
Abstract
There are global challenges in addressing the oil spill treatment. Nanofiber has become a great potential in the oil spill cleaning process because of the environmental friendliness, high efficiency, low cost, and stability of the obtained nanofiber mats. This study presents a novel composite fabricated from cellulose acetate (CA) and cellulose nitrate (CN) nanofibers with the incorporation of carbon black (CA-CN/CB) for efficient oil removal. This nanofiber composite was fabricated in one-step electrospinning of 10% CA and CN solution with different concentrations of carbon black (CB). The morphology and fiber diameter of the CA-CN/CB nanofiber composite were analyzed using scanning electron microscopy (SEM), and they appeared to be smooth, uniform fibers without beads. The average fiber diameter was in nano-meter size and increased with the increasing CB amount in the composite, ranging from 327 to 755 nm. The FTIR results indicated the presence of CA and CN as characteristic peaks of C = O for CA and O-NO2 for CN. The nanofibers mats of the CA-CN, CA-CN/CB0.7, CA-CN/CB1.5, and CA-CN/CB2.2 composites had Brunauer–Emmett–Teller (BET) surface area of 15.29, 38.40, 4.08, and 6.17 m2 g−1, respectively. Under optimal conditions, CA-CN/CB nanofiber mats absorb more than their weight oil in just 30 min. The adsorption result showed that loading 1.5% of CB to CA-CN mats (CA-CN/CB1.5) was more favorable for oil adsorption. The CA-CN/CB1.5 nanofiber showed its reusability for oil adsorption. The Freundlich isotherm model was the most appropriate model among other isotherm models, including Langmuir and Temkin, with a value of correlation coefficient (R2) equal to or closer to unity, and this result was confirmed by the data obtained from studying different error function models. The adsorption kinetics showed that oil adsorption into CA-CN/CB1.5 nanofiber follows a pseudo-second-order kinetics model with R2 close to unity.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
Oil spills and oily wastewater have become a significant challenge around the world that has caused catastrophic damage to aquatic life and ecosystems [1,2,3]. Exploration, transportation, storage, and use of oil in the food and petrochemical industries can all result in oil spills across land and water [4, 5]. Motor oil is made out of base oils that have been strengthened with numerous additives, such as detergents, dispersants, and multi-grade oils, and is a petroleum derivative of hydrocarbons. It is used to lubricate internal combustion engines, reduce friction and dust, protect against wear and tear, and clean, anti-corrode, and cool them [6].
Motor oil breaks down into organic compounds by the force of temperature during its use that are toxic, and it also picks up a number of impurities from engine wear such as metal particles, gums, ash and carbon residue, most of which are highly toxic and cause chemical changes in the oil. Large quantities of used engine oil are disposed of into water through fountains and wells and also swept from the ground with rainwater that affects society environmentally, economically, and socially [7,8,9,10]. Waste oil harms aquatic organisms’ ecology, reduces biological activity, reduces biodiversity, impairs stout aquatic mammals, causes dehydration, and, in some situations, can cause kidney failure when oil reaches the lungs [1, 11].
Several oil–water separation methods include chemical methods such as sedimentation in a centrifugal field [12], adsorption, in situ burning and dispersion, a biological method by employing microorganisms, and physical methods such as booms and skimmers that were widely used for oil–water separation [13]. However, these approaches may not be efficient because all chemical procedures are expensive and unfriendly to the environment, while biological methods take time and their performance is strongly influenced by temperature, pH, oxygen level, organic species, and other factors [14]. Sorbents are the most effective and cheapest method, which has been used to clean up oil spills on beaches. As a result, a global challenge exists to produce a rapid, efficient, and cost-effective sorbent material capable of removing or capturing oil spills before or after release into surface waters [15]. Hence, eco-friendly natural biomass such as cellulose offers many advantages, such as cost-effectiveness and excellent biodegradability [16, 17].
Synthetic organic, inorganic, and natural organic adsorbents are the major types of adsorbents used for oil/water removal [18]. A high capacity for absorption, oil recovery and retention, oleophilicity-hydrophobicity, reusability, and biodegradability are all features of an excellent oil adsorbent. Granules, woven and nonwoven fibers, and particles are all examples of adsorbents [14, 15]. Because of its versatility, low cost, and promise for applications in a variety of industries, electrospinning is commonly employed for nanofiber synthesis [19, 20]. Electrospun nanofibers have been successfully produced using a variety of polymers, including natural, synthetic, and hybrid materials [21]. Cellulose is the world’s first naturally abundant bio-based material [22]. It is non–toxic, biodegradable, and biocompatible, as well as organic, renewable, and environmentally friendly [23]. Cellulose has a large specific surface area, a large Young’s modulus, a high degree of crystallinity, polymerization, and tensile strength [24]. Due to the hydrophilicity of cellulose and its low solubility in water and most common solvents, cellulose-derived materials such as CA and CN are the ideal candidates for electrospinning cellulose nanofibers [25].
CA is a moderately biodegradable polymer that has a negatively charged surface made by esterifying cellulose with a variety of degrees of substitution (DS), the most common of which is 2.5 [26, 27]. CA has good solubility in common solvents, so it can be easily spun into nanofiber by the electrospinning technique. It is also a non-toxic, obtained from renewable origins, hydrophobic, low-cost, and biodegradable polymer with outstanding qualities [28, 29]. CA has a wide range of applications in fibers, membranes, films, lacquers, packaging, adhesives, photographic films, gas separation, reverse osmosis, dialysis, cigarette filters, coatings, and inks [16, 30], desalination [31, 32], and medical applications [21].
Cellulose nitrate is a cellulose derivative made by treating cellulose with strong nitric acid and replacing the –OH groups in the cellulose molecule with –ONO2 groups [33, 34]. It has applications in photographic, X-ray film, protective and decorative coatings, metal coatings, coatings for stone, printing inks, metal, ceramics, plastics, lacquers, adhesives, automobile surface primer coatings, refinishing lacquers, high energy particle detectors, sensitive detectors of neutrons, and clear topcoat finishes for wood furniture [35].
In comparison to synthetic or inorganic sorbents, cellulose-based nanofiber composites represent promising potential as oil absorbents because they are natural, non-toxic, biodegradable, renewable-origins, environmentally benign, cost-effective, biocompatible, and exhibit high oil absorption capacity with moderately easy disposal. Furthermore, cellulose nanofiber composites have a high structural flexibility and porosity [36]. The incorporation of carbon black into cellulose nanofiber provides an effective blend for oil sorption. Its porous features, like surface area, pore volume, and pore size distribution, boost sorption capabilities. Moreover, it improves the fiber properties as it is mostly used to reinforce rubber in industrial processes [37, 38]. In this study, we fabricated a novel composite nanofiber of cellulose acetate with cellulose nitrate incorporated with different loads of carbon black for oil/water separation. The resultant blended nanofiber composite was successfully prepared by using a one-step electrospinning technique. Also, the new composites showed efficient oil sorbent materials for both light machine oil (LMO) and heavy machine oil (HMO) with high oil sorption capacity and high oil retention time.
2 Experimental
2.1 Materials
CA (Mn: 50,000, acetyl content: 29–46%) was obtained from the company of ALPHA Chemie, India, and cellulose nitrate (Nitrocellulose) (CN, 12.1% N content) was obtained as 75% wet with isopropanol/ethanol from Abozabal Co., Egypt, and was dried just before its used. 95% pure n-hexane was obtained from the company M-TEDIA, and Carbon black (CAS Number: 1333–86-4) was obtained from the company Sigma-Aldrich, USA. Polyethylene glycol (PEG, Mwt = 200) was obtained from the company ACROS Organic; tetrahydrofuran (CAS Number: 109–99-9; anhydrous, ≥ 99.9%; inhibitor-free), dimethylformamide (CAS Number: 68–12-2; anhydrous, 99.8%), and ethyl alcohol were obtained from the company Sigma-Aldrich, USA. The HMO (Fully Synthetic Motor Oil 5 W-50, Rally formula) and LMO were obtained from Exxon Mobil, Egypt.
