
Abstract
Background
Tumor-associated macrophages (TAMs) are associated with unfavorable patient prognosis in many cancer types. However, TAMs are a heterogeneous cell population and subsets have been shown to activate tumor-infiltrating T cells and confer a good patient prognosis. Data on the prognostic value of TAMs in colorectal cancer are conflicting. We investigated the prognostic effect of TAMs in relation to tumor-infiltrating T cells in colorectal cancers.
Methods
The TAM markers CD68 and CD163 were analyzed by multiplex fluorescence immunohistochemistry and digital image analysis on tissue microarrays of 1720 primary colorectal cancers. TAM density in the tumor stroma was scored in relation to T cell density (stromal CD3+ and epithelial CD8+ cells) and analyzed in Cox proportional hazards models of 5-year relapse-free survival. Multivariable survival models included clinicopathological factors, MSI status and BRAFV600E mutation status.
Results
High TAM density was associated with a favorable 5-year relapse-free survival in a multivariable model of patients with stage I–III tumors (p = 0.004, hazard ratio 0.94, 95% confidence interval 0.90–0.98). However, the prognostic effect was dependent on tumoral T-cell density. High TAM density was associated with a good prognosis in patients who also had high T-cell levels in their tumors, while high TAM density was associated with poorer prognosis in patients with low T-cell levels (pinteraction = 0.0006). This prognostic heterogeneity was found for microsatellite stable tumors separately.
Conclusions
This study supported a phenotypic heterogeneity of TAMs in colorectal cancer, and showed that combined tumor immunophenotyping of multiple immune cell types improved the prediction of patient prognosis.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
The immune tumor microenvironment (iTME) is an integral part of cancer ecology and plays an important role in tumor development [1]. The iTME consists of diverse cell types and differentiation states, and the cellular composition has a strong prognostic effect in many cancer types [2]. Tumor infiltrating T cells, in particular CD8+ T cells, are associated with a favorable prognosis [3], whereas tumor-associated macrophages (TAMs) are generally associated with an unfavorable prognosis [3,4,5].
In healthy tissue, macrophages have the ability to sense and respond to tissue injury by clearing damaged cells, as well as by recruiting and activating T cells that can help restoring tissue integrity [6]. It is therefore a paradox that TAMs have been shown to promote tumor growth and development of metastases. This can occur through mechanisms such as angiogenesis, production of tumor growth factors, and immunosuppression [5, 7, 8]. TAMs may also have a negative impact on the efficacy of diverse cancer therapies, including immune checkpoint inhibitors, radiotherapy and chemotherapy [9,10,11].
However, TAMs are a heterogeneous cell population, and specific subpopulations can have anti-tumorigenic properties. Macrophages expressing FOLR2 can prime effector CD8+ T cells in the tumor stroma and confer a better survival in breast cancer patients [12]. Subsets of TAMs produce the T-cell attracting chemokine CXCL9 and have positive prognostic associations across several cancer types [13,14,15]. Furthermore, pharmacological activation of TAMs can drive potent anti-tumor immunity together with activated CD8+ T cells in mouse models [16, 17]. Close interaction between T cells and TAMs therefore appears to be important for creation of an anti-tumor microenvironment.
Colorectal cancers (CRCs) have a complex and highly diverse iTME. This is partly determined by genomic factors. In particular, tumors with DNA mismatch repair deficiency and microsatellite instability (MSI) have dense immune cell infiltrations. These hypermutations are also associated with a favorable survival among patients with non-metastatic cancers. However, tumor-infiltrating T cells have prognostic associations beyond the MSI phenotype. This has been thoroughly demonstrated with the Immunoscore [18] and other immunohistochemistry-based approaches [19]. It has also been shown that regulatory T cells can counteract the positive prognostic effect of cytotoxic T cells [19], highlighting heterogeneity of the iTME. Data on the prognostic effect of TAMs in CRC are conflicting, with reports of both a favorable and an unfavorable prognostic effect [20,21,22,23,24,25]. This potentially reflects the relative presence of subpopulations of TAMs with pro-tumorigenic versus anti-tumorigenic properties [5, 7, 12, 13].
The combination of computational and spatial analysis in digital pathology, with a particular focus on immune cell patterns like macrophages and lymphocytes, provides an intricate understanding of the intra-tumoral immune response, and its implication on survival [26, 27]. This can improve the depth and accuracy of diagnosis and prognosis for cancer patients.
We hypothesized that immunophenotyping of CRCs based on markers for both TAMs and tumor-infiltrating T cells would resolve the prognostic effect. To investigate this we used multiplex fluorescence immunohistochemistry and digital image analyses on tissue microarrays (TMAs) of a single-hospital series of 1720 patients.
