
Abstract
The search for renewable sources of energy has resulted in the focus of the use of biomass and biomass-derived platform chemicals for the generation of energy. One such chemical—furfuryl alcohol—which has several industrial applications can be obtained from furfural via hydrogenation using catalysts. Novel N^S (pzyraolyl-thio) and N^Se (pyrazolyl-selenium) bidentate ligands and their corresponding palladium(II) complexes were synthesized and evaluated as catalysts for the hydrogenation of furfural. The catalysts are active for the solventless chemoselective hydrogenation of furfural with TON around 1600 obtained. The role of the sulfur or selenium donor atoms as well as the presence of bulky groups on the catalysts influencing catalytic activity was investigated. Bulky groups present were found to slightly increase catalytic activity while the large size of selenium as compared to sulfur was not found to significantly impact catalytic activity. The complexes were found to hydrogenate furfural by homogenous means with likely palladium-bicarbonate intermediate species being observed. The recyclability of the homogenous catalysts by heterogenization using a supported ionic liquid phase (SILP) approach was investigated.
Graphical abstract







Similar content being viewed by others

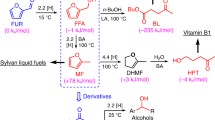
Data availability
The datasets generated during and/or analyzed during the current study are available from the corresponding author upon reasonable request.
References
Lynd LR, Wyman CE, Gerngross TU (1999) Biocommodity engineering. Biotechnol Prog 15:777–793. https://doi.org/10.1021/bp990109e
Ahmed MM, Nasri NS, Hamza DU (2012) Biomass as a renewable source of chemicals for industrial applications. Int J Eng Sci Technol 4:721–730
Briens C, Piskorz J, Berruti F (2008) Biomass valorization for fuel and chemicals production - a review. Int J Chem React Eng 6:1–52. https://doi.org/10.2202/1542-6580.1674
Climent MJ, Corma A, Iborra S (2014) Conversion of biomass platform molecules into fuel additives and liquid hydrocarbon fuels. Green Chem 16:516–547. https://doi.org/10.1039/C3GC41492B
Fuente-Hernandez A, Lee R, Nicolas B, Zamboni I (2017) Reduction of furfural to furfuryl alcohol in liquid phase over a biochar-supported platinum catalyst. Energies 10:286–296. https://doi.org/10.3390/en10030286
Fuente-Hernández A, Corcos P, Beauchet R, Lavoie J (2013) Biofuels and co-products out of hemicelluloses. In: Fang Z (ed) Liquid. InTech Open, Gaseous and Solid Biofuels - Conversion Techniques, pp 3–46
Mariscal R, Ojeda M, Maireless-Torres P et al (2016) Furfural: a renewable and versatile platform molecule for the synthesis of chemicals and fuels. Energy Environ Sci 9:1144–1189. https://doi.org/10.1039/c5ee02666k
Liu X, Zhang B, Fei B et al (2017) Tunable and selective hydrogenation of furfural to furfuryl alcohol and cyclopentanone over Pt supported on biomass-derived porous heteroatom doped carbon. Faraday Discuss. https://doi.org/10.1039/C7FD00041C
Zhang H, Canlas C, Jeremy Kropf A et al (2015) Enhancing the stability of copper chromite catalysts for the selective hydrogenation of furfural with ALD overcoating (II) - comparison between TiO2 and Al2O3 overcoatings. J Catal 326:172–181. https://doi.org/10.1016/j.jcat.2015.03.017
Corma A, Iborra S, Velty A (2007) c. Chem Rev 107:2411–2502. https://doi.org/10.1021/cr050989d
Chen X, Zhang L, Zhang B, et al (2016) Highly selective hydrogenation of furfural to furfuryl alcohol over Pt nanoparticles supported on g-C 3 N 4 nanosheets catalysts in water. Nat Publ Gr 1–13.https://doi.org/10.1038/srep28558
Elliott DC, Hart TR (2009) Catalytic hydroprocessing of chemical models for bio-oil. Energy Fuels 23:631–637. https://doi.org/10.1021/ef8007773
