
Abstract
Based on density functional theory (DFT), the electronic, structural, and optical properties of bismuth oxyfluoride (BiOF) are investigated by using various exchange–correlation functionals. The local density approximation (LDA), Perdew-Burke-Ernzerhof generalized gradient approximation (PBE-GGA), Perdew-Burke-Ernzerhof generalized gradient approximation for solids (PBEsol), and Wu-Cohen (WC) functionals are implemented in Wien2k software. Modified Becke-Johnson (mBJ) and unmodified Becke-Johnson (umBJ) potentials are also applied to obtain an enhanced band gap. Spin–orbit coupling effect (SOC) is also taken into consideration due to the heavy Bi atom. When employing the PBE+umBJ potential, BiOF exhibits the most enhanced band gap energy, with a direct band gap energy of 3.72 eV. Moreover, optical properties such as dielectric functions, absorption coefficients, and optical conductivities are also calculated for photon energies up to ̴̴ 14 eV. In comparison to other available theoretical and experimental findings, the calculated results are in good agreement.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
Density-functional theory (DFT) is commonly used as a powerful tool to calculate and understand the electronic structure of materials due to its reasonable accuracy and computational efficiency [1, 2]. The main idea behind DFT is that electron density is the fundamental quantity that determines a system’s characteristics and that the system’s total energy could be expressed in terms of the functional of electron density rather than by the wavefunction of each electron. Wien2k is one of the most effective theoretical frameworks for performing DFT calculations to investigate the electronic structure and other properties of various materials [3]. It is a full-potential, linear augmented plane wave (LAPW) method that solves the Schrödinger equation using a variety of approximations and provides several options for exchange–correlation (XC) functionals, allowing users to select the best one for their research objectives [4]. The most commonly used XC functionals in Wien2k are local density approximation (LDA) [5] and the variants of the generalized gradient approximation (GGA) that are Perdew-Burke-Ernzerhof (PBE) [6] and its revised version for solids (PBEsol) [7] and Wu-Cohen [8]. LDA is the simplest XC functional, which is based on the assumption that the XC energy density is a function of the local electron density. PBE functional considers both the local electron density and its gradient, which provides more accurate results than LDA for various materials. The WC functional is a modified version of the PBE that takes into account the Laplacian of the electron density in addition to the gradient of the electron density. Depending on the system being studied, WC functional can give more accurate results than PBE. Although many researchers have studied the structural and electronic properties of BiOX (X = Cl, Br, F, I), investigations of the structural and electronic properties of BiOF, particularly those conducted using the Wien2k code, are scarce, which prompted us to carry out the current study. Li et al. previously reported crystal and electronic structures of BiOF and its oxygen vacancy BiO7/8F by using the PBE exchange–correlation functionals in the CASTEP code [9]. They discovered that the band gap for the BiOF is 3.12 eV, less than the experimental value of 3.64 eV [10]. To construct electronic structures of BiOF photocatalyst with and without the Bi 5d states, Huang et al. used PBE functionals in the CASTEP code [11]. According to their findings, BiOF has a direct band gap of 3.12 eV and 3.22 eV with and without using Bi 5d states. Canpolat et al. investigated the crystal structure of the BiOF under high hydrostatic pressure using the Siesta method [12]. In their study, pressure–volume relationships and structural transitions were studied by applying PBE functional. In this work, the structural, electronic, and optical properties of the BiOF, as well as the effect of the different XC functionals, were investigated using first-principles calculations. umBJ and mBJ potentials and SOC effects on the band structures were also determined. The calculated results could be worthwhile for completing the theoretical and experimental works for the BiOF.