2 Materials and methods
2.1 Patients and tumor tissue microarrays
Formalin-fixed and paraffin-embedded samples from the primary tumors of two independent cohorts of patients from a single hospital that were treated by surgical resection for stage I–IV CRC were collected from the diagnostic biobank at Oslo University Hospital, Norway (n = 1720; Supplemental Table 1). This included 1429 (83%) patients diagnosed with TNM stage I–III CRC (locoregional disease) and 288 (17%) patients diagnosed with stage IV (distant metastatic disease, missing data for 3 patients). Patient treatment and follow-up were according to standard national guidelines. Clinicopathological data were extracted from the patients’ medical records and registered in a uniform database. Follow-up data for cancer relapse and survival was complete for at least 5 years for all patients except two; data from one patient is missing and one is censored at 4.2 years.
TMAs were constructed from a single tissue core of the central tumor area of blocks selected by an expert pathologist for representativeness, as previously described [19]. Norwegian series 1 (NS1) included patients treated between 1993 and 2003 (n = 922) and the TMAs were constructed from 0.6 mm diameter cores. Norwegian series 2 (NS2) included patients treated between 2003 and 2012 (n = 798) and the TMAs consisted of 1.0 mm diameter cores. MSI status, BRAFV600E mutations, KRAS mutations [28,29,30,31] and T cell markers (CD3 and CD8) have previously been scored [19]. There were no major differences in the distributions of clinicopathological factors or molecular markers between the two series (Supplemental Table 1). We have shown that intraepithelial CD8 and stromal CD3 scores on these TMAs provided similar prognostic power to separate reports on the Immunoscore in multivariable models of stage I–III colon cancers [18], although the Immunoscore considers both the central region and the invasive front of each tumor. This supported representativeness of the TMAs for prognostic analyses of the iTME [19].
2.2 Multiplex fluorescence immunohistochemistry
A multi-color multiplex immunohistochemistry stain was performed on 4 μm thick sections of the TMAs. The staining was performed using antibodies against CD163 (clone EPR14643, Abcam, diluted 1:1000) visualized with Opal 520, CD68 (clone KP1, DAKO/Agilent, diluted 1:3000) visualized with Opal 690, a cocktail of antibodies targeting the epithelial cancer cells (E-cadherin [clone 36, BD-biosciences, diluted 1:20,000], cytokeratin C-11 [Abcam, diluted 1:4000], cytokeratin Type I/II [Thermo Fisher Scientific, diluted 1:2000]) visualized with Opal 570, CD206 (Clone E2L9N, Cell Signaling, diluted 1:1200) visualized with Opal 620, and also included incubation with DAPI for staining of cell nuclei prior to mounting. The stains were carried out using a multiplex kit (NEL810001KT) together with Opal 620 (FP1495001KT, both from PerkinElmer/Akoya, Marlborough, MA, USA). The Opal protocol (PerkinElmer/Akoya) was followed with the exception that slide deparaffinization, antigen retrieval, and antibody stripping were all performed in a PT-link module (DAKO/Agilent, Santa Clara, CA, USA). Slide deparaffinization and initial antigen retrieval were performed at the same time by placing the slides in EnVision FLEX Target retrieval solution, Low pH (DAKO/Agilent) preheated to 65 °C, heating for 20 min at 97 °C and cooling back down to 65 °C. Antibody stripping was performed in the PT-link solution with high/low pH buffers from Akoya, as specified in Supplemental Table 2. During these cycles, slides were placed in solutions preheated to 80 °C, heated to 97 °C for 20 min and cooled back down to 80 °C. All reagents used are specified in Supplemental Table 3. The multiplex protocol was established in “test-TMAs” of samples from several tissue and cancer types prior to staining the NS1 and NS2 TMAs, following a previously described process [32]. In short, this included antibody testing and determination of the optimal antibody titer by chromogenic DAB-based immunohistochemistry, verification of staining patterns in single-plex fluorescence-based immunohistochemistry, verification of complete stripping/denaturing of the antibodies between detection rounds, and verification of staining patterns in multiplex fluorescence immunohistochemistry. Assessments of CD68 and CD163 staining provided by the Nordic immunohistochemical Quality Control (NordiQC; available at http://www.nordiqc.org) were a valuable resource during protocol optimization. The KP1 clone is one of the monoclonal antibodies recommended by NordiQC for staining CD68 by immunohistochemistry.
