoid arthritis. Rheumatology (Oxford). 2012;51(Suppl 6):vi28–36.
Chen YF, Jobanputra P, Barton P, et al. A systematic review of the effectiveness of adalimumab, etanercept and infliximab for the treatment of rheumatoid arthritis in adults and an economic evaluation of their cost-effectiveness. Health Technol Assess. 2006;10:iii–iv, xi–xiii, 1–229.
Wislowska M, Jakubicz D. Preliminary evaluation in rheumatoid arthritis activity in patients treated with TNF-alpha blocker plus methotrexate versus methotrexate or leflunomide alone. Rheumatol Int. 2007;27:641–7.
Gladman DD, Antoni C, Mease P, Clegg D, Nash P. Psoriatic arthritis: epidemiology, clinical features, course, and outcome. Ann Rheum Dis. 2005;64(Suppl 2):ii14–7.
Kyle S, Chandler D, Griffiths CEM, et al. Guideline for anti-TNF-α therapy in psoriatic arthritis. Rheumatology (Oxford). 2005;44:390–7.
Mease PJ, Goffe BS, Metz J, VanderStoep A, Finck B, Burge DJ. Etanercept in the treatment of psoriatic arthritis and psoriasis: a randomised trial. Lancet. 2000;356:385–90.
Mease PJ, Gladman DD, Ritchlin CT, et al. Adalimumab for the treatment of patients with moderately to severely active psoriatic arthritis: results of a double-blind, randomized, placebo-controlled trial. Arthritis Rheum. 2005;52:3279–89.
Antoni CE, Kavanaugh A, Kirkham B, et al. Sustained benefits of infliximab therapy for dermatologic and articular manifestations of psoriatic arthritis: results from the infliximab multinational psoriatic arthritis controlled trial (IMPACT). Arthritis Rheum. 2005;52:1227–36.
Antoni C, Krueger GG, de Vlam K, et al. Infliximab improves signs and symptoms of psoriatic arthritis: results of the IMPACT 2 trial. Ann Rheum Dis. 2005;64:1150–7.
Kavanaugh A, McInnes I, Mease P, et al. Golimumab, a new human tumor necrosis factor alpha antibody, administered every four weeks as a subcutaneous injection in psoriatic arthritis: twenty-four-week efficacy and safety results of a randomized, placebo-controlled study. Arthritis Rheum. 2009;60:976–86.
Navarro-Sarabia F, Ariza-Ariza R, Hernandez-Cruz B, Villanueva I. Adalimumab for treating rheumatoid arthritis. Cochrane Database Syst Rev. 2005:CD005113.
Ritchlin C, Haas-Smith SA, Hicks D, Cappuccio J, Osterland CK, Looney RJ. Patterns of cytokine production in psoriatic synovium. J Rheumatol. 1998;25:1544–52.
Hohler T, Kruger A, Schneider PM, et al. A TNF-α promoter polymorphism is associated with juvenile onset psoriasis and psoriatic arthritis. J Invest Dermatol. 1997;109:562–5.
Ash Z, Gaujoux-Viala C, Gossec L, et al. A systematic literature review of drug therapies for the treatment of psoriatic arthritis: current evidence and meta-analysis informing the EULAR recommendations for the management of psoriatic arthritis. Ann Rheum Dis. 2012;71:319–26.
Weger W. Current status and new developments in the treatment of psoriasis and psoriatic arthritis with biological agents. Br J Pharmacol. 2010;160:810–20.
Rozenblit M, Lebwohl M. New biologics for psoriasis and psoriatic arthritis. Dermatol Ther. 2009;22:56–60.
Maksymowych WP, Inman RD, Gladman D, Spondyloarthritis Research Consortium of Canada (SPARCC), et al. Canadian Rheumatology Association Consensus on the use of anti-tumor necrosis factor-alpha directed therapies in the treatment of spondyloarthritis. J Rheumatol. 2003;30:1356–63.
van der Heijde D, Sieper J, Maksymowych WP, et al. 2010 Update of the international ASAS recommendations for the use of anti-TNF agents in patients with axial spondyloarthritis. Ann Rheum Dis. 2011;70:905–8.
Braun J, Landewe R, Hermann KG, et al. Major reduction in spinal inflammation in patients with ankylosing spondylitis after treatment with infliximab: results of a multicenter, randomized, double-blind, placebo-controlled magnetic resonance imaging study. Arthritis Rheum. 2006;54:1646–52.
van der Heijde D, Landewe R, Einstein S, et al. Radiographic progression of ankylosing spondylitis after up to two years of treatment with etanercept. Arthritis Rheum. 2008;58:1324–31.