2 Computational Details
In the present work, first-principle calculations were performed for the BiOF compound as implemented in the Wien2k software [13]. This software has the capability to calculate the electronic, structural, optical, and many other properties of the materials. The optimization of the electronic structure has been performed using the LDA and three flavors of generalized gradient approximation: PBE, PBEsol, and WC functionals. To get an accurate value of band gap energy, umBJ and mBJ potentials are used. The key distinction between mBJ and umBJ lies in the inclusion of an empirical parameter within mBJ. This parameter aims to improve performance in specific scenarios, but umBJ maintains a purely theoretical foundation [14]. It is widely accepted that the SOC effect has a significant influence on the electronic properties of a material, particularly if the material contains heavy elements. Because BiOF contains a heavy element, Bi, the SOC effect was also included in the calculations. During the calculation process, the configuration of the valence electrons of the atom is Bi 6s26p3, O 2s22p4, and F 2s22p5. The Brillouin zone was sampled with a Monkhorst–Pack k-point grid of 11 × 11 × 7 (representing 84 k-points in the irreducible Brillouin zone) for the self-consistent (SCF) calculations. The plane wave cut off RMTKmax = 7, and the maximum l value (lmax) for partial waves used in atomic spheres is kept at 10. The energy cutoff is set to −6Ry(−81.63 eV), the energy convergence threshold is less than 10−5 Ry, and the charge convergence threshold is 10−4e. Fourier expansion cutoff Gmax = 12. First, the BiOF structure is optimized with the tetragonal space group P4/nmm (PbFCl structure type) by using LDA, PBE, PBEsol, and WC functionals for stable ground-state structural properties. The structural properties, such as the lattice constant a (bohr), bulk modulus B (GPa), and its pressure derivative B′, were obtained to calculate the electronic structures of the BiOF. To optimize the crystal structure of BiOF, the previous experimental coordinates were used to provide a good initial guess for the atomic positions. The following optimization is performed to obtain the optimal lattice constants in the ground state (0 K, 0 Pa): the fundamental energy \({E}_{0}\) was calculated for a series of volume values V, and then the volume \({V}_{0}\) corresponding to minimal fundamental energy is determined. The fundamental energy \({E}_{0}\) and the equilibrium volume V0 are calculated using the Murnaghan equation [15]:
where \({E}_{0}\) is the fundamental energy at the equilibrium volume \({V}_{0}\), \({B}_{0}\), and \({B}_{0}^{\prime}\) are the bulk modulus and its first derivative.
3 Results and Discussion
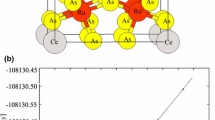
First, the geometry optimization was carried out using the experimental coordinates, which are much better than random starting points, and can significantly speed up the optimization process. As a result of the optimization, the volume corresponding to the minimum energy was determined, and the lattice constants were calculated. The energy against the unit cell volume calculated by LDA, PBE, PBEsol, and WC functionals for BiOF is depicted in Fig. 1.

Volume optimization for BiOF using LDA, PBE, PBEsol, and WC exchange–correlation functionals
The equilibrium volume \({V}_{0}\), fundamental energy \({E}_{0}\), and optimized lattice constants in all exchange–correlation funtionals are presented in Table 1.
The calculated results show that the minimal energy for the BiOF compound was obtained under PBE. While the optimized lattice constants under PBE are a = b = 7.156 Bohr and c = 11.890 Bohr, the optimized lattice constants for LDA are a = b = 7.000 Bohr and c = 11.632 Bohr, which differ by 2.18% from the PBE. PBEsol and WC functionals produce similar results, although they deviate by 1.3% from PBE. Consequently, optimized lattice constants for all functionals are consistent with the previous experimental results [16, 17].