2.3 Digital image analysis
Stained TMAs were multispectrally imaged using the Vectra 3 system (PerkinElmer/Akoya). A single 20 × (0.5 μm/pixel) image was taken for each sample of the NS1 TMAs, and a 2 × 2 image field was captured for each sample of the NS2. Images were spectrally unmixed, including removal of tissue autofluorescence from the Opal fluorophore signal values, and analyzed by tissue and cell segmentation in inForm software v. 2.3.0 (PerkinElmer/Akoya). The tissue segmentation algorithm was trained on manually input ground truth labels for epithelial cancer cell regions, tumor stroma and background (empty regions on the glass slide). During algorithm training we found areas of high erythrocyte density to be (incorrectly) labeled as epithelial cancer regions. Thus, we also included an erythrocyte region which was subsequently combined with the tumor stroma region during downstream marker scoring according to tumor region in R (described below). Only signals from the epithelial antibody cocktail (stained by Opal 570), DAPI and tissue autofluorescence were used for training. Batch-analysis algorithms were optimized based on a subset of the samples for each of the NS1 and NS2 TMAs. For NS1, images from 16 samples were used to make the batch-analysis algorithm. Five ground truth annotations were made for the epithelial cancer region, seven for tumor stroma, six for background and three for high-density erythrocyte regions during tissue segmentation training. For NS2, images from 15 samples were used. Seven ground truth annotations were made for the epithelial cancer region, 14 for stroma, 14 for background and three for high-density erythrocyte regions. Tissue segmentation training was performed with medium pattern scale, and the analyses were output with “fine” segmentation resolution. The minimum segment size was set to 1000 pixels. Both algorithms achieved ≥98% training accuracy based on the ground truth labels. For cell segmentation, the same settings were used for both series/algorithms, and individual nuclei were first segmented with the counterstaining approach based on DAPI signals. Minimum size was set to 80 pixels, with a typical size of 320 pixels. Minimum signal was set to 0.24, splitting was set at 2.26 and growing was set to 0.35. Membrane signal was used to aid segmentation. For cytoplasm segmentation, the inner distance to nucleus was set to 1 pixel, outer distance to 6 pixels and minimum size was set to 20 pixels. For membrane segmentation, Opal 690 full scale count was set to 18, Opal 520 to 10, Opal 570 to 30 and Opal 620 to 30. Distance to membrane (for maximum cell size determination) was set to 12 pixels. All images were manually inspected in the review tab in inForm after batch analysis of the two TMA-cohorts, and tissue folds and necrotic regions were manually marked and excluded from the image analysis. A flow-chart for the digital image analysis and marker scoring pipeline is outlined in Supplemental Fig. 1.
2.4 Marker scoring
Data tables with raw mean fluorescence intensity values per marker per cell for each TMA core were exported from inForm and further processed in R (v. 3.6.3). Each individual cell in each individual TMA core was scored as positive/negative for CD68 and CD163 based on the mean nuclear signal intensity of their corresponding fluorophores in each patient series (normalized counts, total weighting in inForm software; Supplemental Table 4). The nuclear signal was used since marker signals in this cell segment were sufficient to score cells as positive or negative, and nuclear segmentation was more consistent than cytoplasmic and membrane segmentation. The data on CD206 were not included in the current study due to poor technical quality.
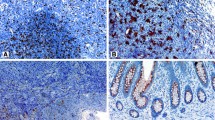
Tumor-infiltration with macrophages was most prominent in stromal regions, outside of the cytokeratin positive regions marking the tumor epithelium (Fig. 1A). Macrophage infiltration was therefore scored in the stromal tissue compartment of each TMA core and normalized by dividing by the stromal tissue area (in mm2) of the individual core.

Prognostic effect of TAMs in stage I–III CRC. A Three TMA cores stained against CD68 (green), CD163 (red) and cytokeratin (purple) illustrate tumors with low (left), intermediate (middle) and high (right) numbers of CD68+ TAMs. B Kaplan-Meier plot of 5-year RFS according to the stromal density of CD68+ TAMs (high, intermediate, low) among patients treated by complete resection for stage I–III CRC (the two patient cohorts NS1 and NS2 combined; n = 1096). C Kaplan-Meier plots for the same analysis as in B, but here stratified according to the stromal densities of CD68+CD163- (left) and CD68+CD163+ (right) TAMs. Hazard ratios (HR), p-values and 95% confidence intervals (CI) are from Cox proportional hazards analyses and p-values. *Significant values p < 0.05 (Color figure online)
Tissue cores with poor technical quality upon manual image inspection were excluded from downstream analysis (n = 191). Additional samples (n = 69) were excluded according to the following data quality control criteria: less than 5% malignant epithelial tissue; fewer than 100 malignant epithelial cells; fewer than 50 stromal cells; or a total tissue area of less than 150,000 pixels (0.0375 mm2).