Saber TP, Veale DJ. Psoriatic arthritis management update—biotherapeutic options. J Rheumatol Suppl. 2009;83:65–8.
Glintborg B, Ostergaard M, Dreyer L, et al. Treatment response, drug survival, and predictors thereof in 764 patients with psoriatic arthritis treated with anti-tumor necrosis factor alpha therapy: results from the nationwide Danish DANBIO registry. Arthritis Rheum. 2011;63:382–90.
Saad AA, Symmons DP, Noyce PR, et al. Risks and benefits of tumor necrosis factor-alpha inhibitors in the management of psoriatic arthritis: systematic review and metaanalysis of randomized controlled trials. J Rheumatol. 2008;35:883–90.
Genovese MC, Mease PJ, Thomson GT, et al. Safety and efficacy of adalimumab in treatment of patients with psoriatic arthritis who had failed disease modifying antirheumatic drug therapy. J Rheumatol. 2007;34:1040–50.
Mease PJ, Kivitz AJ, Burch FX, et al. Etanercept treatment of psoriatic arthritis: safety, efficacy, and effect on disease progression. Arthritis Rheum. 2004;50:2264–72.
Vander Cruyssen B, De Keyser F, Kruithof E, Mielants H, Van den Bosch F. Comparison of different outcome measures for psoriatic arthritis in patients treated with infliximab or placebo. Ann Rheum Dis. 2007;66:138–40.
FitzGerald O, Helliwell P, Mease P, et al. Application of composite disease activity scores in psoriatic arthritis to the PRESTA data set. Ann Rheum Dis. 2012;71:358–62.
Mease PJ. Measures of psoriatic arthritis: Tender and Swollen Joint Assessment, Psoriasis Area and Severity Index (PASI), Nail Psoriasis Severity Index (NAPSI), Modified Nail Psoriasis Severity Index (mNAPSI), Mander/Newcastle Enthesitis Index (MEI), Leeds Enthesitis Index (LEI), Spondyloarthritis Research Consortium of Canada (SPARCC), Maastricht Ankylosing Spondylitis Enthesis Score (MASES), Leeds Dactylitis Index (LDI), Patient Global for Psoriatic Arthritis, Dermatology Life Quality Index (DLQI), Psoriatic Arthritis Quality of Life (PsAQOL), Functional Assessment of Chronic Illness Therapy-Fatigue (FACIT-F), Psoriatic Arthritis Response Criteria (PsARC), Psoriatic Arthritis Joint Activity Index (PsAJAI), Disease Activity in Psoriatic Arthritis (DAPSA), and Composite Psoriatic Disease Activity Index (CPDAI). Arthritis Care Res (Hoboken). 2011;63:S64–85.
Gottlieb A, Menter A, Mendelsohn A, et al. Ustekinumab, a human interleukin 12/23 monoclonal antibody, for psoriatic arthritis: randomised, double-blind, placebo-controlled, crossover trial. Lancet. 2009;373:633–40.
Mease PJ, Gladman DD, Keystone EC. Alefacept in combination with methotrexate for the treatment of psoriatic arthritis: results of a randomized, double-blind, placebo-controlled study. Arthritis Rheum. 2006;54:1638–45.
Thorlund K, Druyts E, Avina-Zubieta JA, et al. Anti-tumor necrosis factor (TNF) drugs for the treatment of psoriatic arthritis: an indirect comparison meta-analysis. Biologics. 2012;6:417–27.
McGonagle D, Marzo-Ortega H, O’Connor P, et al. Histological assessment of the early enthesitis lesion in spondyloarthropathy. Ann Rheum Dis. 2002;61:534–7.
McGonagle D, Wakefield RJ, Tan AL, et al. Distinct topography of erosion and new bone formation in achilles tendon enthesitis: implications for understanding the link between inflammation and bone formation in spondylarthritis. Arthritis Rheum. 2008;58:2694–9.
Sterry W, Ortonne JP, Kirkham B, et al. Comparison of two etanercept regimens for treatment of psoriasis and psoriatic arthritis: PRESTA randomised double blind multicentre trial. BMJ. 2010;340:c147.
Mease PJ, Ory P, Sharp JT, et al. Adalimumab for long-term treatment of psoriatic arthritis: 2-year data from the Adalimumab Effectiveness in Psoriatic Arthritis Trial (ADEPT). Ann Rheum Dis. 2009;68:702–9.
van den Broek M, Lems WF, Allaart CF. BeSt practice: the success of early-targeted treatment in rheumatoid arthritis. Clin Exp Rheumatol. 2012;30:S35–8.