2.2 Formation of CA-CN/CB solution
CA-CN polymer with a concentration of the 10% (w/v) was prepared by dissolving CA-CN with a ratio (3:2) (Wt/Wt) in THF/DMF with a ratio of (4:1) (v/v). To avoid the surface tension of polymer solution, PEG was added to the polymer solution (1:1) (wt:wt, as the ratio of PEG to CA). To obtain CA-CN/CB electrospun composite, CB was added to the polymer solution with three different concentrations of (0.7, 1.5, and 2.2) wt% CB of the total wt. Complete dissolution of the polymer in the common solvents occurred by continuous stirring for 2 h at room temperature (RM), followed by ultrasonically for 2 h at RM. Continue to combine the polymer solutions for 24 h without stirring to induce the release of air bubbles from the solution after it has been mixed [17, 39].
2.3 Electrospinning of CA-CN/CB solutions
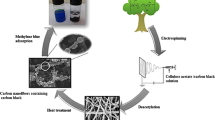
Using electrospinning equipment (Bioinicia, FLUIDNATEK LE-50, Spain), the CA-CN/CB solutions were electrospun. A syringe (20 mL) with a blunt-end needle was used to dose the solutions. Using a syringe pump, it was fed at a steady rate of 10 mL/min. The distance between the needle tip and the collector was kept constant at 10 cm, while the spinning voltage was + 26 kV and − 10 kV. The fiber samples were collected on a square aluminum collector. In order to verify that all PEG was removed from the produced fibers, they were washed with distilled water and then with ethanol, followed by drying for 24 h at 50 °C in an oven to achieve the desired result [40] as shown in Fig. 1.

Scheme for the fabrication of CA-CN/CB nanofiber composite and its application in oil removal
2.4 Characterization of CA-CN/CB composite
The fabricated fibers were investigated by FT-IR spectroscopy to determine the chemical functionality of fiber samples. The FT-IR spectroscopy was analyzed in the wavenumber range between 400 and 4000 cm−1 using Bruker VERTEX 70 attached to the Platinum ATR unit (model V-100) using 16 scans. The specific surface area was examined by BELSORP (Mini II, BEL Japan, Inc.). The Brunauer–Emmett–Teller (BET) model was used to determine it from the N2 adsorption isotherm. N2 adsorption–desorption isotherms at 77 K were used to test the nanofiber composite samples. The total pore volume was measured depending on the volume of N2 adsorbed at a relative pressure of 0.95. Using SEM, the physical characteristics and morphology of the produced fibers were studied (JEOL, Model JSM 6360LA, Japan). Prior to observation, the composite nanofiber samples were coated with a tiny layer of gold to protect them from damage. Using Image-G Program, the average fiber diameter (AFD) was measured. The SDT650-Simultaneous Thermal Analyzer apparatus was used to conduct thermal stability tests from 50 to 900 °C, with 10 °C per minute as a ramping temperature under a nitrogen gas environment (100 mL/min). About 10 mg of each composite nanofiber sample was placed in a silica crucible and pyrolyzed using the conditions described above [17, 41]. For density measurement, samples of nanofiber mats with 2 cm × 2 cm dimensions were used and the thickness of the nanofiber was calculated by digital micrometer and the mass was measured by an analytical balance.
The following Eq. (1) is used to calculate the density (ρ) (g/cm3) of the nanofiber mats
where a, b, and h are the length, width, and thickness of the sample (cm), respectively, and m represents the sample mass (g) [42].
2.5 Water uptake capacity
The swelling and ability to absorb water of electrospun CA, CA-CN, CA-CN/CB0.7, CA-CN/CB1.5, and CA-CN/CB2.2 composite nanofiber mats were determined to better understand the diffusion of the medium inside the mats of composite nanofiber, which is important for oil removal applications. About 0.6 g of as-spun fiber mats were submerged in three mediums (pH 3, 6, and 11) at RT for 24 h. To establish the swelling behavior of the samples, they were gently dried using filter paper to remove excess water, then weighed to define WS, then dried in a vacuum oven at 50 °C to a constant mass, and reweighed to determine the dry weight (Wd). Equation (2) was applied to calculate the product's percentage of swelling ratio [43]:
where WS and Wd are the weights of the swollen and dried nanofibers, respectively.
2.6 Determining the adsorption and retention capacity of oil (Static test)
0.5 g of each sample of the CA-CN, CA-CN/CB0.7, CA-CN/CB1.5, and CA-CN/CB2.2 composite of nanofiber was placed into 30 mL of machine oil (HMO or LMO) for 1 h at RT. The samples were then weighed after being placed on a wire mesh and drained for 5 min to remove any loosely adhering oil. The samples were elevated to drain on a wire mesh with free oil trickling out for 24 h to determine oil retention capacity (ORC). All of these procedures were repeated three times in order to obtain an average value as the final result. A standard approach described in ASTM 726–06 [44] “Standard Test Method for Sorbent Performance of Adsorbents” was applied to determine the oil absorption capacity (OSC) and retention of the adsorbents. The following Eqs. (3, 4) were used to compute the adsorption capacity (SQ) and retention capacity (RQ) of the HMO and LMO [45]:
where Wi is the sample beginning mass before sorption (g), W5 is the oil mass (g) of a wetted sample after 5 min dripping time, and W24 is the oil mass (g) of a wetted sample after 24 h dripping time.
2.7 Sorption capacity measurement (oil/water solution) (dynamic test)
Batch experiments were performed by using HMO and LMO to study the selectivity of the structured fiber for absorbing spilt oil from water as described by Lim and Huang [46] and Rengasamy et al. [47]. 50 mL of seawater with a certain amount of machine oil and structured fiber was poured into a 250-mL jar. After being placed in a shaker at 200 rpm for varied time periods (5, 10, 15, 30, and 60 min), the solution was removed. Following the collection of the samples, they were allowed to drain for 5 min on a mesh of wire with free oil dropping out and weighted. Following that, samples were carried to the piston, where they were squeezed through the press five times to remove the oil from the samples. To aid in the extraction of the oil, a few milliliters of n-hexane (15 mL, the prepared composite is insoluble in n-hexane) were added to the pressing solution before pressing began. The solution was transferred to a graduated tube of a centrifuge for analysis of water content using the ASTM D1696-91 centrifuge technique [48, 49]. The oil-to-water selectivity was calculated according to Eq. (5).
where WS (water + oil + adsorbent) is the mass of saturated adsorbent, Ww is the adsorbed weight of water, and Wi is the original dry mass of the adsorbent [50].
2.8 Reusability test
Reusability is an important parameter for the selection of adsorbent materials. For a static test, composite samples (0.5 g) were immersed for 1 h at RT in oil (30 mL). Then, the samples were drained for 5 min and weighted. The oil was separated from oil-saturated samples by centrifuge and then washed with 3 mL of hexane and centrifuged for further use of samples. After washing, the samples were placed at 60 °C for 24 h in an oven. This step was repeated for 4 cycles. For the dynamic test, the same method was repeated for recycling the water/oil system. The OSC of the composite after each cycle was measured and reported using 0.4 g of composite fiber in seawater (50 mL) and an oil thickness layer of 5 mm of HMO and 7 mm of LMO for 15 min for HMO and 1 h for LMO, and the OSC of a composite sample after each cycle was measured and reported.
2.9 Adsorption isotherm
It is important to understand how adsorption isotherms depict the distribution of the oil molecules between the liquid and solid phases at the equilibrium state of the adsorption process. Adsorption isotherms also characterize the interaction with adsorbents, which is important in maximizing the efficiency with which adsorbents are used.
According to the LIM theory, the adsorbent has certain homogenous locations where adsorption occurs. The maximal adsorption capacity was calculated using the LIM model equation. The LIM non-linear equation explains the saturated monolayer isotherm as followed Eq. (6) [51]:
where Ce (mg L−1) represents the equilibrium concentration, qe (mg g−1) represents the mass of oil adsorbed, Qm (mg g−1) represents the maximum adsorption capacity for a complete monolayer, and Ka (L mg−1) represents adsorption equilibrium constant related to the apparent energy of sorption. The linear equation for LIM can be represented as Eq. (7) [52].