2.5 Survival analyses
The study follows the Reporting recommendations for tumor marker prognostic studies (REMARK; Supplemental Table 5). The primary endpoint was 5-year relapse-free survival (RFS), analyzed as the time from surgery to recurrence or death from any cause as defined by Punt et al. [33]. Analyses of RFS were performed in patients with a free resection margin >1 mm and no residual tumor (R0 status) and therefore excluded stage IV cancers. Patients who received pre-operative chemoradiotherapy (n = 32), had synchronous primary CRCs (n = 29), or were registered with an RFS of 0 (n = 4) were also excluded, leaving 1096 patients with stage I–III CRC and good technical quality of immunohistochemistry data for analyses. Survival analysis of patients with stage IV CRC was performed with 5-year overall survival (OS) as the endpoint, since the majority of these patients did not receive radical treatment. Complete 5-year OS data was available for all patients and was calculated from the time of surgery of the primary tumor (n = 248 patients with good technical quality of immunohistochemistry data). Uni- and multivariable Cox proportional hazards models of immune cell infiltration scores were calculated with the coxph function in the R package survival (v. 3.1-8). Potentially confounding variables included in multivariable analysis of CD68 (Table 1) were pT and pN stage, tumor location, MSI status, BRAFV600E mutational status, patient age and sex. Multivariable survival analysis was stratified by cohort (NS1 and NS2). Patients were excluded from multivariable analysis if data was missing for any of the variables included. The assumptions of proportional hazards were tested using the cox.zph function in the survival package. Kaplan-Meier plots were made using the survminer package (v. 0.4.9), while statistics presented in the plots are based on the Cox proportional hazards model, as described above.
2.6 Statistics
All statistical analyses were performed in Stata and RStudio v. 1.1.383 with R v. 3.6.3. All statistical tests were two-sided and p-values less than 0.05 were considered significant. Group comparisons were performed using Kruskal-Wallis and Wilcoxon tests, while correlations were measured by the Pearson correlation coefficient.
3 Results
3.1 High density of tumor-associated macrophages is associated with better prognosis in stage I–III microsatellite stable CRC
The density of TAMs in the stromal compartment of CRCs was evaluated by multiplex fluorescence immunohistochemistry of a pan-TAM marker (CD68) and a marker for a TAM sub-population (CD163; Fig. 1A) on TMAs of primary tumors from 1720 stage I–IV CRCs (collected in two time periods at a single hospital; Supplemental Table 1). Samples (n = 260, 15%) with poor technical quality evaluated upon manual image inspection (large tissue folds or necrotic regions) or that didn’t meet the data quality control parameters (criteria specified in the Materials and Methods section) were excluded, leaving 1460 samples for further analysis. 44% of CD68+ TAMs co-expressed CD163 (Supplemental Table 1). The density of TAMs decreased with each progressive cancer stage (Supplemental Fig. 2), and locoregional cancers (stage I–III) had a higher density of CD68+ TAMs than cancers with distant metastasis (stage IV) irrespective of CD163 expression (Supplemental Fig. 3).
Tumors were stratified into three equally sized groups based on the stromal density of CD68. The group with low density had less than 268 CD68+ cells per mm2 of tumor stroma, the intermediate group had between 268 and 707, and the group with high density had above 707 CD68+ cells per mm2 of tumor stroma (maximum density was 7010 CD68+ cells per mm2). Analysis of 5-year RFS in patients treated by complete resection for stage I–III CRC showed that high infiltration of CD68+ TAMs was associated with a significantly better survival than lower infiltration, both when examining the two cohorts together and separately (Fig. 1B and Supplemental Fig. 4). This was not dependent on the expression of CD163, since high density of both CD68+CD163− and CD68+CD163+ TAMs were associated with better 5-year RFS than intermediate/low densities (Fig. 1C). Further analyses were therefore performed based on CD68+ TAMs. The density of CD68+ TAMs was prognostic also in a multivariable analysis with clinicopathological and genetic prognostic markers (Table 1). There was no difference in the prognostic effect of CD68 according to whether patients with stage III cancers were treated with adjuvant chemotherapy or not (pinteraction = 0.9, with CD68 as continuous variable). However, TAM densities and their prognostic value differed according to tumoral MSI status. MSI tumors had a significantly higher TAM density than microsatellite stable (MSS) tumors (Fig. 2A), and the prognostic value of CD68high was exclusive to patients with MSS tumors, although the interaction test was not significant (pinteraction = 0.8, with CD68 as continuous variable) (Fig. 2B). There was no difference in the 5-year overall survival of patients with stage IV cancers according to the density of CD68 (Supplemental Fig. 5).