Wei S, Cui H, Wang J et al (2011) Preparation and activity evaluation of NiMoB/γ-Al2O3 catalyst by liquid-phase furfural hydrogenation. Particuology 9:69–74. https://doi.org/10.1016/j.partic.2010.05.009
Chen X, Li H, Luo H, Qiao M (2002) Liquid phase hydrogenation of furfural to furfuryl alcohol over Mo-doped Co-B amorphous alloy catalysts. Appl Catal A Gen 233:13–20
Yuan Q, Zhang D, Van HL et al (2015) Selective liquid phase hydrogenation of furfural to furfuryl alcohol by Ru/Zr-MOFs. J Mol Catal A Chem 406:58–64. https://doi.org/10.1016/j.molcata.2015.05.015
Han S, Chen WT, Gao ZT et al (2022) Mechanochemical-assisted synthesis of nitrogen-doped carbon supported cobalt catalysts for efficient and selective hydrogenation of furfural. Catal Letters. https://doi.org/10.1007/s10562-022-04042-y
An Z, Li J (2022) Recent advances in the catalytic transfer hydrogenation of furfural to furfuryl alcohol over heterogeneous catalysts. Green Chem 24:1780–1808. https://doi.org/10.1039/d1gc04440k
Puthiaraj P, Kim K, Ahn WS (2019) Catalytic transfer hydrogenation of bio-based furfural by palladium supported on nitrogen-doped porous carbon. Catal Today 324:49–58. https://doi.org/10.1016/j.cattod.2018.07.033
Ouyang W, Yepez A, Romero AA, Luque R (2018) Towards industrial furfural conversion: selectivity and stability of palladium and platinum catalysts under continuous flow regime. Catal Today 308:32–37. https://doi.org/10.1016/j.cattod.2017.07.011
Yan K, Jarvis C, Lafleur T et al (2013) Novel synthesis of Pd nanoparticles for hydrogenation of biomass-derived platform chemicals showing enhanced catalytic performance. RSC Adv 3:25865–25871. https://doi.org/10.1039/c3ra43619e
Wang Y-Q, Lu S-M, Zhou Y-G (2005) Palladium-catalyzed asymmetric hydrogenation of functionalized ketones. Org Lett 7:3235–3238. https://doi.org/10.1021/ol051007u
Chen Q-A, Ye Z-S, Duan Y, Zhou Y-G (2013) Homogeneous palladium-catalyzed asymmetric hydrogenation. Chem Soc Rev 42:497–511. https://doi.org/10.1039/C2CS35333D
Tukacs JM, Bohus M, Dibo G, Mika LT (2017) Ruthenium-catalyzed solvent-free conversion of furfural to furfuryl alcohol. RSC Adv 7:3331–3335. https://doi.org/10.1039/C6RA24723G
Paganelli S, Piccolo O, Pontini P et al (2015) Aqueous-phase hydrogenation and hydroformylation reactions catalyzed by a new water-soluble [rhodium ]– thioligand complex. Catal Today 247:64–69. https://doi.org/10.1016/j.cattod.2014.05.038
Strassberger Z, Mooijman M, Ruijter E et al (2010) A facile route to ruthenium – carbene complexes and their application in furfural hydrogenation. Appl Organomet Chem 24:142–146. https://doi.org/10.1002/aoc.1584
Townsend TM, Kirby C, Ruff A, O’Connor AR (2017) Transfer hydrogenation of aromatic and linear aldehydes catalyzed using Cp*Ir(pyridinesulfonamide)Cl complexes under base-free conditions. J Organomet Chem 843:7–13. https://doi.org/10.1016/j.jorganchem.2017.05.004
Wu W-P, Xu Y-J, Chang S-W et al (2016) pH-regulated aqueous catalytic hydrogenation of biomass carbohydrate derivatives by using semisandwich iridium complexes. ChemCatChem 8:3375–3380. https://doi.org/10.1002/cctc.201601009
Appavoo D, Omondi B, Guzei IA et al (2014) Bis(3,5-dimethylpyrazole) copper(II) and zinc(II) complexes as efficient initiators for the ring opening polymerization of ??-caprolactone and d, l-lactide. Polyhedron 69:55–60. https://doi.org/10.1016/j.poly.2013.11.011
Amenuvor G, Obuah C, Nordlander E, Darkwa J (2016) Novel pyrazolylphosphite- and pyrazolylphosphinite-ruthenium(II) complexes as catalysts for hydrogenation of acetophenone. Dalt Trans 45:13514–13524. https://doi.org/10.1039/c6dt02164f
Jasiński D, Meredith J, Kirwan K (2018) The life cycle impact for platinum group metals and lithium to 2070 via surplus cost potential. Int J Life Cycle Assess 23:773–786. https://doi.org/10.1007/s11367-017-1329-4