The FIM equation is predicated on the premise of a heterogeneous surface with a non-uniform distribution of the adsorption heat on the surface, as shown in Fig. 1. With the use of an exponential equation, we can estimate the amount of adsorbate that is absorbed onto the surface of the composite nanofibers. FIM makes the assumption that as the adsorbate concentration rises, the adsorbate concentration on the adsorbent surface will climb in proportion. A number of 1/n less than one implies a conventional LIM, whereas a value of 1/n more than one shows cooperative adsorption. In Eq. (8), the FIM is shown in its non-linear form [53]:
where KF and 1/n are the system’s FIM constants, reflecting the oil’s adsorption capability and intensity on the adsorbent, respectively. Equation (8) can be rewritten in logarithmic form as Eq. (9).
It was assumed in the Tempkin isotherm model [54] that because of the interactions between the adsorbed molecules, the heat of adsorption of all molecules in the layer decreases linearly with coverage and that adsorption is characterized by an evenly distributed distribution of binding energies, up to maximum binding energy [55]. The Temkin isotherm model (TIM) is often employed in Eq. (10) below [56,57,58], which reads as follows:
Equation (10) of the TIM can be linearized to Eq. (11):
where β = (RT)/b, T is the absolute temperature in Kelvin, and R is the universal gas constant of 8.314 J/mol K. The heat of adsorption is proportional to the constant b [59, 60].
2.9.1 Best-fit isotherm model
Different error functions were used to find the most appropriate isotherm model for the practical data. Where the symbol N refers to the number of experimental data points and P refers to the number of parameters in the isotherm model, the error functions were applied in the manner described below. The average percentage errors (APE) produced using Eq. (12) were used to determine the degree to which the experimental and projected values of adsorption capacity utilized for constructing isotherm curves were in agreement [61].
HYBRID is a hybrid fractional error function that remunerates low concentrations by balancing absolute deviation versus fractional error and is more trustworthy than other error functions as determined by Eq. (13) [62, 63].
The chi-square error, X2, is given as Eq. (14) [64].
The sum of the squares of the errors (ERRSQ) is given by the following Eq. (15) [64].
According to the following Eq. (16), Marquart’s percentage standard deviation (MPSD) is calculated [64].
The following Eq. (17) calculates the absolute error sum (EABS), which is the sum of all absolute errors [64].
The root-mean-square errors (RMSEs) are calculated using the following Eq. (18) [64].
2.9.2 Kinetics studies
Adsorption kinetics is used to explain the rate of oil intake, which is responsible for regulating the equilibrium time. In order to determine the optimal experimental processes for full-scale batch operation, it is necessary to understand the kinetics of oil absorption. The kinetic parameters are essential for estimating the sorption rate as well as for delineating and modeling of the experiments involved in the sorption. Therefore, the kinetics of oil sorption on the nanofiber mats was explored using pseudo-first-order (PFO) Eqs. (19) [65] and pseudo-second-order (PSO) kinetic models [66], which were derived from the results of the research. For the purpose of describing the congruence between experimental and model-predicted data, the correlation coefficients (R2, values ≅ 1) were chosen. The greater their value, the more appropriate the model was for the dynamics of crude oil sorption. The Lagergren PFO model [65] was used to investigate the kinetic data since it is the oldest known model for explaining sorption rate based on sorption capacity. It is usually stated mathematically as the following Eq. (19):
where qe and qt (mg/g) are the equilibrium and adsorption capacities at time (t), respectively, and k1 is the PFO sorption rate constant (L min−1). Equation (20) was rearranged using the boundary constraints t = 0 to t = t and qt = 0 to qt = qt as well as the boundary conditions t = 0 to t = t and qt = 0 to qt = qt.
Ho et al. [66] proposed a PSO model for adsorption kinetics, which was expressed as Eq. (21):
The PSO rate constant of adsorption is K2 (g mg−1 min−1). Integrating Eq. (21) for the boundary conditions q = 0 to q = qt at t = 0 to t = t is simplified since Eq. (21) may be rearranged and linearized to Eq. (22).
The K2 was utilized to derive the initial sorption rate, h, which was calculated using Eq. (23) below.
3 Results and discussion
3.1 Nanofiber composite characterization
3.1.1 Scanning electron microscope
The morphologies and fiber diameter distribution were characterized by SEM images of the CA-CN, CA-CN/CB0.7, CA-CN/CB1.5, and CA-CN/CB2.2 composite nanofiber mats and are presented in Fig. 2a–d. The image of SEM exhibited the polymer matrix of composite nanofiber with no beads formed and smooth uniform fibers. The fiber diameter was on a submicron scale, which increases with increasing carbon black content, where AFD for CA-CN was 327 nm, CA-CN/CB0.7 was 636 nm, CA-CN/CB1.5 was 722 nm, and CA-CN/CB2.2 was 755 nm, which was obtained as a mean value of 20 images analyzed using the Image j software. From the obtained images, it was clearly found that the carbon particles adhered to the surface of the fibers in a flat shape, so that the carbon particles did not appear alone, but appeared in a perfectly consistent and regular distribution [67].

Photos of SEM for a the CA-CN nanofiber mats, b the CA-CN/CB0.7, c the CA-CN/CB1.5, and d the CA-CN/CB2.2 composites (Images were taken at 15 kV)
3.1.2 FT-IR analysis
The FT-IR analysis of CA, CN, CB, and their composites is shown in Fig. 3. All of the CA-CN composite nanofiber samples, including the CA-CN/CB0.7 composite, CA-CN/CB1.5 composite, and CA-CN/CB2.2 composite samples, showed the same beaks as a broad peak of about 3393 cm−1, which was allocated to the O–H group. The O-NO2 of cellulose nitrate was identified as the source of the asymmetric stretching peak detected at 1640 cm−1. The stretching vibration of the ester’s C = O atoms is accountable for the absorption peak at 1737 cm−1. Bending vibrations in the C–O and C–H directions are accountable for the peaks at about 1229 and 1367 cm−1, respectively. It was determined that C–O stretching vibration in CA had a significant band at 1050 cm−1. Carbon black (CB) exhibited three peaks in the infrared spectrum, with the first at 850 cm−1 ascribed to the carbon skeleton (C–C), the second at 1531 cm−1 allocated to stretching (C–C), and the third at 2395 cm−1 originating from CO2. When comparing the FTIR analysis of the CA-CN/CB composite fibrous membrane with that of the CA-CN composite fibrous membrane, it is found that there is no difference in the peaks [68].

FT-IR analysis of a CA, b CN, c CA-CN, d CA-CN/CB0.7, e CA-CN/CB1.5, f CA-CN/CB2.2
3.1.3 Surface area analysis
Figure 4 depicts the adsorption–desorption isotherm and pore size analysis of the CA-CN, CA-CN/CB0.7, CA-CN/CB1.5, and CA-CN/CB2.2 composites, as well as the adsorption–desorption and pore size analysis curves of the CA and CN materials. From the IUPAC classification of adsorption–desorption isotherm models, type V defined a low energy, homogenous solid surface with mesoporosity that was present in all of the samples examined. The nitrogen adsorption–desorption graph shows that at relative pressures between 0.2 and 0.9, the amount of nitrogen adsorbed rose fast, indicating that the adsorbent has a smaller exterior surface area and is made up of mesoporous or microporous materials. The isotherm hysteresis loop at pressures relatively near to the saturation vapor pressure indicates that nanofiber mats with slit-like pores are present, and this is supported by the results. As a result of BET measurements, surface parameters such as specific surface area, mean pore diameter, and total pore volume of CA-CN and CA-CN/CB composite were determined. A comparison of all samples' textural characteristics is shown in Table 1.