Density and prognostic effect of CD68+ TAMs relative to MSI status. A Box plots of TAM densities (CD68+ per mm2, log2 transformed) in primary MSS and MSI tumors of stage I–IV CRCs. P-value was estimated by the Wilcoxon test. B Kaplan-Meier plots of 5-year RFS according to the stromal density of CD68+ TAMs in MSS and MSI tumors separately for patients treated by complete resection of stage I–III CRC. Hazard ratios (HR), p-values and 95% confidence intervals (CI) are from Cox proportional hazards analyses and p-values. *Significant values p < 0.05
3.2 Prognostic value of TAMs depends on the number of tumor-infiltrating T cells
The density of TAMs correlated with the density of stromal CD3+ and epithelial CD8+ T cells in CRCs (Pearson’s correlation 0.4 and 0.3, respectively, p < 0.0001; Supplemental Fig. 6). We have previously shown that low levels of these T-cell populations are associated with a poor survival in this patient cohort [19]. Here, we performed a stratified analysis of TAM density according to T cells. CD68high was associated with a favorable 5-year RFS among patients with Thigh tumors (epithelial CD8high and stromal CD3high; Fig. 3A). In contrast, CD68high was associated with a poor survival among patients with Tlow tumors (epithelial CD8low and stromal CD3low; pinteraction = 0.0006). TAMs had no prognostic effect in patients with mixed levels of the two T-cell populations (stromalCD3high epithelialCD8low and stromalCD3low epithelialCD8high; Supplemental Fig. 7). The prognostic heterogeneity of CD68high according to high and low T-cell density was consistent among patients with MSS tumors separately (pinteraction = 0.0007; Fig. 3B), while a similar analysis of MSI tumors was inconclusive due to low patient numbers in several of the subgroups (Supplemental Fig. 8).

Prognostic heterogeneity of CD68+ TAMs relative to tumor-infiltrating T cells. A Kaplan-Meier plots of 5-year RFS according to the stromal density of CD68+ TAMs in stage I–III CRCs with high (stromal CD3high and epithelial CD8high) and low (stromal CD3low and epithelial CD8low) T-cell densities separately. B Kaplan-Meier plots from the same analysis as in A, but including MSS tumors only. Hazard ratios (HR), p-values and 95% confidence intervals (CI) are from Cox proportional hazards analyses and p-values. *Significant values p < 0.05
4 Discussion
This study shows that integrated immunophenotyping based on TAMs and tumor-infiltrating T cells can specify the prognostic heterogeneity of TAMs in locoregional CRCs. While high TAM density was a positive prognostic factor in general, the prognostic effect was dependent on the presence of T cells, and high TAM densities appeared to confer a particularly poor patient survival when found in tumors with low T-cell densities. This apparent dichotomy may, at least in part, be explained by a bidirectional interaction between TAMs and T cells. An iTME rich in T cells contains activated and IFN-γ producing T cells [34]. Macrophages become highly phagocytic and efficient at antigen presentation in response to IFN-γ [35]. IFN-γ activated TAMs may thus attack and phagocytose malignant cells and provide anti-tumor activity through their capacity to stimulate T cells and produce chemokines that attract T cells [13, 14, 36]. Thus, TAM activation may create a positive feedback loop for further recruitment and activation of T cells. In contrast, in a T-cell deprived iTME, it is likely that pro-tumorigenic TAMs will dominate and promote angiogenesis, produce tumor growth factors and suppress other immune cells [5, 7, 8]. A larger panel of TAM markers to delineate various populations, along with integrated spatial analysis with T-cell populations is needed to confirm this dichotomy.
Multiple studies have investigated the prognostic value of CD68+ TAMs in CRC, several of which have reported that high numbers of TAMs are associated with better patient prognosis [7, 37,38,39]. This is in contrast to most other solid tumor types, and the definite reasons for this disparity are yet to be completely understood. Intestinal macrophages are constantly replenished from blood monocytes and interact with commensal bacteria and the dynamic intestinal niche [40,41,42]. Intestinal macrophages and TAMs in CRC may therefore have different properties from TAMs in other cancers.
The prognostic effect of TAMs was exclusive to patients with MSS tumors. The lack of a prognostic impact in the MSI setting may be associated with the good survival of these patients in general. However, the favorable prognostic effect of MSI is attributed to a high infiltration of T cells in response to the thousands of neoantigens produced in these mismatch repair deficient tumors. Our study was not sufficiently powered to discern the prognostic heterogeneity of TAMs in relation to both T-cell densities and MSI status. Nonetheless, approximately 85% of all primary CRCs have the MSS phenotype, and our results are representative for the majority of patients with locoregional CRC.
The heterogeneous prognostic effect suggests that TAMs could be important targets for anti-tumor therapy in CRC [36]. Macrophages are extremely plastic cells that differentiate in response to cues from the microenvironment [43]. This plasticity can be exploited therapeutically, and several studies have shown that macrophages can be reprogrammed in vivo [16, 17, 44]. A better understanding of which TAM phenotypes constitute the CD68high populations in the T-cell rich versus T-cell depleted iTME of CRC will be important to identify new therapeutic targets.
A limitation of this study is that TMAs only include a small part of the original tissue sample and offer a restricted view of the iTME in the tumor compared to whole tissue sections. There are immunological differences within a tumor. However, our study demonstrates that the immunological trends within a tumor, represented with a large sample size from central tumor regions, have predictive value. Another limitation is that the macrophage and T-cell markers were analyzed on serial sections. Other techniques for even higher-plex fluorescence immunohistochemistry, like CODEX and BLEACH &STAIN, could be valuable to analyze these and additional markers within the same tissue section, and would allow for even more advanced spatial analyses. These approaches do, however, have drawbacks, such as increased cost, standardization challenges, and the requirement for significant data analysis in addition to their technological complexity. Serial tissue section is a conventional and economical technique with well-established guidelines for regular pathological diagnostic assessment.