The electronic band structures of BiOF shown in Fig. 2 were calculated using the LDA, PBE, PBEsol, and WC functionals. The band structures having the same energy range of −6 and 8 eV were calculated and presented for each functional to make a better comparison. The Fermi energy is set at 0 eV. The results of band gaps and band gap types for different functionals are listed in Table 2. When the LDA functional is used, the conduction band minimum (CBM) is located at point Z, but the valance band maximum (VBM) is not located exactly at point Z. Rather, it lies between the Z and R points. This means that BiOF exhibits a near-direct band gap of 3.15 eV when LDA functional is used. In the case of the PBE functional, VBM and CBM are both located at the Z high symmetry point, indicating that BiOF has a direct band gap of 3.29 eV. Using the PBEsol functional resulted in a near-direct band gap of 3.19 eV because CBM is located at the Z point, while VBM is not located at the Z point (located between the Z and R points). Finally, the result obtained via the WC functional indicates that BiOF has a near-direct band gap of 3.21 eV, as well. Similarly, VBM lies between the Z and R high symmetry points, and CBM is located at the Z high symmetry point. In the literature, it is stated that BiOF has a direct band gap with an experimental value of 3.64 eV [9,10,11]. Considering the above results, the PBE functional is the most effective of the functionals used. The LDA, PBEsol, and WC functionals produced a near-direct band gap. The results obtained by these functionals do not perfectly match previously observed experimental findings. This clearly illustrates how essential it is to take into account several methods for the accurate estimation of electronic properties. When the band gaps are compared with the experimental finding, there is about 9.7% underestimation in the case of PBE functional, which is the best result. LDA, PBEsol, and WC functionals underestimate the band gap by about 13.5%, 12.3%, and 11.8%, respectively. LDA, PBE, PBEsol, and WC functionals alone, to the best of our knowledge, are insufficient for predicting the semiconductor band gap [18, 19]. With these approximations, sometimes the band gap of the materials is underestimated with respect to the experimental values, such that instead of insulating material, it results in metallic material. The Wien2k code includes mBJ and umBJ potentials to reproduce the experimental band gap of semiconductors with increased accuracy. Therefore, to enhance the band gap results, the umBJ and mBJ potentials are applied to the LDA, PBE, PBEsol, and WC functionals. The band gap types remain the same when umBJ potential is added to the LDA, PBE, PBEsol, and WC functionals. The band gaps are 3.79 eV, 3.77 eV, and 3.78 eV for the LDA, PBEsol, and WC functionals, and there are about 4.17%, 3.62%, and 3.62% overestimations. PBE functional, on the other hand, has a direct band gap of 3.72 eV with an overestimation of only 2.28%. In the case where the mBJ potential is applied to the LDA, PBE, PBEsol, and WC functionals, BiOF has a near-direct band gap for all the functionals, and the band gap values are highly overestimated, which is 30% more than the experimental findings. The band gap energies can be found in Table 2. According to the results, one can conclude that the umBJ potential is more effective than the mBJ potential for the BiOF, especially when the PBE functional is used. The spin–orbit coupling (SOC) effect is also crucial for determining the electronic band structure and band gaps in heavy element compounds. The more heavy elements the material contains, the greater the effect of SOC on the material’s electronic band structure and band gaps [9]. Due to the heavy Bi atoms in BiOF, the SOC effect has to be considered when calculating the electronic band structure. The band structures for the PBE, PBE+umBJ, and PBE+umBJ+SOC have been shown in Fig. 3. After umBJ or mBJ potential is applied to the LDA, PBE, PBEsol, and WC functionals, the SOC effect is also added to check if more accurate results can be obtained for the electronic band structure of BiOF.

The band structure of BiOF obtained using equilibrium lattice constants calculated by a LDA, b PBE, c PBEsol, and d Wu-Cohen

Band structure of BiOF using a PBE, b PBE+umBJ, and c PBE+umBJ+SOC functionals
Band gap types remain the same for all the functionals with and without SOC effect. According to the results, the umBJ potential provides the most accurate outcome, being only one to predict the direct band gap. The valence band maximum remains nearly the same after applying the umBJ and umBJ+SOC to the PBE, but the conduction band minimum increases. The increment on the band gap energy is greater with umBJ than with umBJ+SOC. The decrease in the band gap energy when the SOC effect is taken into consideration is a well known phenomenon [20, 21]. Figure 4 clearly illustrates the effect of umBJ potential and SOC on the electronic structure of BiOF. To sum up, the PBE functional works better than LDA, PBEsol, and WC functionals for the BiOF. Adding the umBJ potential to the PBE functional corrects the underestimation of the band gap from 9.7 to 2.28%. To account for the SOC effects arising from the presence of the heavy element Bi in BiOF, which is not directly captured by the umBJ potential, seperate SOC calculations were performed in conjunction with DFT calculations. The result obtained by including the umBJ and SOC effect differs by 8.24% from the experimental band gap. Since the most enhanced band gap is achieved by the PBE functional, the total and partial densities of states (TDOS and PDOS) of the BiOF have been plotted to explore the contribution of the atoms in the formation of the electronic structures. Figure 5 shows the TDOS and PDOS of the BiOF when PBE, PBE+umBJ, and PBE+umBJ+SOC functionals are used. As presented in Fig. 5, TDOS and PDOS are comparable for all three functionals. TDOS clearly shows that BiOF has a semiconductor nature; calculated band gaps for PBE, PBE+umBJ, and PBE+umBJ+SOC functionals are 3.29 eV, 3.72 eV, and 3.34 eV, respectively. These results clearly show that BiOF has a semiconductor characteristic with a wide band gap. The valence band top of BiOF can be examined in three parts. On the left and right sides of the VBM, the largest contribution comes from O atoms, while in the middle part, most of the contribution comes from the F atom. The contribution from the Bi atom is the least for all three parts. The conduction band bottom of the BiOF mainly consists of two parts. In both parts, while the Bi atom dominates these two portions, the F atom contributes very little.