a The curve of the N2 gas adsorption–desorption isotherms. b The curve of BET analysis. c The curve of BJH adsorption analysis. d The curve of BJH desorption analysis of the CA-CN, CB, CA-CN/CB0.7, CA-CN/CB1.5, and CA-CN/CB2.2 composites nanofibers
From the sample’s surface area results, it is found that increasing the content of CB in the composite nanofiber up to 0.7% CB load to CA-CN leads to a decrease in the surface area of CA-CN. After that, it is decreasing with a further increase of CB to 2.2% CB content, which may be due to the surface non-uniformities of the CA-CN/CB. The largest surface area was 15.29 m2/g for CA-CN which is higher than CA nanofiber that was 7.65 m2/g [69]. CA-CN, CA-CN/CB0.7, CA-CN/CB1.5, and CA-CN/CB2.2 had a surface area (SBET) of 15.29, 3.84, 4.08, and 6.17 m2/g, respectively. Similarly, the average pore diameters of the CA-CN, CA-CN/CB0.7, CA-CN/CB1.5, and CA-CN/CB2.2 composite nanofiber mats were determined to be 5.655, 12.078, 11.712, and 7.868 nm, respectively. The CA-CN, CA-CN/CB0.7, CA-CN/CB1.5, and CA-CN/CB2.2 composite nanofiber had a mesopore volume distribution of 1–10 Å, whereas the carbon black had a mesopore volume range of 1–20 Å [70].
3.1.4 Thermal stability
Using a TGA in a nitrogen environment, the thermal stability of the composite was investigated. TGA curves for various CB loadings of CA-CN composite nanofiber are depicted in Fig. 5a. All of the samples went through three steps of thermal transitions, with the first thermal step (i.e., 50–80 °C) resulting in mass losses of 5.947, 12.48, 12.56, and 12.04% for the CA-CN, CA-CN/CB0.7, CA-CN/CB1.5, and CA-CN/CB2.2 composites, respectively, corresponding to moisture loss, and the second thermal step (i.e., 80–200 °C) resulting in weight losses of 37.38, 29.05, 31.55, and 33.79% for the CA-CN, CA-CN/CB0.7, CA-CN/CB1.5, and CA-CN/CB2.2 composites, respectively, represents the thermal degradation of CN chains [71]. The third thermal stage, which occurred at 200–400 °C and resulted in weight losses of 45.4%, 45.99%, 46.98%, and 44.07% for the CA-CN, CA-CN/CB0.7, CA-CN/CB1.5, and CA-CN/CB2.2 composites, respectively. According to the experimental results, the residues of the samples after a thermal breakdown at 900 °C, the residues of the samples were 11.273, 12.48, 8.91, and 9.41% for CA-CN, CA-CN/CB0.7, CA-CN/CB1.5, and CA-CN/CB2.2, respectively.

a The curves of TGA. b The curves of DSC thermal analyses for the CA-CN, CA-CN/CB0.7, CA-CN/CB1.5, and CA-CN/CB2.2 composites nanofibers applying a temperature ranging from 50 to 900 °C under flow of N2 (100 mL/min) gas
DSC (differential scanning calorimetry) is a technique for obtaining data on temperature transitions that do not result in a weight shift [32]. The heat flow displayed in Fig. 4b depicts the thermal degradation of CA-CN and CA-CN/CB composite nanofibers. Clearly, all samples had three peaks on their DSC curves; one crystallization endopeak corresponded to a crystallization temperature of 68.97, 79.42, 99.08, and 90.8 °C for the CA-CN, CA-CN/CB0.7, CA-CN/CB1.5, and CA-CN/CB2.2 composites, respectively, which is attributable to water loss from the composite sample. Similarly, the cellulose nitrate melting temperature for the CA-CN, CA-CN/CB0.7, CA-CN/CB1.5, and CA-CN/CB2.2 composites, respectively, exhibited cellulose nitrate degradation temperatures of 194.28, 192.3, 195.62, and 192.62 °C. Moreover, the second exo-melting peak is for cellulose acetate degradation temperatures at 535.4, 523.72, 647.76, and 559.9 °C for the CA-CN, CA-CN/CB0.7, CA-CN/CB1.5, and CA-CN/CB2.2 composites, respectively. It is shown in Fig. 5b that by adding CB to cellulose acetate mats, the observed maximum degradation temperatures increased to 195.62 and 647.76 °C for CN and CA, respectively, as CB supports CA-CN fibers. As seen in the SEM photos, the hybrid mat has a point bonded structure due to the homogeneous mixing of CA-CN and CB. A higher concentration of CB in the CA nanofibers induced a lower onset temperature of decomposition for CA/CB composite nanofibers.
3.1.5 Swelling behavior
Figure 6 represents the CA-CN composite nanofiber and its carbon composite fibrous mat swelling behavior. Three different pH (3, 6, and 11) media were used to investigate the swelling property of CA-CN and carbon black composite fibrous mats. The swelling tests were carried out for 24 h, and the maximum swelling ratio of CA-CN mats was 238% at pH 6. The maximum swelling ratio of CA-CN/CB composite nanofiber mats showed higher swelling percentage values at pH 6 (226, 271.43, and 270%) for CA-CN/CB0.7, CA-CN/CB1.5, and CA-CN/CB2.2 composites, respectively. Subsequently, the highest swelling ratio was obtained at a near-neutral and an alkaline medium, whereas at acidic pH 3, the swelling ratio was significantly low. That is, as previously reported, the alkaline condition promoted swelling, whereas the acidic condition lowered swelling, as reported before for cellulose nanofiber aerogel [72] and bacterial cellulose nanofiber/sodium alginate composite [73].

At ambient temperature, the swelling equilibrium behaviors of the CA-CN, CA-CN/CB0.7, CA-CN/CB1.5, and CA-CN/CB2.2 composite nanofibers were investigated at pH 3, 6, and 11
The swelling ratio of the CA-CN nanofiber without carbon black was lower than the CA-CN/CB composites, which may be due to carbon black allowing water molecules to penetrate into the network of the nanofibers, which leads to an increase in the inter-space between molecules of the polymer, so the swelling ratio increases by increasing the percentage of CB [43]. The attempt to measure the contact angle failed because the prepared composites are fiber and there is no flat surface to measure the contact angle.
3.1.6 Density measurement
Table 2 gives the calculated thickness and density of the CA-CN/CB nanofiber mats. The result showed that the thickness and density of CA-CN/CB nanofiber mats are higher than CA-CN without CB, and also, increasing CB content to the nanofiber lead to increase the density of the nanofiber. These results are confirmed with the result obtained from SEM as the diameter of the nanofiber increase with increasing CB content as CB inter the fiber pore.
3.2 Oil adsorption and retention capacity
As reported in Table 3, the results of OSC and ORC tests on HMO and LMO are provided in the same way. The oil adsorption maximum capacity reached 13.67 g/g for CA-CN/CB2.2, which means that it adsorbed oil more than 13 times its own weight. The fabricated fibers exhibit excellent oil retention capacity, where more than 81% of the adsorbed oil is retained after 24 h dripping. The best oil adsorption was obtained for CA-CN fiber with a 2.2% CB load. On the other hand, more oil retention was achieved with a 1.5% CB load to CA-CN fiber, so it is clearly observed that adding more CB would decrease the ORC of the fibers but increase the oil adsorption capacity [74]. The result showed that the oil sorption capacity increased with increasing carbon black content as carbon inter the inter space between the polymer so allow the penetration of oil in the fiber with further increased of carbon content it lead to clogged the pore so, the oil sorption capacity decrease as discuses in the swelling test. By comparison of the sorption capacities for both HMO and LMO, the HMO absorbed more than the LMO into the fabricated fiber. This is related to oil viscosity as HMO is heavier than LMO of the same unit volume, as it prevents the escape of the absorbed oil from the pores of fabricated fibers [75].
3.3 Batch adsorption experiments
CA-CN and its carbon black composite were evaluated for their affinity for machine oil adsorption capacity according to the effectiveness of numerous variables such as sorbent dose, contact time, and thickness of the oil.