Vural Gürsel I, Noël T, Wang Q, Hessel V (2015) Separation/recycling methods for homogeneous transition metal catalysts in continuous flow. Green Chem 17:2012–2026. https://doi.org/10.1039/C4GC02160F
Cole-Hamilton DJ (2003) Homogeneous catalysis - new approaches to catalyst separation, recovery, and recycling. Science 1702–1706.https://doi.org/10.1126/science.1081881
Riisager A, Fehrmann R, Haumann M, Wasserscheid P (2006) Supported ionic liquids: versatile reaction and separation media. Top Catal 40:91–102. https://doi.org/10.1007/s11244-006-0111-9
Haumann M (2020) Continuous catalytic processes with supported ionic liquid phase (SILP) materials. In: Commer Appl Ionic Liquids 49–67
Roa AE, Campos J, Paneque M et al (2015) Synthesis of new heteroscorpionate iridium(i) and iridium(iii) complexes. Dalt Trans 44:6987–6998. https://doi.org/10.1039/c5dt00482a
Singh VV, Kumar U, Tripathi SN, Singh AK (2014) Shape dependent catalytic activity of nanoflowers and nanospheres of Pd4S generated via one pot synthesis and grafted on graphene oxide for Suzuki coupling. Dalt Trans 43:12555–12563. https://doi.org/10.1039/c4dt01396d
Sharma NK, Joshi H, Singh VV et al (2013) Accepted manuscript. Dalt Trans 42:3908–3918. https://doi.org/10.1039/C2DT31975F
Major MM, Rövid G, Balogh S et al (2019) Double stereoselective coordination of chiral N, S ligands: synthesis, coordination chemistry and utilization in Pd-catalyzed allylic alkylation. Appl Organomet Chem 33:1–14. https://doi.org/10.1002/aoc.4726
Kumar U, Dubey P, Vati SV et al (2014) Multi-phase semicrystalline microstructures drive exciton dissociation in neat plastic semiconductors. RSC Adv 4:41659–41665. https://doi.org/10.1039/b000000x
Pal S (2018) Pyridine: a useful ligand in transition metal complexes. In: Pyridine. 13
Zargari N, Jung E, Lee JH, Jung KW (2017) Carbon dioxide hydrogenation: efficient catalysis by an NHC-amidate Pd(II) complex. Tetrahedron Lett 58:3330–3332. https://doi.org/10.1016/j.tetlet.2017.07.040
Anderson GK, Lin M, Sen A, Gretz E (2007) Bis(benzonitrile)dichloro complexes of palladium and platinum. John Wiley & Sons, Hoboken, NJ, USA, NJ, USA
Van WJL, Omondi B, Appavoo D et al (2012) Solvent-free synthesis of 3,5-Di-Tert-butylpyrazole and 3,5-di-substituted-butylpyrazol-1-ylethanol. J Chem Res 36:474–477
Bruker AXS Inc (2012) SADABS, SAINT Softw. Ref. Manuals Bruker AXS Inc U. Madison WI
Acknowledgements
We would like to thank Sasol SA, UJ Center, for the synthesis and catalysis and the UJ Faculty of Science Spectrum for the use of facilities.
Funding
This work was supported by the National Research Foundation of South Africa (NRF) (Grant Numbers: 117989, 112943, and 99269) and The Oppenheimer Memorial Trust (Grant Number: OMT Ref. 21759/01).
Author information
Authors and Affiliations
Contributions
All authors contributed to the study conception and design. Material preparation, data collection, and analysis were performed by Winifred D. Anyomih, Novisi K. Oklu, and Edward Ocansey. The first draft of the manuscript was written by Edward Ocansey, and all authors commented on the previous versions of the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
We dedicate this work to the memory of Novisi K. Oklu.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Appendix
Appendix
1.1 Footnotes
Electronic supporting information (ESI) is available free of charge. CCDC numbers 1843223 and 1,848,203 contain the supplementary crystallographic data for C1 and C2, respectively. This data can be obtained free of charge from The Cambridge Crystallographic Data Centre from the Director, CCDC, 12 Union Road, Cambridge, CB2 1EZ, UK (fax: + 44 1223 336,063; deposit@ccdc.cam.ac.uk or https://www.ccdc.cam.ac.uk/structures/.