Integrated analysis of multiple immune cell markers on tissue sections can be compatible with standard diagnostics, as equipment for simultaneous in situ evaluation of three immune markers is available in many pathology laboratories today. Alternatively, tumor-infiltrating T cells and TAMs can be stained on serial tissue sections, as was performed in this study. There is a need to validate the superior prognostic power and to develop the optimal assay for combined evaluation of T cell and TAM counts in independent cohorts.
5 Conclusions
We define prognostic heterogeneity of TAMs in CRC and suggest that combined quantitation of both T cells and TAMs can increase the prognostic power of iTME tests compared to examining T cells alone.
Data availability
All data generated or analyzed during this study are included in this published article. Immunohistochemistry data of macrophage markers will be made available upon reasonable request to the corresponding author.
Abbreviations
- CRC:
-
Colorectal cancer
- iTME:
-
Immune tumor microenvironment
- TAMs:
-
Tumor-associated macrophages
- TMA:
-
Tissue microarray
- MSI:
-
Microsatellite instable
- MSS:
-
Microsatellite stable
- RFS:
-
Relapse-free survival
References
M. Binnewies, E.W. Roberts, K. Kersten, V. Chan, D.F. Fearon, M. Merad, et al., Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat. Med. 24(5), 541–550 (2018)
V. Thorsson, D.L. Gibbs, S.D. Brown, D. Wolf, D.S. Bortone, T.H. Ou Yang, et al., The immune landscape of cancer. Immunity 48(4), 812–30 e14 (2018)
D. Bruni, H.K. Angell, J. Galon, The immune contexture and immunoscore in cancer prognosis and therapeutic efficacy. Nat. Rev. Cancer 20(11), 662–680 (2020)
T. Xue, K. Yan, Y. Cai, J. Sun, Z. Chen, X. Chen, et al., Prognostic significance of CD163+ tumor-associated macrophages in colorectal cancer. World J. Surg. Oncol. 19(1), 186 (2021)
L. Cassetta, S. Fragkogianni, A.H. Sims, A. Swierczak, L.M. Forrester, H. Zhang, et al., Human tumor-associated macrophage and monocyte transcriptional landscapes reveal cancer-specific reprogramming, biomarkers, and therapeutic targets. Cancer Cell 35(4), 588–602 e10 (2019)
M.D. Park, A. Silvin, F. Ginhoux, M. Merad, Macrophages in health and disease. Cell 185(23), 4259–4279 (2022)
B. Ruffell, L.M. Coussens, Macrophages and therapeutic resistance in cancer. Cancer Cell 27(4), 462–472 (2015)
Y. Liu, Q. Zhang, B. Xing, N. Luo, R. Gao, K. Yu, et al., Immune phenotypic linkage between colorectal cancer and liver metastasis. Cancer Cell 40(4), 424–37 e5 (2022)
A. Mantovani, F. Marchesi, A. Malesci, L. Laghi, P. Allavena, Tumour-associated macrophages as treatment targets in oncology. Nat. Rev. Clin. Oncol. 14(7), 399–416 (2017)
S.L. Shiao, B. Ruffell, D.G. DeNardo, B.A. Faddegon, C.C. Park, L.M. Coussens, TH2-polarized CD4(+) T cells and macrophages limit efficacy of radiotherapy. Cancer Immunol. Res. 3(5), 518–525 (2015)
J. Xu, J. Escamilla, S. Mok, J. David, S. Priceman, B. West, et al., CSF1R signaling blockade stanches tumor-infiltrating myeloid cells and improves the efficacy of radiotherapy in prostate cancer. Cancer Res. 73(9), 2782–2794 (2013)
R. Nalio Ramos, Y. Missolo-Koussou, Y. Gerber-Ferder, C.P. Bromley, M. Bugatti, N.G. Nunez, et al., Tissue-resident FOLR2(+) macrophages associate with CD8(+) T cell infiltration in human breast cancer. Cell 185(7), 1189–207 e25 (2022)
Y. Qu, J. Wen, G. Thomas, W. Yang, W. Prior, W. He, et al., Baseline frequency of inflammatory Cxcl9-expressing tumor-associated macrophages predicts response to avelumab treatment. Cell Rep. 32(1), 107873 (2020)
M. Pascual-Garcia, E. Bonfill-Teixidor, E. Planas-Rigol, C. Rubio-Perez, R. Iurlaro, A. Arias, et al., LIF regulates CXCL9 in tumor-associated macrophages and prevents CD8(+) T cell tumor-infiltration impairing anti-PD1 therapy. Nat. Commun. 10(1), 2416 (2019)