3.3.1 Contact time effect
The impact of adsorption on the OSC was studied at different contact times. 0.4 g of each sample was tested with a 5-mm oil thickness of HMO and a 7-mm oil thickness of LMO. To measure the maximum OSC of the CA-CN, CA-CN/CB0.7, CA-CN/CB1.5, and CA-CN/CB2.2 composite nanofibers for HMO and LMO using batch adsorption studies in oil/water mixtures, batch experiments were used, and the results are shown in Fig. 7. In both HMO and LMO, the samples demonstrated a fast adsorption rate within the first 5 min of testing, and the maximum oil sorption capacity for both HMO and LMO was achieved within 30 min of testing for the CA-CN/CB2.2 composite nanofiber, yielding 19.29 and 16.36 g/g, respectively, for the compound. This experiment clearly demonstrates that the OSC increases marginally with contact time during the first 30 min of the experiment, followed by a somewhat slow process for all sorbents after that. This is due to the oil on the exterior surface of the fibers gradually seeping into the hollow lumen as the fibers are stretched. This finding is congruent, especially for HMO adsorption, with the observations of Wang et al. [76], who made the same observation.

Impact of reaction time on the sorption capacity of a HMO and b LMO onto the CA-CN, CA-CN/ CB0.7, CA-CN/CB1.5, and CA-CN/CB2.2 composites at RT
3.3.2 Impact of sorbent dosage
The amount of mass of the adsorbent is a significant consideration in large-scale industrial applications of adsorbent in oil removal. The consequence of adsorbent dose of the CA-CN/CB0.7, CA-CN/CB1.5, and CA-CN/CB2.2 composites was investigated using 0.1, 0.2, 0.3, 0.4, and 0.5 g of the adsorbent material at a fixed initial amount of oil. Figure 8 discusses that there is a progressive increase in the HMO and LMO sorption capacity with increasing the composite amount from 0.1 to 0.4 g, and then there is a slight increase from 0.4 to 0.5 g. Where, the maximum adsorption capacity for HMO was 19.29 g/g for CA-CN/CB2.2 and 14.05 g/g for LMO. These results were attributed to the fact that the number of contact sites available for the adsorption process per unit gram of the CA-CN composite and its carbon black composites [77].

Impact of sorbent mass on the adsorption capacity of a HMO and b LMO onto CA-CN/CB0.7, CA-CN/CB1.5, and CA-CN/CB2.2 at RT
3.3.3 Impact of the thickness of oil
Experiments with varying oil thicknesses were carried out in order to determine whether or not the amount of oil had an influence on the OSC of the CA-CN, CACN/CB0.7, CACN/CB1.5, and CACN/CB2.2 composites. 0.4 g of the composite material was added to the oil/seawater system using different thicknesses of HMO (3, 4, 5, and 6) mm and LMO (3, 5, 7, and 10) mm. Figure 9 shows the impact of prolongation of HMO and LMO thickness on the adsorption capacity. The progressive increase of adsorption capacity was observed with increasing the amount of HMO and LMO and maximum values reached 19.73 g/g for CA-CN/CB1.5 for HMO. Also, the maximum adsorption capacity for LMO reached 16.36 g/g for CA-CN/CB1.5 and then tended to be constant with a further increase of oil amount up to 5 mm of HMO thickness layer and 7 mm of LMO thickness layer, at constant seawater (50 ml). This could be due to the adsorbent sites being saturated with oil. Increasing the amount of oil available provides a critical driving force for overcoming all ORC between the water and solid phases, resulting in increased adsorption as a result. As the beginning concentration of oil rises, so does the number of collisions between the oil and the composites, which results in increased adsorption capacity [78].

Impact of oil thickness layer on the adsorption capacity of a HMO and b LMO onto the CA-CN/CB0.7, CA-CN/CB1.5, and CA-CN/CB2.2 composite nanofiber
3.4 Reusability
The reusability study of the fabricated fibers investigated the performance of the adsorbent regarding its efficiency in removing spilled oil after a certain number of cycles. The efficiency of the reusability of the CA-CN/CB composites was examined by using the centrifuge method using hexane extraction for oil-saturated samples. This method has been found to be economical and practical for the recovery of oil from the fiber samples and also provides feasibility for reusing the adsorbent materials. Figure 10 and Table 4 propose the use of the centrifuge technique to investigate the variations in OSC by CA-CN/CB0.7, CA-CN/CB1.5, and CA-CN/CB2.2 composites after a number of reusability cycles. The results showed that the CA-CN/CB nanofiber composites exhibited a high uptake of the oil in the first cycle and were followed by a decrease in the oil adsorption capacity. This may be due to oil remaining in the composite nanofiber structure or deterioration of the composite fiber structure during the washing process. The sorption capacity of oil onto the fabricated nanofiber composites remained constant in the last 2 cycles. The reusability test results showed that the CA-CN/CB nanofiber composites are promising oil-adsorbing materials as they can be applied for recycling several times by using the centrifuge method with the aid of hexane extraction [79].

The reuse efficiency (OSC and ORC) of different CA-CN/CB composites nanofibers for a HMO and b LMO was tested for four cycles with each type of oil
3.5 Isotherm study
The importance of adsorption isotherms reveals the specific relationship between equilibrium concentrations of the CA-CN/CB0.7, CA-CN/CB1.5, and CA-CN/CB2.2 composite nanofiber in the bulk and the oil amount at the solid surface. For single-solute systems, the Langmuir (LIM) and Freundlich isotherm models (FIM) are the most widely accepted surface adsorption models. The relationship between the amount of adsorbent and the liquid phase concentration was investigated using the LIM, FIM, and TIM equations. The correlation coefficient (R2) is used to compare the applicability of the isotherm equations. Table 5 shows that the adsorption data of oil fitted with the LIM as shown in Figs. 11 and 12 performs better than other isotherm models as indicated by higher (R2) ≥ 0.990 [80]. The maximum monolayer capacity Qm of the CA-CN/CB0.7, CA-CN/CB1.5, and CA-CN/CB2.2 composites obtained from linear LIM was 18.05, 21.23, and 19.72 g/g, respectively, for the removal of HMO, and it reached 16.81, 18.98, and 19.31 g/g, respectively, for the removal of LMO. The value of heterogeneity factor (nf) obtained from FIM > 1 described in Table 4 means that the adsorption process of oil on the CA-CN/CB0.7, CA-CN/CB1.5, and CA-CN/CB2.2 composites is a favorable physical process [81]. This finding was supported by data from several error functions, as shown in Table 6. According to the data, the TIM has the lowest accuracy, whereas the FIM and LIM have the highest fit. However, the error functions analyzed produced varying findings for each isotherm model, necessitating a comparison of the isotherm models for each error function individually.

a LIM, b FIM, and c TIM for adsorption of HMO over the CA-CN/CB0.7, CA-CN/CB1.5, and CA-CN/CB2.2 composites

a LIM, b FIM, and c TIM for adsorption of LMO over the CA-CN/CB0.7, CA-CN/CB1.5, and CA-CN/CB2.2 composites
3.6 Adsorption kinetic studies
Kinetic studies investigated the factors affecting the adsorption process, including diffusion control, mass transfer, and chemical reaction. Kinetic parameters play an essential role in determining adsorption rates and modeling adsorption processes. The kinetic models used to establish the correlation coefficients for the adsorption of oil by the CA-CN/CB0.7, CA-CN/CB1.5, and CA-CN/CB 2.2 composite nanofiber were PFO [65] and PSO [66], and the experimental data was compared to the model-predicted values (R2). According to Table 6, if the estimated qe of all the data is not equal to the experimental qe, the reaction is unlikely to be first-order and has a low correlation coefficient. When pore-diffusion acts as a limiting factor in the adsorption process, the relationship between the beginning oil concentration and the rate of adsorption is not linear. The results indicated that the adsorption of oil onto CA-CN/CB0.7, CA-CN/CB1.5, and CA-CN/CB2.2 is not a first-order reaction. While the kinetics of adsorption indicated that the PSO kinetic model was more fit to the adsorption of oil into fabricated fibers as the calculated correlation coefficients (R2) ≥ 0.963. The results of Fig. 13 clearly show that the estimated qe values are largely equal to the experimental qe values. The values of the initial sorption rate (h), which represents the rate of initial sorption, are virtually enhanced as the initial oil amount increases, whereas the PSO rate constant (k2) decreases as the oil amount increases (Table 7).