1.2 Experimental section
1.2.1 Materials and methods
All reactions were carried out in air unless otherwise stated. All solvents used were reagent grade, purchased from Sigma-Aldrich, and dried under nitrogen before use. 2-Hydroxyethyl hydrazine, pentane-2,4-dione, 2,2,6,6-tetramethylheptane-3,5-dione, thionyl chloride, thiophenol, diphenyl diselenide, sodium borohydride, sodium hydride, and sodium hydroxide were purchased from Sigma-Aldrich and used without further purification. PdCl2 was purchased from Heraeus South Africa and used as received. [PdCl2(MeCN)2] [42], 2-(3,5-di-tert-butyl-1H-pyrazol-1-yl)ethan-1-ol [43], and 2-(3,5-dimethyl-1H-pyrazol-1-yl)ethan-1-ol [43] were synthesized according to literature procedures. NMR spectra were recorded on Bruker 400 MHz NMR spectrometer (1H at 400 MHz and 13C{1H} at 100 MHz). Spectrometer chemical shifts were reported relative to the internal standard tetramethylsilane (δ 0.00 ppm) and referenced to the residual proton and carbon signals at 7.24 and 77.0 ppm, respectively, of CDCl3. Infrared spectra were obtained neat using a PerkinElmer Spectrum BX II fitted with an ATR probe. Melting points were obtained using a Gallenkamp Digital Melting-point Apparatus 5A 6797. Elemental analysis was performed on a Thermos Scientific FLASH 2000 CHNS-O Analyzer. Mass spectrometry was performed using Waters Synapt G2 mass spectrometer with both ESI positive and Cone Voltage 15 V.
1.2.2 X-ray structure determination
Single crystal X-ray diffraction data were collected on a Bruker APEXII diffractometer with Mo Kα (λ = 0.71073 Å) radiation and a detector to a crystal distance of 4.00 cm. The initial cell matrix was obtained from three series of scans at different starting angles. Each series consisted of 12 frames collected at intervals of 0.5° in a 6° range with an exposure time of about 10 s per frame. The reflections were successfully indexed by an automated indexing routine built in the APEXII program suite. The data were collected using the full sphere data collection routine to survey the reciprocal space to the extent of a full sphere to a resolution of 0.75 Å. Data were harvested of which 9674 (C1) and 4203 (C2) reflections were used [44]. The data integration and reduction were processed with SAINT software. A multi-scan absorption correction was applied to the collected reflections with SADABS using XPREP. Structures were solved by the direct method using the program SHELXS-97 and were refined on F2 by the full-matrix least-squares technique using the SHELXL-97 program package. All non-hydrogen atoms were refined with anisotropic displacement coefficients. All hydrogen atoms were included in the structure factor calculations at idealized positions and were allowed to ride on the neighboring atoms with relative isotropic displacement coefficients.
1.3 Synthesis of ligand precursors: 3,5-di-tert-butyl-1-(2-chloroethyl)-1H-pyrazole (1) and 1-(2-chloroethyl)-3,5-dimethyl-1H-pyrazole (2)
1.3.1 General experimental procedure
Into a, 1 eq of 2-(3,5-alkyl-1H-pyrazol-1-yl)ethan-1-ol (1 eq.) was added to a round bottom flask containing 50 mL dichloromethane, and the mixture was cooled to 0–5 °C using ice bath. Thionyl chloride (4 eq.) was then added drop-wise to the reaction mixture and slowly warmed up to room temperature. The reaction mixture was further stirred at room temperature for 16 h. The solvent was then removed via rotary evaporation followed by quenching the content of the reaction mixture with 1 M NaHCO3 solution. The product was extracted from the aqueous phase using 4 × 20 mL portions of chloroform. The collected chloroform portions were dried over anhydrous Na2SO4, and removal of the solvent via rotary evaporation yielded the pure product which was used in next step reaction without further purification.