Breedveld FC, Weisman MH, Kavanaugh AF, et al. The PREMIER study: a multicenter, randomized, double-blind clinical trial of combination therapy with adalimumab plus methotrexate versus methotrexate alone or adalimumab alone in patients with early, aggressive rheumatoid arthritis who had not had previous methotrexate treatment. Arthritis Rheum. 2006;54:26–37.
Emery P, Breedveld FC, Hall S, et al. Comparison of methotrexate monotherapy with a combination of methotrexate and etanercept in active, early, moderate to severe rheumatoid arthritis (COMET): a randomised, double-blind, parallel treatment trial. Lancet. 2008;372:375–82.
Jones G, Sebba A, Gu J, et al. Comparison of tocilizumab monotherapy versus methotrexate monotherapy in patients with moderate to severe rheumatoid arthritis: the AMBITION study. Ann Rheum Dis. 2010;69:88–96.
van der Heijde D, Klareskog L, Rodriguez-Valverde V, et al. Comparison of etanercept and methotrexate, alone and combined, in the treatment of rheumatoid arthritis: two-year clinical and radiographic results from the TEMPO study, a double-blind, randomized trial. Arthritis Rheum. 2006;54:1063–74.
Klareskog L, van der Heijde D, de Jager JP, et al. Therapeutic effect of the combination of etanercept and methotrexate compared with each treatment alone in patients with rheumatoid arthritis: double-blind randomised controlled trial. Lancet. 2004;363:675–81.
Maini R, St Clair EW, Breedveld F, et al. Infliximab (chimeric anti-tumour necrosis factor alpha monoclonal antibody) versus placebo in rheumatoid arthritis patients receiving concomitant methotrexate: a randomised phase III trial. ATTRACT Study Group. Lancet. 1999;354:1932–9.
Weinblatt ME, Keystone EC, Furst DE, et al. Adalimumab, a fully human anti-tumor necrosis factor alpha monoclonal antibody, for the treatment of rheumatoid arthritis in patients taking concomitant methotrexate: the ARMADA trial. Arthritis Rheum. 2003;48:35–45.
Ritchlin CT. Therapeutic considerations in spondyloarthritis patients who fail tumour necrosis factor antagonists. Best Pract Res Clin Rheumatol. 2010;24:683–92.
Sandborn WJ. Optimizing anti-tumor necrosis factor strategies in inflammatory bowel disease. Curr Gastroenterol Rep. 2003;5:501–5.
Heiberg MS, Koldingsnes W, Mikkelsen K, et al. The comparative one-year performance of anti-tumor necrosis factor alpha drugs in patients with rheumatoid arthritis, psoriatic arthritis, and ankylosing spondylitis: results from a longitudinal, observational, multicenter study. Arthritis Rheum. 2008;59:234–40.
Rodgers M, Epstein D, Bojke L, et al. Etanercept, infliximab and adalimumab for the treatment of psoriatic arthritis: a systematic review and economic evaluation. Health Technol Assess. 2011;15:i–xxi, 1–329.
Sandborn WJ, Hanauer SB, Katz S, et al. Etanercept for active Crohn’s disease: a randomized, double-blind, placebo-controlled trial. Gastroenterology. 2001;121:1088–94.
Akobeng AK, Zachos M. Tumor necrosis factor-alpha antibody for induction of remission in Crohn’s disease. Cochrane Database Syst Rev. 2004;(1):CD003574.
Behm BW, Bickston SJ. Tumor necrosis factor-alpha antibody for maintenance of remission in Crohn’s disease. Cochrane Database Syst Rev. 2008;(1):CD006893.
Goulabchand R, Mouterde G, Barnetche T, Lukas C, Morel J, Combe B. Effect of tumour necrosis factor blockers on radiographic progression of psoriatic arthritis: a systematic review and meta-analysis of randomised controlled trials. Ann Rheum Dis. 2013 (Epub ahead of print).
Cargill M, Schrodi SJ, Chang M, et al. A large-scale genetic association study confirms IL12B and leads to the identification of IL23R as psoriasis-risk genes. Am J Hum Genet. 2007;80:273–90.
McInnes IB, Kavanaugh A, Gottlieb AB, et al. Ustekinumab in patients with active psoriatic arthritis: results of the phase 3, multicenter, double-blind, placebo-controlled PSUMMIT I Study. Ann Rheum Dis. 2012;71:107.