Plot t/qt versus t of the PSO model for removal of a HMO and b LMO by CA-CN/CB0.7, CA-CN/CB1.5, and CA-CN/CB2.2
3.7 Comparison study for removal of machine oil by CA-CN/CB nanofiber composite with other adsorbents
The maximum oil sorption capacity obtained by the fabricated nanofiber composite was 21.23 g/g. The obtained result was compared to previous materials reported in other literature, as seen in Table 8. This comparison showed that the fabricated CA-CN/CB nanofiber composite exhibits excellent efficiency for oil sorption as it absorbs four times more than carbonized cotton husks and about twenty times more than Kapok fiber. As a result, the fabricated CA-CN/CB nanofiber composite has the potential to be used as an adsorbent for oil removal.
4 Conclusion
The fabricated fibers have diameters on a submicron scale that increase with increasing the content of carbon black. It was clearly found that carbon black particles adhered well to the surface of the fabricated fibers. It is clear from the surface area analysis that increasing CB content in the fabricated nanofibers results by in a decrease in the surface area. All sample fiber mats exhibited three steps in the TGA analysis. While in the DSC analysis, a higher concentration of CB in the fabricated fibers induced a lower onset temperature of decomposition for composite nanofibers. The swelling ratio of the composite nanofiber without carbon black was lower than the composites with CB, which may be due to the carbon black allowing water molecules to penetrate into the nanofiber network, which increased the interspace between the polymer molecules. The HMO adsorbed more than LMO into the fabricated fiber and the oil adsorption capacity improved marginally with increasing contact time during the first 30 min. Under the optimum circumstances, the CA-CN/CB composite mats adsorbed oil 13 times more than their own weight in only 30 min. The adsorption result showed that a 1.5% carbon black load to CA-CN mats was more favorable for the adsorption of machine oil. The reusability study of the fabricated fibers proved the performance of the composite adsorbent regarding its efficiency to remove spilled oil after a certain number of cycles. The TIM has the lowest accuracy, while the FIM and LIM have the best fit. The adsorption kinetics indicated that the PSO kinetic model was fit for the sorption of oil into a fabricated composite nanofiber. Therefore, according to this study, CA-CN/CB composites have a lot of promise to be exploited as efficient adsorbent materials for HMO and LMO. They are considered promising materials as they are biodegradable, durable, renewable origin, and reusable sorbent materials.
Data availability
The datasets used in this investigation are accessible for review upon request from the corresponding author of the paper.
References
El Nemr A (2005) Petroleum contamination in warm and cold marine environment. Nova Science Publishers, Inc. Hauppauge New York. [ISBN 1–59454–615–0] 150pp
Deshpande R, Sundaravadivelu D, Techtmann S, Conmy R, Santodomingo J, Campo P (2018) Microbial degradation of Cold Lake Blend and Western Canadian Select Dilbits by freshwater enrichments. J Hazard Mater 352:111–120. https://doi.org/10.1016/j.jhazmat.2018.03.030
Lou L, Kendall RJ, Smith E, Ramkumar SS (2020) Functional PVDF/rGO/TiO2 nanofiber webs for the removal of oil from water. Polymer 186:122028
Jamaly S, Giwa A, Hasan SW (2015) Recent improvements in oily wastewater treatment: progress, challenges, and future opportunities. J Environ Sci (China) 37:15–30
Cheng H, Li Z, Li Y, Shi Z, Bao M, Han C, Wang Z (2020) Multi-functional magnetic bacteria as efficient and economical pickering emulsifiers for encapsulation and removal of oil from water. J Coll Interf Sci 560:349–358
Kannan CS, Kumar KSM, Hussain MS, Priya ND, Saravanan K (2014) Studies on reuse of re-refined used automotive lubricating oil. Res J Eng Sci 3(6):8–14
Xu C, Santschi PH, Quigg A, Chin WC (2019) Impact of exposure of crude oil and dispersant (Corexit) on aggregation of extra cellular polymeric substances. Sci Total Environ 657:1535–1542
Xu EU, Khursigara AJ, Li S, Esbaugh AJ, Dasgupta S, Volz DC, Schlenk D (2019) mRNA-miRNA-Seq reveals neuro-cardio mechanisms of crude oil toxicity in red drum (Sciaenopsocellatus). Environ Sci Technol 53(6):3296–3305
Han Y, Nambi IM (2018) Environmental impacts of the Chennai oil spill accident — a case study. Sci Total Environ 626:795–806
Boulais M, Vignier J, Loh AN, Lay CR, Morris JM, Krasne MO, Volety A (2018) Sub lethal effects of oil-contaminated sediment to early life stages of the Eastern oyster, Crassostrea virginica. Environ Pollut 243:743–751
Bajpai SK, Chanpuria A, Dubey S (2020) Magnetically driven poly(sulfur/oil) composite as an efficient oil adsorbent. Part-I: Synthesis, characterization and preliminary oil removal study. Environ Nanotech Monit Manag 13:1002933
Huang Q, Mao F, Han X, Yan J, Chi Y (2014) Migration of emulsified water droplets in petroleum sludge during centrifugation. Energy Fuels 28:4918–4924
Zapata K, Carrasco-Marín F, Arias JP, Castelo-Quibén J, Franco CA, Rojano B, Cortés FB (2020) Novel biomaterial design based on Pseudomonas stutzeri–carbon xerogel microspheres for hydrocarbon removal from oil-in-saltwater emulsions: a new proposed treatment of produced water in oilfields. J Water Process Eng 35:101222
Rafieian F, Hosseini M, Jonoobi M, Yu Q (2018) Development of hydrophobic nanocellulose-based aerogel via chemical vapor deposition for oil separation for water treatment. Cellulose 25(8):4695–4710
Wu H, Wang Z-M, Kumagai A, Endo T (2019) Amphiphilic cellulose nanofiber-interwoven graphene aerogel monolith for dyes and silicon oil removal Composites. Sci Technol 171:190–198
Yang G, Kong H, Chen Y, Liu B, Zhu D, Guo L, Wei G (2022) Recent advances in the hybridization of cellulose and carbon nanomaterials: interactions, structural design, functional tailoring, and applications. Carbohydr Polym 279:118947. https://doi.org/10.1016/j.carbpol.2021.118947
Elmaghraby NA, El Nemr A, Omer AM, Kenawy E-R, Gaber M (2022) Novel electrospun composites nanofibers prepared from cellulose acetate/carbon black as efficient sorbents for machine oil from aquatic environment. J Iran Chem Soc 19:3013–3027. https://doi.org/10.1007/s13738-022-02510-1
Lichinga KN, Luanda A, Sahini MG (2022) A novel alkali-surfactant for optimization of filtercake removal in oil–gas well. J Petrol Explor Prod Technol 12:2121–2134. https://doi.org/10.1007/s13202-021-01438-1
Agarwal S, Greiner A, Wendorff JH (2013) Functional materials by electrospinning of polymers. Prog Polym Sci 38:963–991
Bhardwaj N, Kundu SC (2010) Electrospinning: a fascinating fiber fabrication technique. Biotechnol Adv 28:325–347