-
(1)
Appearance, yellowish solid; solubility, soluble in chloroform, DCM, insoluble in water. yield = 1.49 g, 81%; melting point: 42 °C. FT-IR (vmax/cm−1): 1739 (C = N). 1H NMR (400 MHz, CDCl3, 30 °C) (ppm) = 5.77 (s, 1H, Hpz), 4.39 (t, JHH = 7.6 Hz, HCH2, 2H), 3.87 (t, JHH = 7.6 Hz, HCH2, 2H), 1.33 (s, HtBu, 9H), 1.23 (s, HtBu, 9H), 13C{1H} NMR (100 MHz, CDCl3, 30 °C) (ppm) = 160.50; 151.29; 99.54; 51.51; 41.87; 31.91;31.12; 30.49; 30.25; ESI–MS [M + H]+ = 243.1630 (100%); elemental analysis; anal calcd for C13H23ClN2: C, 64.31; H, 9.55, N, 11.54% found C, 64.35; H, 9.25; N, 11.74%.
-
(2)
1-(2-Chloroethyl)-3,5-dimethyl-1H-pyrazole; appearance, yellowish oil; solubility, soluble in chloroform, DCM, insoluble in water. Yield = 1.71 g, 84%; 1H NMR (400 MHz, CDCl3, 30 °C) (ppm) = 5.81 (s, 1H, Hpz), 4.28 (t, JHH = 6.4 Hz, HCH2, 2H), 3.86 (t, JHH = 6.4 Hz, HCH2, 2H), 2.28 (s, HCH3, 3H), 2.21 (s, HCH3, 3H), 13C{1H} NMR (100 MHz, CDCl3, 30 °C) (ppm) = 148.34; 139.76; 105.12; 49.69; 43.02; 13.50; 11.08; ESI–MS [M + H]+ = 159.0678 (100%); elemental analysis; anal calcd for C7H11ClN2: C, 53.00; H, 6.99, N, 17.66% found C, 52.66; H, 7.21; N, 17.99%.
1.4 Synthesis of ligands (L1-L4)
1.4.1 Synthesis of ligands L1 and L3: General experimental procedure
A solution of NaBH4 (121 mg, 3.2 mmol) in 2 mL of 5% aqueous NaOH was added dropwise to a solution of diphenyldiselenide (500 mg, 1.60 mmol) in ethanol (15 mL) under argon till the solution become colorless. This indicates the formation of sodium phenylsalenolate (PhSeNa). Then a solution of halide (1 or 2) (3.2 mmol) in 10 mL ethanol was added dropwise to the reaction mixture and stirred further for 24 h at room temperature. After the reaction time had elapsed, the solvent was evaporated on a rotary evaporator, and the residue obtained was dissolved in dichloromethane (20 mL) and washed with water (3 × 7 mL). The organic layer was collected and dried over anhyd. MgSO4. The removal of solvent on the rotary evaporator yields the product.
3,5-Di-tert-butyl-1-(2-(phenylselanyl)ethyl)-1H-pyrazole (L1) appearance, white solid; solubility, soluble in chloroform, DCM, insoluble in water. Yield = 0.91 g, 71%; melting point: 75 °C. FT-IR (vmax/cm−1): 1534 (C = N). 1H NMR (400 MHz, CDCl3, 30 °C) (ppm) = 7.58–7.56 (m, 2H, HPh), 7.26–7.24 (m, 3H, HPh), 5.72 (s, 1H, Hpz), 4.32 (t, JHH = 6 Hz, HCH2, 2H), 3.28 (t, JHH = 6 Hz, HCH2, 2H), 1.23 (s, HtBu, 9H), 1.16 (s, HtBu, 9H), 13C{1H} NMR (100 MHz, CDCl3, 30 °C) (ppm) = 160.08; 150.77; 133.70; 133.55; 129.12; 128.98; 128.78; 127.41; 99.23; 50.90; 31.93; 30.98; 30.57; 30.08; 27.22; ESI–MS [M-Pz]+ = 184.9867 (15%); elemental analysis; anal calcd for C19H28N2Se: C, 62.80; H, 7.77, N, 7.71% found C, 62.65; H, 7.44; N, 7.59%.