Comparison of band structures for BiOF using PBE, PBE+umBJ, and PBE+umBJ+SOC functionals

Total density of states (TDOS) and atomic contributions to TDOS for BiOF using PBE, PBE+umBJ, and PBE+umBJ+SOC functionals
Beyond the electronic structure analysis, the optical properties of BiOF are also investigated in detail. Calculating the complex dielectric function, which describes a material’s response to an electric field oscillating at a specific frequency \((\omega)\), is one of the most effective approaches for studying the optical properties of materials. The total dielectric function is described as [22]:
Here, the real part \(\varepsilon_{1}(\omega)\) represents the polarizability of the material, which essentially means its ability to store electrical energy by an external electric field. The imaginary part \(\varepsilon_{2}(\omega)\) describes energy loss caused by various mechanisms such as the material’s absorption and scattering of light. Figure 6 presents the calculated real and imaginary parts of the dielectric function for BiOF within PBE, PBE+umBJ, and PBE+umBJ+SOC functionals in the xx and zz directions for the photon energy up to 14 eV. The static values \(\varepsilon_{1}(0)\), maximum-minimum values of \(\varepsilon_{1}(\omega)\), and their corresponding energies are given in Table 3 for PBE, PBE+umBJ, and PBE+umBJ+SOC, including both xx and zz directions. As shown in Table 3, the maximum and minimum values of \(\varepsilon_{1}(\omega)\) move towards the higher energy regions when PBE+umBJ, and PBE+umBJ+SOC functionals are used. In the static limit, optical spectra show anisotropy in two directions with differences of 0.48, 0.42, and 0.43 for PBE, PBE+umBJ, and PBE+umBJ+SOC, respectively. The imaginary part \(\varepsilon_{2}(\omega)\) has a very low value under 3.30 eV for PBE, under 3.80 eV for PBE+umBJ, and under 3.40 eV for PBE+umBJ+SOC, which is consistent with the calculated band gap energies.

The calculated real \(\varepsilon_{1}(\omega)\) and imaginary \(\varepsilon_{2}(\omega)\) part of the dielectric constant for BiOF within PBE, PBE+umBJ, and PBE+umBJ+SOC functionals a \(\varepsilon_{1}(\omega)\) for xx directions, b \(\varepsilon_{1}(\omega)\) for zz direction, c \(\varepsilon_{2}(\omega)\) for xx directions, and d \(\varepsilon_{2}(\omega)\) for zz direction
The absorption coefficient indicates how efficiently a material absorbs the electromagnetic wave at a certain wavelength or frequency [23]. Figure 7 illustrates the calculated absorption coefficient of the BiOF as function of incident photon energy in the xx and zz directions. Below the band gap energy of 3.29 eV, 3.72 eV, and 3.34 eV for the PBE, PBE+umBJ, and PBE+umBJ+SOC, respectively, the absorption coefficient is very low. The absorption coefficient rapidly increases with increasing incident photon energy. It has a maximum between 8.0 and 10.0 eV in the xx direction and a maximum between 8.5 and 9.0 eV in the zz direction for the PBE, PBE+umBJ, and PBE+umBJ+SOC. The absorption coefficients decrease rapidly in the xx direction above 9 eV and in the zz direction above 10 eV, respectively. Optical conductivity (σ), which describes how a material interacts with light, of BiOF is shown in Fig. 8 for the PBE, PBE+umBJ, and PBE+umBJ+SOC for both polarizations. The optical conductivities increase rapidly around the band gap energies for both polarizations. The photon energies corresponding to the first three peaks and the maximum value of the optical conductivities obtained with PBE, PBE+umBJ, and PBE+umBJ+SOC are presented in Table 4. As presented in Fig. 8, the maximum conductivity of the BiOF lies in the UV light spectrum.