Fahmi MZ, Prasetya RA, Dzikri MF, Sakti SCW, Yuliarto B (2020) Irzaman and Ferdiansjah, MnFe2O4 nanoparticles/cellulose acetate composite nanofiber for controllable release of naproxen. Mater Chem Phys 250:123055
Seddiqi H, Oliaei E, Honarkar H, Jin J, Geonzon LC, Bacabac RG, Klein-Nulen J (2021) Cellulose and its derivatives: towards biomedical applications. Cellulose 28:1893–1931. https://doi.org/10.1007/s10570-020-03674-w
Oliver S, Muralidhara HB, Venkatesh K, Guna VK, Gopalakrishna K, Kumar KY (2016) Potential applications of cellulose and chitosan nanoparticles/composites in wastewater treatment: a review. Carbohydr Polym 153:600–618
Nandi S, Guha P (2018) A review on preparation and properties of cellulose nanocrystal-incorporated natural biopolymer. J Pack Tech Res 2:149–166
Bochek AM (2003) Effect of hydrogen bonding on cellulose solubility in aqueous and nonaqueous solvents. Russ J Appl Chem 76:1711–1719
Sultana N, Rahman R (2022) Electrospun nanofiber composite membranes based on cellulose acetate/nano-zeolite for the removal of oil from oily wastewater. Emerg Mater 5:145–153. https://doi.org/10.1007/s42247-021-00326-y
Puls J, Wilson SA, Hölter D (2011) Degradation of cellulose acetate-based materials: a review. J Polym Environ 19:152–165. https://doi.org/10.1007/s10924-010-0258-0
Ramesh S, Shanti R, Morris E (2013) Characterization of conducting cellulose acetate based polymer electrolytes doped with “green” ionic mixture. Carbohydr Polym 91(1):14–21
Liu Y, Wang Q, Lub Y, Deng H, Zhou X (2020) Synergistic enhancement of cytotoxicity against cancer cells by incorporation of rectorite into the paclitaxel immobilized cellulose acetate nanofibers. Int J Biol Macromol 152:672–680
Cheng HN, Dowd MK, Selling GW, Biswas A (2011) Synthesis of cellulose acetate from cotton byproducts. Carbohydr Polym 80:449–452
EL-Ashhab F, Sheha L, Abdalkhalek M, Khalaf HA (2013) The influence of gamma irradiation on the intrinsic properties of cellulose acetate polymers. J Assoc Arab Univ Basic Appl Sci 14(1):46–50
El Nemr A, Eleryan A, Mashaly M, Khaled A (2021) Comparative study of synthesis of cellulose propionate from different sources using NIS as a New Catalyst. Polym Bull 78:4369–4386. https://doi.org/10.1007/s00289-020-03313-1
Chai H, Duan QL, Jiang L, Sun JH (2019) Effect of inorganic additive flame retardant on fire hazard of polyurethane exterior insulation material. J Therm Anal Calorim 135(5):2857–2868
Chai H, Duan Q, Jiang L, Gong L, Chen H, Sun J (2019) Theoretical and experimental study on the effect of nitrogen content on the thermal characteristics of nitrocellulose under low heating rates. Cellulose 26(2):763–776
Gao X, Jiang L, Xu Q, Wu W-Q, Mensah RA (2020) Thermal kinetics and reactive mechanism of cellulose nitrate decomposition by traditional multi kinetics and modeling calculation under isothermal and non-isothermal conditions. Ind Crops Prod 145:112085
Chin SF, Romainor ANB, Pang SC (2014) Fabrication of hydrophobic and magnetic cellulose aerogel with high oil absorption capacity. Mater Lett 115:241–243
Gao J, Xiong Y, Zhang Q, Jiang Y, Wang J, Zou S, Fu M, Wu J, Hu Y, Ye D (2020) 3D geometric modeling analysis of contact probability effect in carbon black oxidation over MnOx-CeO2 catalysts. Chem Eng J 398:125448
Kato H, Nakamura A, Horie M, Endoh S, Fujita K, Iwahashi H, Kinugasa S (2011) Preparation and characterization of stable dispersions of carbon black and nano-diamond in culture medium for in vitro toxicity assessment. Carbon 49:3989–3997
Celebioglu A, Uyar T (2011) Electrospun porous cellulose acetate fibers from volatile solvent mixture. Mater Lett 65:2291–2294
Salihu G, Goswami P, Russell S (2012) Hybrid electrospun nonwovens from chitosan/cellulose acetate. Cellulose 19:739–749
Filho GR, de Assunção RMN, Vieira JG, Meireles CS, Cerqueira DA, Barud HS, Ribeiro SJL, Messaddeq Y (2007) Characterization of methylcellulose produced from sugar cane bagasse cellulose: crystallinity and thermal properties. Polym Degrad Stabil 92:205–210
Le Quoc P, Solovieva AY, Uspenskaya MV, Olekhnovich RO, Sitnikova VE, Strelnikova IE, Kunakova AM (2021) High-porosity polymer composite for removing oil spills in cold regions ACS. Omega 31:20512–20521
El Nemr A, Serag E, El-Maghraby A, Fathy SA, Abdel Hamid FF (2019) Manufacturing of pH sensitive PVA/PVP/MWCNT and PVA/PEG/MWCNT nanocomposites: an approach for significant drug release. J Macromol Sci A 56(8):781–793. https://doi.org/10.1080/10601325.2019.1607377
ASTM F726-06 (2006) Standard test method for sorbent performance of adsorbents, ASTM International, West Conshohocken. https://doi.org/10.1520/F0726-06; www.astm.org
Likon M, Remškar M, Ducman V, Šveg F (2013) Populus seed fibers as a natural source for production of oil super absorbents. J Environ Manag 114:158–167
Lim T-T, Huang X (2007) Evaluation of kapok (Ceiba pentandra (L.) Gaertn.) as a natural hollow hydrophobic–oleophilic fibrous sorbent for oil spill cleanup. Chemosphere 66:955–963
Rengasamy R, Das D, Praba Karan C (2011) Study of oil sorption behavior of filled and structured fiber assemblies made from polypropylene, kapok and milkweed fibers. J Hazard Mater 186:526–532
ASTM (1993) Annual Book of American Society for Testing and Materials (ASTM) standard section, standard method of test for water and sediment in crude oil and fuel oils by centrifuge, D1796–91, 17, Philadelphia
Husseien M, Amer A, El-Maghraby A, Hamedallah NA (2009) Comprehensive characterization of corn stalk and study of carbonized corn stalk in dye and gas oil sorption. J Anal Appl Pyrol 86:360–363
Alaa El-Din G, Amer A, Malsh G, Hussein M (2017) Study on the use of banana peels for oil spill removal. Alex Eng J 57:2061–2068
Langmuir I (1916) The constitution and fundamental properties of solids and liquids. J Am Chem Soc 38:2221–2295
Longhinotti E, Pozza F, Furlan L, Sanchez MDND, Klug M, Laranjeira MCM, Favere VT (1998) Adsorption of anionic dyes on the biopolymer chitin. J Brazil Chem Soc 9:435–440
Freundlich HMF (1906) Über die adsorption in lösungen, Zeitschrift für PHysikalische. Chemie (Leipzig) 57(A):385–470
Temkin MJ, Pyzhev V (1940) Acta Physiochim. URSS 12:217–222
Kavitha D, Namasivayam C (2007) Experimental and kinetic studies on methylene blue adsorption by coir pith carbon. Bioresour Technol 98:14–21
Aharoni C, Ungarish M (1977) Kinetics of activated chemisorption. Part 2. Theoretical models. J Chem Soc Faraday Trans 73:456–464
Aharoni C, Sparks DL (1991) Kinetics of soil chemical reactions — a theoretical treatment. In: Sparks DL, Suarez DL (eds) Rate of soil chemical processes. Soil Science Society of America, Madison, pp 1–18
Wang XS, Qin Y (2005) Equilibrium sorption isotherms for of Cu2+ on rice bran. Process Biochem 40:677–680
Pearce CI, Lioyd JR, Guthrie JT (2003) The removal of color from textile wastewater using whole bacterial cells: a review. Dyes Pigments 58:179–196