3,5-Dimethyl-1-(2-(phenylselanyl)ethyl)-1H-pyrazole (L3) appearance, yellowish oil; solubility, soluble in chloroform, DCM, insoluble in water. Yield = 1.21 g, 77%; FT-IR (vmax/cm−1): 1734 (C = N). 1H NMR (400 MHz, CDCl3, 30 °C) (ppm) = 7.59–7.57 (m, 2H, HPh), 7.28–7.24 (m, 3H, HPh), 5.77 (s, 1H, Hpz), 4.18 (t, JHH = 7.2 Hz, HCH2, 2H), 3.24 (t, JHH = 7.6 Hz, HCH2, 2H), 2.14 (s, HCH3, 3H), 2.09 (s, HCH3, 3H), 13C{1H} NMR (100 MHz, CDCl3, 30 °C) (ppm) = 147.73; 138.88; 132.82; 131.51; 129.20; 104.99; 57.87; 48.54; 27.39; 27.06; 26.72; 18.43; 13.46; ESI–MS [M + H]+ = 281.0552 (100%); elemental analysis; anal calcd for C13H16N2Se: C, 55.92; H, 5.78, N, 10.03% found C, 59.77; H, 5.62; N, 9.66%.
1.5 Synthesis of ligands L2 and L4
1.5.1 General experimental procedure
NaH (217 mg, 9.07 mmol) was added portion-wise to a stirring solution of thiophenol (500 mg, 4.54 mmol) in 10 mL DMF maintained between 0 and 5 °C. The reaction mixture was then allowed to stir at room temperature for 1 h. Subsequently, a 10-mL DMF solution of halide (1 or 2) (4.54 mmol) was added drop-wise to the reaction mixture and stirred further for 24 h at room temperature. After the reaction time had elapsed, 10 mL water was added and the product was extracted with 4 × 20 mL portions of chloroform. The organic layer was collected and dried over anhyd. MgSO4 and removal of solvent using a rotary evaporator yielded the product.
3,5-Di-tert-butyl-1-(2-(phenylthio)ethyl)-1H-pyrazole (L2) appearance, white solid; solubility, soluble in chloroform, DCM, insoluble in water. Yield = 1.2 g, 73%; melting point: 57 °C. FT-IR (vmax/cm−1): 1566 (C = N). 1H NMR (400 MHz, CDCl3, 30 °C) (ppm) = 7.30–7.26 (m, 2H, HPh), 7.21–7.18 (m, 3H, HPh), 5.74 (s, 1H, Hpz), 4.28 (t, JHH = 8 Hz, HCH2, 2H), 3.36 (t, JHH = 8 Hz, HCH2, 2H), 1.25 (s, HtBu, 9H), 1.21 (s, HtBu, 9H); 13C{1H} NMR (100 MHz, CDCl3, 30 °C) (ppm) = 160.06; 150.89; 135.12; 135.00; 130.26; 128.95; 128.77; 126.54; 99.17; 50.01; 33.78; 31.89; 30.95; 30.54; 30.08; ESI–MS [M-Pz]+ = 184.9867 (15%); elemental analysis; anal calcd for C19H28N2S: C, 72.10; H, 8.92, N, 8.85% found C, 71.69; H, 8.55; N, 8.55%.
3,5-Dimethyl-1-(2-(phenylthio)ethyl)-1H-pyrazole (L4) appearance, yellowish oil; solubility, soluble in chloroform, DCM, insoluble in water. Yield = 1.44 g, 82%; FT-IR (vmax/cm−1): 1583 (C = N). 1H NMR (400 MHz, CDCl3, 30 °C) (ppm) = 7.26–7.24 (m 2H, HPh), 7.19–7.16 (m, 3H, HPh), 5.71 (s, 1H, Hpz), 4.14 (t, JHH = 7.2 Hz, HCH2, 2H), 3.31 (t, JHH = 6 Hz, HCH2, 2H), 2.17 (s, HCH3, 3H), 2.12 (s, HCH3, 3H), 13C{1H} NMR (100 MHz, CDCl3, 30 °C) (ppm) = 147.58; 139.03; 134.95; 129.29; 128.92; 126.28; 104.82; 47.55; 36.31; 33.59; 31.20; 13.32: 10.79; ESI–MS [M + H]+ = 233.1104 (100%); ELEMENTAL analysis; anal calcd for C13H16N2S: C, 67.20; H, 6.94, N, 12.06%, S 13.80%; found C, 66.94; H, 6.54; 11.88%, S, 13.55%.