R. Bill, P. Wirapati, M. Messemaker, W. Roh, B. Zitti, F. Duval, et al., CXCL9:SPP1 macrophage polarity identifies a network of cellular programs that control human cancers. Science 381(6657), 515–524 (2023)
M. Binnewies, J.L. Pollack, J. Rudolph, S. Dash, M. Abushawish, T. Lee, et al., Targeting TREM2 on tumor-associated macrophages enhances immunotherapy. Cell Rep. 37(3), 109844 (2021)
J.L. Guerriero, A. Sotayo, H.E. Ponichtera, J.A. Castrillon, A.L. Pourzia, S. Schad, et al., Class IIa HDAC inhibition reduces breast tumours and metastases through anti-tumour macrophages. Nature 543(7645), 428–432 (2017)
F. Pages, B. Mlecnik, F. Marliot, G. Bindea, F.S. Ou, C. Bifulco, et al., International validation of the consensus immunoscore for the classification of colon cancer: a prognostic and accuracy study. Lancet 391(10135), 2128–2139 (2018)
C.H. Bergsland, M. Jeanmougin, S.H. Moosavi, A. Svindland, J. Bruun, A. Nesbakken, et al., Spatial analysis and CD25-expression identify regulatory T cells as predictors of a poor prognosis in colorectal cancer. Mod. Pathol. 35(9), 1236–1246 (2022)
S. Goerdt, O. Politz, K. Schledzewski, R. Birk, A. Gratchev, P. Guillot, et al., Alternative versus classical activation of macrophages. Pathobiology 67(5-6), 222–226 (1999)
P. Allavena, A. Sica, C. Garlanda, A. Mantovani, The yin-yang of tumor-associated macrophages in neoplastic progression and immune surveillance. Immunol. Rev. 222, 155–161 (2008)
A. Sica, P. Larghi, A. Mancino, L. Rubino, C. Porta, M.G. Totaro, et al., Macrophage polarization in tumour progression. Semin. Cancer Biol. 18(5), 349–355 (2008)
C. Lackner, Z. Jukic, O. Tsybrovskyy, G. Jatzko, V. Wette, G. Hoefler, et al., Prognostic relevance of tumour-associated macrophages and von Willebrand factor-positive microvessels in colorectal cancer. Virchows Arch. 445(2), 160–167 (2004)
J. Forssell, A. Oberg, M.L. Henriksson, R. Stenling, A. Jung, R. Palmqvist, High macrophage infiltration along the tumor front correlates with improved survival in colon cancer. Clin. Cancer Res. 13(5), 1472–1479 (2007)
Q. Zhou, R.Q. Peng, X.J. Wu, Q. Xia, J.H. Hou, Y. Ding, et al., The density of macrophages in the invasive front is inversely correlated to liver metastasis in colon cancer. J. Transl. Med. 8, 13 (2010)
I.P. Nearchou, B.M. Gwyther, E.C.T. Georgiakakis, C.G. Gavriel, K. Lillard, Y. Kajiwara, et al., Spatial immune profiling of the colorectal tumor microenvironment predicts good outcome in stage II patients. Npj Digit. Med. 3, 71 (2020)
A. Nestarenkaite, W. Fadhil, A. Rasmusson, S. Susanti, E. Hadjimichael, A. Laurinaviciene, et al., Immuno-interface score to predict outcome in colorectal cancer independent of microsatellite instability status. Cancers (Basel) 12(10), 2902 (2020)
M.A. Merok, T. Ahlquist, E.C. Royrvik, K.F. Tufteland, M. Hektoen, O.H. Sjo, et al., Microsatellite instability has a positive prognostic impact on stage II colorectal cancer after complete resection: results from a large, consecutive Norwegian series. Ann. Oncol. 24(5), 1274–1282 (2013)
H.M. Vedeld, M. Merok, M. Jeanmougin, S.A. Danielsen, H. Honne, G.K. Presthus, et al., CpG island methylator phenotype identifies high risk patients among microsatellite stable BRAF mutated colorectal cancers. Int. J. Cancer 141(5), 967–976 (2017)
J. Smeby, A. Sveen, M.A. Merok, S.A. Danielsen, I.A. Eilertsen, M.G. Guren, et al., CMS-dependent prognostic impact of KRAS and BRAFV600E mutations in primary colorectal cancer. Ann. Oncol. 29(5), 1227–1234 (2018)
C.H. Bergsland, J. Bruun, M.G. Guren, A. Svindland, M. Bjornslett, J. Smeby, et al., Prediction of relapse-free survival according to adjuvant chemotherapy and regulator of chromosome condensation 2 (RCC2) expression in colorectal cancer. ESMO Open 5(6), e001040 (2020)
N. Lopes, C.H. Bergsland, M. Bjornslett, T. Pellinen, A. Svindland, A. Nesbakken, et al., Digital image analysis of multiplex fluorescence IHC in colorectal cancer recognizes the prognostic value of CDX2 and its negative correlation with SOX2. Lab. Invest. 100(1), 120–134 (2020)