The calculated optical absorption coefficient of BiOF within PBE, PBE+umBJ, and PBE+umBJ+SOC functionals a for xx directions and b for zz directions

The calculated optical conductivity σ (1/Ω.cm) of BiOF within PBE, PBE+umBJ, and PBE+umBJ+SOC functionals a for xx direction and b for zz direction
4 Conclusions
To summarize, using the first-principles DFT computations, the effect of the various exchange–correlation functionals on the electronic, structural, and optical properties of BiOF was investigated. LDA, PBE, PBEsol, and WC functionals were used to accomplish structural optimization, and as a result, structural properties such as equilibrium volumes, ground state energies, lattice parameters, and pressure derivatives were determined. The results indicate that the optimized lattice constants for BiOF are in consistent with the available experimental results. Furthermore, the electronic properties were calculated using LDA, PBE, PBEsol, and WC functionals. It can be concluded that the PBE approximation produced the most enhanced band gap result when compared to others. It is found that BiOF has a direct band gap when using PBE functionals; on the other side, BiOF has a near-direct band gap when LDA, PBEsol, and WC functionals are applied. The band gap differs by 9.7% from the experimental band gap when PBE functional was used. To achieve more enhanced band gap, umBJ and mBJ potentials were employed to all functionals. The band gaps were improved when umBJ potential was employed to the LDA, PBE, PBEsol, and WC functionals. The best band gap energy, which differs only 2.28% from the experimental band gap, was obtained with PBE+umBJ functionals. In the case of mBJ potential, the band gap worsened and was observed to be at least 30% different from the experimental band gap. In addition, since BiOF contains a heavy Bi atom, it is recommended to take into account the SOC effect. Considering the SOC effect, PBE+umBJ+SOC provided the best band gap, which differs by 4.56% from experimental results. Lastly, TDOS and PDOS were generated for PBE, PBE+umBJ, and PBE+umBJ+SOC functionals, in which the most enhanced band gaps were observed. Considering the results at the high simetry point Z, VBM is about the same for all three functionals, but CBM moves upward in energy when PBE+umBJ and PBE+umBJ+SOC are applied, resulting in a larger band gap. The optical properties of BiOF were studied for both in xx and zz directions. Dielectric function, absorption coefficient, and optical conductivity were explored for the photon energies up to 14 eV. All the optical features of BiOF showed anisotropy in the xx and zz directions. Hopefully, these findings will contribute to a deeper understanding of the structural, electronic, and optical properties of BiOF, as well as develop a more accurate computational model of BiOF.
Data Availability
Data will be made available on reasonable request.
References
W. Kohn, A.D. Becke, R.G. Parr, Density functional theory of electronic structure. J. Phys. Chem. 100(31), 12974–12980 (1996)
W. Kohn, Nobel lecture: Electronic structure of matter wave functions and density functionals. Rev. Mod. Phys. 71(5), 1253–1266 (1999)
K. Schwarz, P. Blaha, Solid state calculations using WIEN2k. Comput Mater. Sci. 28(2), 259–273 (2003)
D.J. Singh, Planewaves, pseudopotentials, and the LAPW method (Kluwer Academic Publisher, Boston, 1994)
W. Kohn, L.J. Sham, Self-consistent equations including exchange and correlation effects. Phys. Rev. 140(4A), A1133–A1138 (1965)
J.P. Perdew, K. Burke, M. Ernzerhof, Generalized gradient approximation made simple. Phys. Rev. Lett. 78(7), 1396 (1996)
J.P. Perdew et al., Restoring the density-gradient expansion for exchange in solids and surfaces. Phys. Rev. Lett. 100(13), 136406 (2008)