1.5.2 General experimental procedure-synthesis of palladium complexes C1–C4
To a stirring solution of ligand (1 eq.) in 10 mL DCM, PdCl2MeCN2 (1 eq) was added and the reaction mixture was stirred at room temperature for 24 h. After the reaction time had elapsed, the volume of the solvent was reduced to approximately 2 mL using a rotary evaporator followed by the addition of 10 mL of hexane or diethyl ether. This resulted in the precipitation of the product. The product was filtered off, washed with ether (3 × 5 mL) to remove any impurity, and dried in vacuo.
κ2-N^Se-{3,5-di-tert-butyl-1-(2-(phenylselanyl)ethyl)-1H-pyrazole}dichloropalladium(II) (C1) appearance, yellow solid; solubility, soluble in chloroform, DCM, insoluble in water, hexane and diethyl ether. Yield = 111 mg, 81%; Melting point: 235 °C (Decomp.). FT-IR (vmax/cm−1): 1799 (C = N). 1H NMR (400 MHz, CDCl3, 30 °C) (ppm) = 7.93–7.91 (m, 2H, HPh), 7.21–7.19 (m, 3H, HPh), 6.25 (t, JHH = 13.2 Hz, HCH2, 1H) 6.00 (s, 1H, Hpz), 5.34 (d, JHH = 16 Hz, HCH2, 1H), 3.97 (d, JHH = 13.6 Hz, HCH2, 1H), 2.93 (t, JHH = 12.8 Hz, HCH2, 1H), 1.44 (s, HtBu, 9H), 1.37 (s, HtBu, 9H); 13C{1H} NMR (100 MHz, CDCl3, 30 °C) (ppm) = 163.52; 155.77; 130.26; 129.74; 129.88; 128.81; 126.97; 105.44; 53.23; 32.93; 32.33; 31.79; 30.83; 30.22; 27.22; ESI–MS [M-Cl]+ = 504.0155 (75%); elemental analysis; anal calcd for C19H28Cl2N2PdSe: C, 42.20; H, 5.22, N, 5.18% found C, 42.40; H, 5.55; N, 5.00%.
κ2-N^S-{3,5-di-tert-butyl-1-(2-(phenylthio)ethyl)-1H-pyrazole}dichloropalladium(II) (C2) appearance, yellow solid; solubility, soluble in chloroform, DCM, insoluble in water hexane, diethyl ether. Yield = 92 mg, 75%; Melting point: 233 °C (Decomp.). FT-IR (vmax/cm−1): 1788 (C = N). 1H NMR (400 MHz, CDCl3, 30 °C) (ppm) = 7.41–7.39 (m, 2H, HPh), 7.28–7.23 (m, 3H, HPh), 6.21 (m, HCH2, 1H) 6.07 (s, 1H, Hpz), 5.26 (d, JHH = 14.8 Hz, HCH2, 1H), 3.76 (m, HCH2, 1H), 3.04 (m, HCH2, 1H), 1.61 (s, HtBu, 9H), 1.49 (s, HtBu, 9H); 13C{1H} NMR (100 MHz, CDCl3, 30 °C) (ppm) = 161.52; 158.75; 130.46; 130.11; 129.94; 128.61; 127.77; 105.00; 54.13; 33.03; 30.81; 30.21; 27.02; ESI–MS [M-Cl]+ = 456.0703 (20%); elemental analysis; anal calcd for C19H28Cl2N2PdS: C, 46.21; H, 5.72, N, 5.67% found C, 45.81; H, 5.55; N, 5.33%.
κ2-N^Se-{3,5-dimethyl-1-(2-(phenylselanyl)ethyl)-1H-pyrazole}dichloropalladium(II) (C3) (9) appearance, yellow solid; solubility, soluble in chloroform, DCM, insoluble in water hexane, diethyl ether. Yield = 100 mg, 71%; melting point: 243 °C; FT-IR (vmax/cm−1): 1744 (C = N). 1H NMR (400 MHz, CDCl3, 30 °C) (ppm) = 7.63–7.61 (m, 2H, HPh), 7.38–7.32 (m, 3H, HPh), 5.84 (s, 1H, Hpz), 5.40 (m, HCH2, 1H), 5.03 (m, HCH2, 1H), 3.98 (m, HCH2, 1H), 2.80 (m, HCH2, 1H), 2.52 (s, HCH3, 3H), 2.31 (s, HCH3, 3H), 13C{1H} NMR (100 MHz, CDCl3, 30 °C) (ppm) = 147.73; 138.88; 132.82; 131.51; 129.89; 116.42; 101.19; 57.67; 47.34; 28.39; 26.71; 18.63; 12.08; ESI–MS [M-Cl]+ = 420.0940 (20%); elemental analysis; anal calcd for C13H16Cl2N2PdSe: C, 34.20; H, 3.53, N, 6.14% found C, 33.77; H, 3.39; N, 6.54%.