C.J. Punt, M. Buyse, C.H. Kohne, P. Hohenberger, R. Labianca, H.J. Schmoll, et al., Endpoints in adjuvant treatment trials: a systematic review of the literature in colon cancer and proposed definitions for future trials. J. Natl. Cancer Inst. 99(13), 998–1003 (2007)
K. Pelka, M. Hofree, J.H. Chen, S. Sarkizova, J.D. Pirl, V. Jorgji, et al., Spatially organized multicellular immune hubs in human colorectal cancer. Cell 184(18), 4734–52.e20 (2021)
P.J. Murray, T.A. Wynn, Protective and pathogenic functions of macrophage subsets. Nat. Rev. Immunol. 11(11), 723–737 (2011)
C. Baer, M.L. Squadrito, D. Laoui, D. Thompson, S.K. Hansen, A. Kiialainen, et al., Suppression of microRNA activity amplifies IFN-gamma-induced macrophage activation and promotes anti-tumour immunity. Nat. Cell Biol. 18(7), 790–802 (2016)
J. Li, L. Li, Y. Li, Y. Long, Q. Zhao, Y. Ouyang, et al., Tumor-associated macrophage infiltration and prognosis in colorectal cancer: systematic review and meta-analysis. Int. J. Colorectal Dis. 35(7), 1203–1210 (2020)
Z. Yang, M. Zhang, R. Peng, J. Liu, F. Wang, Y. Li, et al., The prognostic and clinicopathological value of tumor-associated macrophages in patients with colorectal cancer: a systematic review and meta-analysis. Int. J. Colorectal Dis. 35(9), 1651–1661 (2020)
Q.W. Zhang, L. Liu, C.Y. Gong, H.S. Shi, Y.H. Zeng, X.Z. Wang, et al., Prognostic significance of tumor-associated macrophages in solid tumor: a meta-analysis of the literature. PLoS One 7(12), e50946 (2012)
C.C. Bain, A. Bravo-Blas, C.L. Scott, E.G. Perdiguero, F. Geissmann, S. Henri, et al., Constant replenishment from circulating monocytes maintains the macrophage pool in the intestine of adult mice. Nat. Immunol. 15(10), 929–937 (2014)
C.C. Bain, A. Schridde, Origin, differentiation, and function of intestinal macrophages. Front. Immunol. 9, 2733 (2018)
D. Domanska, U. Majid, V.T. Karlsen, M.A. Merok, A.R. Beitnes, S. Yaqub, et al., Single-cell transcriptomic analysis of human colonic macrophages reveals niche-specific subsets. J. Exp. Med. 219(3), e20211846 (2022)
Y. Lavin, D. Winter, R. Blecher-Gonen, E. David, H. Keren-Shaul, M. Merad, et al., Tissue-resident macrophage enhancer landscapes are shaped by the local microenvironment. Cell 159(6), 1312–1326 (2014)
M. Molgora, E. Esaulova, W. Vermi, J. Hou, Y. Chen, J. Luo, et al., TREM2 modulation remodels the tumor myeloid landscape enhancing anti-PD-1 immunotherapy. Cell 182(4), 886–900 e17 (2020)
Acknowledgements
Not applicable.
Funding
Open access funding provided by University of Oslo (incl Oslo University Hospital). This study was supported by grants from the South Eastern Norway Health Authority (no. 2021058 to RAL) and by the Norwegian Cancer Society (no. 208336–2019 to AS).
Author information
Authors and Affiliations
Contributions
Conception and design: CHB, JB, RAL. Development of methodology: CHB, JB. Acquisition, analysis and interpretation of data: All authors. Drafting of manuscript: UM, CHB, AS, FLJ. Revisions and final approval of manuscript: All authors. Study supervision: ESB, SY, FLJ, RAL.
Corresponding author
Ethics declarations
Ethics approval and consent to participation
The study was approved by the Regional Committees for Medical and Health Research Ethics South East Norway–REC South East (project number 2005.1629; 2010/1805). The corresponding research biobank is registered (number 2781). Informed written consent was obtained from all patients prior to enrollment. The study was performed in accordance with the Declaration of Helsinki.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Majid, U., Bergsland, C., Sveen, A. et al. The prognostic effect of tumor-associated macrophages in stage I-III colorectal cancer depends on T cell infiltration. Cell Oncol. (2024). https://doi.org/10.1007/s13402-024-00926-w
Accepted:
Published:
DOI: https://doi.org/10.1007/s13402-024-00926-w