Z. Wu, R.E. Cohen, More accurate generalized gradient approximation for solids. Phys. Rev. B 73(23), 235116 (2006)
S. Li, C. Zhang, F. Min, X. Dai, C. Pan, W. Cheng, Electronic structural properties of BiOF crystal and its oxygen vacancy from first-principles calculations. Russ. J. Phys. Chem. A 91(12), 2425–2430 (2017)
W. Su et al., Synthesis and catalytic performances of a novel photocatalyst BiOF. Scripta Materialia 62(6), 345–348 (2010)
W.L. Huang, Q. Zhu, DFT calculations on the electronic structures of BiOX (X = F, Cl, Br, I) photocatalysts with and without semicore Bi 5d states. J. Comput. Chem 30(2), 183–190 (2009). https://doi.org/10.1002/jcc.21055
M. Canpolat, C. Kürkçü, Ç. Yamçıçıer, Z. Merdan, Structural and electronic properties of BiOF with two-dimensional layered structure under high pressure: Ab initio study. Solid State Commun. 288, 33–37 (2019)
P. Blaha, K. Schwarz, F. Tran, R. Laskowski, G.K.H. Madsen, L.D. Marks, WIEN2k: An APW+lo program for calculating the properties of solids. J. Chem. Phys. 152(7), pp (2020)
D. Koller, F. Tran, P. Blaha, Improving the modified Becke-Johnson exchange potential. Phys. Rev. B 85(15), 155109 (2012)
M. Causà, R. Dovesi, C. Pisani, C. Roetti, Electronic structure and stability of different crystal phases of magnesium oxide. Phys. Rev. B 33(2), 1308–1316 (1986)
J.L. Soubeyroux, S.F. Matar, J.M. Reau, P. Hagenmuller, Etude des proprietes structurales et electriques de la solution solide Pb1−xBixOxF2−x. Solid State Ion. 14(4), 337–345 (1984)
S. Singh, R. Sharma, M. Khanuja, A review and recent developments on strategies to improve the photocatalytic elimination of organic dye pollutants by BiOX (X=Cl, Br, I, F) nanostructures. Korean J. Chem. Eng. 35(10), 1955–1968 (2018)
J. Heyd, J.E. Peralta, G.E. Scuseria, R.L. Martin, Energy band gaps and lattice parameters evaluated with the Heyd-Scuseria-Ernzerhof screened hybrid functional. J. Chem. Phys. 123(17), 174101 (2005). https://doi.org/10.1063/1.2085170
F. Tran, P. Blaha, Accurate band gaps of semiconductors and insulators with a semilocal exchange-correlation potential. Phys. Rev. Lett. 102(22), 226401 (2009)
H.T.T. Nguyen et al., Spin–orbit coupling effect on electronic, optical, and thermoelectric properties of Janus Ga2SSe. RSC Adv 10(73), 44785–44792 (2020). https://doi.org/10.1039/D0RA08279A
V. Brousseau-Couture, X. Gonze, M. Côté, Effect of spin-orbit coupling on the zero-point renormalization of the electronic band gap in cubic materials: First-principles calculations and generalized Frohlich model. Phys. Rev. B 107(11), 115173 (2023)
J. Sun, H.-T. Wang, N.-B. Ming, J. He, Y. Tian, Optical properties of heterodiamond B2CN using first-principles calculations. Appl. Phys. Lett. 84(22), 4544–4546 (2004)
D. Sharma, A. Verma, R. Bhandari, S. Kumari, V. Jindal, Ab initio studies of structural, electronic, optical, elastic and thermal properties of Ag-chalcopyrites (AgAlX2: X=S, Se). Mater. Sci. Semicond. Process. 26, 187–198 (2014)
Funding
Open access funding provided by the Scientific and Technological Research Council of Türkiye (TÜBİTAK).
Author information
Authors and Affiliations
Contributions
Yakup Boran is responsible for the following: study conception and design, analysis and interpretation of results, and preparation of manuscript. Hüsnü Kara is responsible for the following: analysis and interpretation of results, reviewing, and editing.
Corresponding author
Ethics declarations
Competing Interests
The author declares no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Boran, Y., Kara, H. A Comprehensive Density Functional Theory Analysis on Structural, Electronic, and Optical Properties of BiOF. Braz J Phys 54, 159 (2024). https://doi.org/10.1007/s13538-024-01523-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s13538-024-01523-w