Kavanaugh A, McInnes IB, Gottlieb AB, et al. Ustekinumab improves arthritis-related and skin-related quality of life in patients with active psoriatic arthritis: patient reported outcomes from randomized and double blinded phase III PSUMMIT I Trial. Paper presented at: American College of Rheumatology Annual Meeting, Washington, DC; 2012.
Ritchlin CT, Gottlieb AB, McInnes IB, et al. Ustekinumab in active psoriatic arthritis including patients previously treated with anti-TNF agents: results of a phase 3, multicenter, double-blind, placebo-controlled study. Paper presented at: American College of Rheumatology Annual Meeting, Washington, DC; 2012.
Wada Y, Lu R, Zhou D, et al. Selective abrogation of Th1 response by STA-5326, a potent IL-12/IL-23 inhibitor. Blood. 2007;109:1156–64.
Krausz S, Boumans MJ, Gerlag DM, et al. Brief report: a phase IIa, randomized, double-blind, placebo-controlled trial of apilimod mesylate, an interleukin-12/interleukin-23 inhibitor, in patients with rheumatoid arthritis. Arthritis Rheum. 2012;64:1750–5.
Johansen C, Usher PA, Kjellerup RB, et al. Characterization of the interleukin-17 isoforms and receptors in lesional psoriatic skin. Br J Dermatol. 2009;160:319–24.
Hueber W, Patel DD, Dryja T, et al. Effects of AIN457, a fully human antibody to interleukin-17A, on psoriasis, rheumatoid arthritis, and uveitis. Sci Transl Med. 2010;2:52ra72.
Genovese MC, Van den Bosch F, Roberson SA, et al. LY2439821, a humanized anti-interleukin-17 monoclonal antibody, in the treatment of patients with rheumatoid arthritis: a phase I randomized, double-blind, placebo-controlled, proof-of-concept study. Arthritis Rheum. 2010;62:929–39.
McInnes I, Sieper J, Braun J, et al. Anti-interleukin 17A monoclonal antibody secukinumab reduces signs and symptoms of psoriatic arthritis in a 24-week multicenter, double-blind, randomized, placebo-controlled trial. Arthritis Rheum. 2011;63:779.
McInnes IB, Sieper J, Braun J, et al. Efficacy and safety of secukinumab, a fully human anti-interleukin-17A monoclonal antibody, in patients with moderate-to-severe psoriatic arthritis: a 24-week, randomised, double-blind, placebo-controlled, phase II proof-of-concept trial. Ann Rheum Dis. 2013 (Epub ahead of print).
Krueger G, Gottlieb A, Sterry W. A multicenter, open-label study of repeat courses of intramuscular alefacept in combination with other psoriasis therapies in patients with chronic plaque psoriasis. J Dermatol Treat. 2008;19:146–55.
Ellis CN, Krueger GG. Treatment of chronic plaque psoriasis by selective targeting of memory effector T lymphocytes. NEJM. 2001;345:248–55.
Brimhall AK, King LN, Licciardone JC, et al. Safety and efficacy of alefacept, efalizumab, etanercept and infliximab in treating moderate to severe plaque psoriasis: a meta-analysis of randomized controlled trials. Br J Dermatol. 2008;159:274–85.
Mease PJ, Reich K. Alefacept with methotrexate for treatment of psoriatic arthritis: open-label extension of a randomized, double-blind, placebo-controlled study. J Am Acad Dermatol. 2009;60:402–11.
Mease P, Genovese MC, Gladstein G, et al. Abatacept in the treatment of patients with psoriatic arthritis: results of a six-month, multicenter, randomized, double-blind, placebo-controlled, phase II trial. Arthritis Rheum. 2011;63:939–48.
Abrams JR, Lebwohl MG, Guzzo CA, et al. CTLA4Ig-mediated blockade of T-cell costimulation in patients with psoriasis vulgaris. J Clin Invest. 1999;103:1243–52.
Canete JD, Celis R, Hernandez V, et al. Synovial immunopathological changes associated with successful abatacept therapy in a case of severe refractory psoriatic arthritis. Ann Rheum Dis. 2010;69:935–6.
Vieira FJ, Callado MR, Vieira WP. Abatacept as an option therapy in difficult to treat psoriatic arthritis. Rheumatol Int. 2010;30:849–50.