Akkaya G, Ozer A (2005) Adsorption of acid red 274 (AR 274) on Dicranella varia: determination of equilibrium and kinetic model parameters. Process Biochem 40(11):3559–3568
Ng JCY, Cheung WH, McKay G (2002) Equilibrium studies of the sorption of Cu(II) ions onto chitosan. J Coll Interfer Sci 255:64–74
Porter JF, McKay G, Choy KH (1999) The prediction of sorption from a binary mixture of acidic des using single-and mixed-isotherm variants of the ideal adsorbed solute theory. Chem Eng Sci 54:5863–5885
Allen SJ, Gan Q, Matthews R, Johnson PA (2003) Comparison of optimized isotherm models for basic dye adsorption by kudzu. Bioresour Technol 88:143–152
Ho YS, Chiu WT, Wang CC (2005) Regression analysis for the sorption isotherms of basic dyes on sugarcane dust. Bioresour Technol 96:1285–1291
Lagergren S (1898) Zur theorie der sogenannten adsorption geloster stoffe. Kungliga Svenska Vetenskapsakademiens Handlingar 24:1–39
Ho YS, Mckay G, Wase DAJ, Foster CF (2000) Study of the sorption of divalent metal ions on to peat. Adsorpt Sci Technol 18:639–650
Chen C, Wang L, Huang Y (2009) Crosslinking of the electrospun polyethylene glycol/cellulose acetate composite fibers as shape-stabilized phase change materials. Mater Lett 63:569–571
Natchimuthu N, Rajalincam P, Radhakrishnan G (1992) Cellulose nitrate-poly (vinyl chloride-co-vinyl acetate)-polyurethane ternary IPNs: FT-IR and morphological studies. J Appl Polym Sci 44:981–986
Goetz LA, Jalvo B, Rosal R, Mathew A (2016) Superhydrophilic anti-fouling electrospun cellulose acetate membranes coated with chitin nanocrystals for water filtration. J Membr Sci 510:238–248
Lawal AA, Hassan MA, Farid MAA, Yasim-Anuar TAT, Yusoff MZM, Zakaria MR, Roslan AM, Mokhtar MN, Shirai Y (2020) Production of biochar from oil palm frond by steam pyrolysis for removal of residual contaminants in palm oil mill effluent final discharge. J Clean Product 265:121643
Nartker S, Drzal LT (2010) Electrospun cellulose nitrate nanofibers. J Nanosci Nanotech 10:5810–5813
Bhandari J, Mishra H, Mishra PK, Wimmer R, Ahmad FJ, Talegaonkar S (2017) Cellulose nanofiber aerogel as a promising biomaterial for customized oral drug delivery. Inter J Nanomedicine 12:2021–2031
Shi X, Zheng Y, Wang G, Lin Q, Fan J (2014) pH and electro-response characteristics of bacterial cellulose nanofiber/sodium alginate hybrid hydrogels for dual controlled drug delivery. RSC Adv 4:47056–47065
Dong T, Xu G, Wang F (2015) Oil spill cleanup by structured natural sorbents made from Cattail fibers. Ind Crops Prod 76:25–33
Abdullah M, Rahmah AU, Man Z (2010) Physicochemical and sorption characteristics of Malaysian Ceiba pentandra (L.) Gaertn. as a natural oil sorbent. J Hazard Mater 177:683–691
Wang J, Zheng Y, Kang Y, Wang A (2013) Investigation of oil sorption capability of PBMA/SiO2 coated kapok fiber. Chem Eng J 223:632–639
Liu F, Ma M, Zang D, Gao Z, Wang C (2014) Fabrication of superhydrophobic/superoleophilic cotton for application in the field of water / oil separation. Carbohydr Polym 103:480–487
Omer AM, Khalifa RE, Tamer TM, Elnouby M, Hamed AM, Ammar YA, Ali AA, Gouda M, Mohy Eldin MS (2019) Fabrication of a novel low-cost superoleophilic nonanyl chitosan-poly (butyl acrylate) grafted copolymer for the adsorptive removal of crude oil spills. Int J Biol Macromol 140:588–599
Teli MD, Valia SP (2013) Acetylation of banana fibre to improve oil absorbency. Carbohydr Polym 92:328–333
Chagas NV, Meira JS, Anaissi FJ, Luiz F, Quináia SP, Felsner ML (2014) Chem Eng Sci 54:5863–5885
Kargi F, Cikla S (2006) Biosorption of zinc(II) ions onto powdered waste sludge (PWS): kinetics and isotherms. Enzyme Microbial Tech 38:705–710
Yang M, Wang J, Chen Y, Gao J (2020) Biochar produced from cotton husks and its application for the adsorption of oil products. IOP Conf Ser Earth Environ Sci 545:012022. https://doi.org/10.1088/1755-1315/545/1/012022
Piperopoulos E, Calabrese L, Mastronardo E, Proverbio E, Milone C (2020) Sustainable reuse of char waste for oil spill recovery foams. Water Air Soil Pollut 231. https://doi.org/10.1007/s11270-020-04671-2
Cai L, Zhang Y, Zhou Y, Zhang X, Ji L, Song W, Zhang H, Liu J (2019) Effective adsorption of diesel oil by crab-shell-derived biochar nanomaterials. Materials 12. https://doi.org/10.3390/ma12020236
Angelova D, Uzunov I, Uzunova S, Gigova A, Minchev L (2011) Kinetics of oil and oil products adsorption by carbonized rice husks. Chem Eng J 172:306–311. https://doi.org/10.1016/j.cej.2011.05.114
Said AEAA, Ludwick AG, Aglan HA (2009) Usefulness of raw bagasse for oil absorption: a comparison of raw and acylated bagasse and their components. Bioresour Technol 100(7):2219–2222. https://doi.org/10.1016/j.biortech.2008.09.060
Radetic M, Ilic V, Radojevic D, Miladinovic R, Jocic D, Jovancic P (2008) Efficiency of recycled wool-based nonwoven material for the removal of oils from water. Chemosphere 70(3):525–530. https://doi.org/10.1016/j.chemosphere.2007.07.005
Rajakovic V, Aleksic G, Radetic M, Rajakovic L (2007) Efficiency of oil removal from real wastewater with different sorbent materials. J Hazard Mater 143(1–2):494–499. https://doi.org/10.1016/j.jhazmat.2006.09.060
Suni S, Kosunen A, Hautala M, Pasila A, Romantschuk M (2004) Use of a by-product of peat excavation, cotton grass fibre, as a sorbent for oil-spills. Mar Pollut Bull 49(11–12):916–921. https://doi.org/10.1016/j.marpolbul.2004.06.015
Khan E, Virojnagud W, Ratpukdi T (2004) Use of biomass sorbents for oil removal from gas station runoff. Chemosphere 57(7):681–689. https://doi.org/10.1016/j.chemosphere.2004.06.028
Sun X (2004) Acetylation of sugarcane bagasse using NBS as a catalyst under mild reaction conditions for the production of oil sorption-active materials. Biores Technol 95(3):343–350. https://doi.org/10.1016/j.biortech.2004.02.025
Funding
Open access funding provided by The Science, Technology & Innovation Funding Authority (STDF) in cooperation with The Egyptian Knowledge Bank (EKB). The Egyptian Science and Technological Development Fund (STDF) provided partial funding for this study (Project Nos. CB-4874, CB-22816 and IG-34795).
Author information
Authors and Affiliations
Contributions
The practical portion was led by Miss Nehad A. Elmaghraby, who also wrote the first draft. Professor Dr. Ahmed El Nemr oversaw the actual work, edited the article, and then submitted it. The work was overseen by Dr. Ahmed M. Omer, Professor Dr. El-Refaie Kenawy, and Professor Dr. Mohamed Gaber.
Corresponding author
Ethics declarations
Consent to participate
Not applicable.
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Elmaghraby, N.A., Omer, A.M., Kenawy, ER. et al. Fabrication of cellulose acetate/cellulose nitrate/carbon black nanofiber composite for oil spill treatment. Biomass Conv. Bioref. (2022). https://doi.org/10.1007/s13399-022-03506-w
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s13399-022-03506-w