κ2-N^S-{3,5-dimethyl-1-(2-(phenylthio)ethyl)-1H-pyrazole}dichloropalladium(II) (C4) appearance, yellow solid; solubility, soluble in chloroform, DCM, insoluble in hexane, diethyl ether and water. Yield = 120 mg, 83%; melting point: 180 °C; FT-IR (vmax/cm−1): 1552 (C = N). 1H NMR (400 MHz, CDCl3, 30 °C) (ppm) = 7.54–7.44 (m 2H, HPh), 7.39–7.35 (m, 3H, HPh), 5.93 (s, 1H, Hpz), 5.70 (m, HCH2, 1H), 5.31 (m, HCH2, 1H), 3.75 (m, HCH2, 1H), 2.85 (m, HCH2, 1H), 2.50 (s, HCH3, 3H), 2.39 (s, HCH3, 3H), 13C{1H} NMR (100 MHz, CDCl3, 30 °C) (ppm) = 152.68; 131.20; 131.03; 130.79; 130.39; 129.98; 108.89; 48.96; 31.571; 22.60; 15.09; 14.10; 12.30; ESI–MS [M-Cl]+ = 372.9749 (100%); elemental analysis; anal calcd for C13H16Cl2N2PdS: C, 38.11; H, 3.94, N, 6.84% found C, 38.55; H, 4.21; 7.00%.
1.5.3 Synthesis of silica support S1
Silica gel 100 was pretreated by heating at 400 °C in an oven for 16 h. To 3.2 g of pretreated silica in a tin-foil coated round bottom flask, 1.63 g of 1-(3-propyltrimethoxysilane)-3-methylimidazolium iodide, 1 mL of 25% NH4O and 10 mL of DCM was added and stirred for 72 h at room temperature. After the reaction period had elapsed, the solvent was removed by rotary evaporation to yield the SILP support which was also stored in tin foil-coated glass vials.
1.5.4 Synthesis of SILP catalyst
One milliliter of DCM was added to 10 mg of C1, in a tin foil-coated glass vial. To the contents of this vial was added 90 mg of SILP pretreated support S1. This was followed by the addition of 90 mg of 1-hexyl-3-methylimidazolium iodide dissolved in 1 mL of DCM. The resulting mixture was then sonicated for 30 min and dried in vacuo to remove the solvent. The material was then dried at 100 °C for 16 h.
1.5.5 General description of furfural hydrogenation
Into a stainless steel reactor, appropriate amount of the catalyst or SILP material was added to the appropriate base with the required amount of furfural. As soon as the required amount of formic acid was added to the contents of the reactor, the reactor was instantly closed and the reaction mixture heated at the required temperature for the required time. After the reaction time had elapsed, the reactor was cooled in ice-cold water after which any generated gases were slowly vented. The contents of the reactor were then analyzed by 1H NMR spectroscopy. In the case of silica-containing SILP materials, 5 mL of water was added to the contents of the reactor after which it was centrifuged at 10,000 rpm for 10 min. Subsequently, the filtrate containing the furfuryl alcohol was separated from the residue. To 0.4 mL aliquot of the filtrate was added 0.2 mL of CDCl3 and 5 µL of DMF (as standard) to determine the amount of product obtained. The rest of the filtrate solutions were analyzed by ICP-OES to determine metal leaching. The filtered catalytic materials were then added to a cleaned reactor and the cycle repeated for catalyst recycling.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Anyomih, W.D., Oklu, N.K., Ocansey, E. et al. Homogenous palladium(II) pyrazolyl complexes and corresponding Pd-SILP material as catalysts for the selective hydrogenation of furfural. Biomass Conv. Bioref. (2022). https://doi.org/10.1007/s13399-022-03449-2
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s13399-022-03449-2