Mease P, Genovese MC, Ritchlin C, et al. Abatacept in psoriatic arthritis: results of a phase II study [abstract]. Arthritis Rheum. 2009;60:1260.
Voll RE, Kalden JR. Do we need new treatment that goes beyond tumor necrosis factor blockers for rheumatoid arthritis? Ann N Y Acad Sci. 2005;1051:799–810.
Schiff M. Abatacept treatment for rheumatoid arthritis. Rheumatology (Oxford). 2011;50:437–49.
Schiff M, Bessette L. Evaluation of abatacept in biologic-naive patients with active rheumatoid arthritis. Clin Rheumatol. 2010;29:583–91.
Emery P, Durez P, Dougados M, et al. Impact of T-cell costimulation modulation in patients with undifferentiated inflammatory arthritis or very early rheumatoid arthritis: a clinical and imaging study of abatacept (the ADJUST trial). Ann Rheum Dis. 2010;69:510–6.
Genant HK, Peterfy CG, Westhovens R, et al. Abatacept inhibits progression of structural damage in rheumatoid arthritis: results from the long-term extension of the AIM trial. Ann Rheum Dis. 2008;67:1084–9.
Genovese MC, Schiff M, Luggen M, et al. Efficacy and safety of the selective co-stimulation modulator abatacept following 2 years of treatment in patients with rheumatoid arthritis and an inadequate response to anti-tumour necrosis factor therapy. Ann Rheum Dis. 2008;67:547–54.
Westhovens R, Kremer JM, Moreland LW, et al. Safety and efficacy of the selective costimulation modulator abatacept in patients with rheumatoid arthritis receiving background methotrexate: a 5-year extended phase IIB study. J Rheumatol. 2009;36:736–42.
Skvara H, Dawid M, Kleyn E, et al. The PKC inhibitor AEB071 may be a therapeutic option for psoriasis. J Clin Invest. 2008;118:3151–9.
Spitaler M, Cantrell DA. Protein kinase C and beyond. Nat Immunol. 2004;5:785–90.
Guo B, Su TT, Rawlings DJ. Protein kinase C family functions in B-cell activation. Curr Opin Immunol. 2004;16:367–73.
Monks CR, Kupfer H, Tamir I, et al. Selective modulation of protein kinase C-theta during T-cell activation. Nature. 1997;385:83–6.
Pfeifhofer C, Gruber T, Letschka T, et al. Defective IgG2a/2b class switching in PKC alpha−/− mice. J Immunol. 2006;176:6004–11.
Mecklenbrauker I, Saijo K, Zheng NY, et al. Protein kinase Cdelta controls self-antigen-induced B-cell tolerance. Nature. 2002;416:860–5.
Chaudhary D, Kasaian M. PKCtheta: a potential therapeutic target for T-cell-mediated diseases. Curr Opin Investig Drugs. 2006;7:432–7.
Wagner J, von Matt P, Faller B, et al. Structure–activity relationship and pharmacokinetic studies of sotrastaurin (AEB071), a promising novel medicine for prevention of graft rejection and treatment of psoriasis. J Med Chem. 2011;54:6028–39.
Evenou JP, Wagner J, Zenke G, et al. The potent protein kinase C-selective inhibitor AEB071 (sotrastaurin) represents a new class of immunosuppressive agents affecting early T-cell activation. J Pharmacol Exp Ther. 2009;330:792–801.
Tan SL, Zhao J, Bi C, et al. Resistance to experimental autoimmune encephalomyelitis and impaired IL-17 production in protein kinase C theta-deficient mice. J Immunol. 2006;176:2872–9.
Murray PJ. The JAK-STAT signaling pathway: input and output integration. J Immunol. 2007;178:2623–9.
Punwani N, Scherle P, Flores R, et al. Preliminary clinical activity of a topical JAK1/2 inhibitor in the treatment of psoriasis. J Am Acad Dermatol. 2012;67:658–64.
Valentino L, Pierre J. JAK/STAT signal transduction: regulators and implication in hematological malignancies. Biochem Pharmacol. 2006;71:713–21.
Kimball AB, Gordon KB, Langley RG, et al. Safety and efficacy of ABT-874, a fully human interleukin 12/23 monoclonal antibody, in the treatment of moderate to severe chronic plaque psoriasis: results of a randomized, placebo-controlled, phase 2 trial. Arch Dermatol. 2008;144:200–7.
Subramaniam SV, Cooper RS, Adunyah SE. Evidence for the involvement of JAK/STAT pathway in the signaling mechanism of interleukin-17. Biochem Biophys Res Commun. 1999;262:14–9.
Wang M, Tan Z, Thomas EK, et al. Conservation of the genomic structure and receptor-mediated signaling between human and rat IL-24. Genes Immun. 2004;5:363–70.
O’Shea J. Targeting the Jak/STAT pathway for immunosuppression. Ann Rheum Dis. 2004;63:ii67–71.
Boy MG, Wang C, Wilkinson BE, et al. Double-blind, placebo-controlled, dose-escalation study to evaluate the pharmacologic effect of CP-690,550 in patients with psoriasis. J Invest Dermatol. 2009;129:2299–302.
Fleischmann R, Kremer J, Cush J, et al. Placebo-controlled trial of tofacitinib monotherapy in rheumatoid arthritis. NEJM. 2012;367:495–507.
Kremer JM, Cohen S, Wilkinson BE, et al. A phase IIb dose-ranging study of the oral JAK inhibitor tofacitinib (CP-690,550) versus placebo in combination with background methotrexate in patients with active rheumatoid arthritis and an inadequate response to methotrexate alone. Arthritis Rheum. 2012;64:970–81.
Kudlacz E, Perry B, Sawyer P, et al. The novel JAK-3 inhibitor CP-690550 is a potent immunosuppressive agent in various murine models. Am J Transplant. 2004;4:51–7.
Quintas-Cardama A, Vaddi K, Liu P, et al. Preclinical characterization of the selective JAK1/2 inhibitor INCB018424: therapeutic implications for the treatment of myeloproliferative neoplasms. Blood. 2010;115:3109–17.
Mesa RA. Ruxolitinib, a selective JAK1 and JAK2 inhibitor for the treatment of myeloproliferative neoplasms and psoriasis. IDrugs. 2010;13:394–403.
Fridman JS, Scherle PA, Collins R, et al. Selective inhibition of JAK1 and JAK2 is efficacious in rodent models of arthritis: preclinical characterization of INCB028050. J Immunol. 2010;184:5298–307.
Vissing S. Consumption of topical treatment in psoriasis patients [Abstract]. Paper presented at: 21st Annual Congress of the EADV, Prague; 2012.
Stump KL, Lu LD, Dobrzanski P, et al. A highly selective, orally active inhibitor of Janus kinase 2, CEP-33779, ablates disease in two mouse models of rheumatoid arthritis. Arthritis Res Ther. 2011;13:R68.
Papp K, Cather JC, Rosoph L, et al. Efficacy of apremilast in the treatment of moderate to severe psoriasis: a randomised controlled trial. Lancet. 2012;380:738–46.
Schett G, Wollenhaupt J, Papp K, et al. Oral apremilast in the treatment of active psoriatic arthritis: results of a multicenter, randomized, double-blind, placebo-controlled study. Arthritis Rheum. 2012;64:3156–67.
Mease PJ, Wei N, Fudman EJ, et al. Safety, tolerability, and clinical outcomes after intraarticular injection of a recombinant adeno-associated vector containing a tumor necrosis factor antagonist gene: results of a phase 1/2 Study. J Rheumatol. 2010;37:692–703.
Schnepp BC, Jensen RL, Chen CL, et al. Characterization of adeno-associated virus genomes isolated from human tissues. J Virol. 2005;79:14793–803.
Mease PJ, Hobbs K, Chalmers A, et al. Local delivery of a recombinant adenoassociated vector containing a tumour necrosis factor alpha antagonist gene in inflammatory arthritis: a phase 1 dose-escalation safety and tolerability study. Ann Rheum Dis. 2009;68:1247–54.
Acknowledgments
This manuscript has been reviewed by the medical advisors at Janssen, Pfizer, Celgene, and Novartis for scientific accuracy. Dr L. Savage is the guarantor for this article, and takes responsibility for the integrity of the work as a whole.
Conflict of interest
Dr L. Savage and Professor D. McGonagle have received honorarium (for education provision) and/or grant funding from several pharmaceutical companies, including Abbvie, Astra-Zeneca, Boehringer Ingelheim, Bristol-Myers Squibb, Celgene, Galderma, GlaxoSmithKline, Janssen-Cilag, Leo, Merck, Sharp & Dohme, Novartis, and Pfizer.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 2.0 International License (https://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Savage, L.J., McGonagle, D.G. The Role of Biological and Small Molecule Therapy in the Management of Psoriatic Arthritis. Biol Ther 3, 61–81 (2013). https://doi.org/10.1007/s13554-013-0010-0
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13554-013-0010-0