
Abstract
The H-factor, a parameter used extensively to analyze and predict the outcome of kraft pulping, is applied to organosolv pretreatment. The total solid yield after organosolv pretreatment fits well with the H-factor. The concept has been extended to apply to the individual biomass polymers using unique values for the activation energy for the depolymerization of the individual biomass polymers, giving the O-factor concept analogous to the P factor used for analyzing prehydrolysis kinetics. The results showed a linear relationship between ln(L0/L) and O-factor at an activation energy of 96 kJ/mol. The best linear fit for mannan and xylan degradation was obtained at O-factor activation energies of 104 kJ/mol and 142 kJ/mol, respectively, and the formation of furfural and 5-HMF gave a good linear fit using an O-factor activation energy of 150 kJ/mol. The O-factor is thus a useful concept for analyzing organosolv pretreatment when the temperature during pretreatment is not constant.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
An increased concern about the environment and limited availability of fossil fuels have led to an urge for alternative, sustainable resources. Several sources are already being utilized, such as solar energy, wind, and hydro energy. Among the alternative sources, biofuels attracted attention of scientific community as well as the industry. Biofuels provide an effective solution to reduce greenhouse gas emission in countries with sufficient biomass resources [5]. The use of ethanol as a biofuel can be traced back to 1826 when it was used to power an internal combustion engine [16]. Ethanol was on high demand during World War II, when countries used it as gasoline booster or as a fuel [39]. Current large-scale ethanol production uses sugars or starch as raw material creates the food or fuel dilemma [28]. In the recent years, ethanol from lignocellulosic biomass, also known as advanced biofuel, has gained attention due to its sustainability as it reduces the food or fuel dilemma. Lignocellulose biomass primarily consists of three main components: cellulose, a highly crystalline homopolymer, hemicellulose, an amorphous polymer consisting of hexose and pentose sugars, and lignin, a complex polymer of phenolic subunits [6]. This complex structure of biomass makes it resistant to enzyme attack, an evolutionary trait of trees to survive in the wild but a challenge in biofuel production. This recalcitrance causes a need for a pretreatment step before enzymes can hydrolyze the polymers into mono-sugars [8]. The pretreatment step removes lignin and part of the hemicellulose so that the cellulose becomes accessible to enzymes.
Several pretreatment methods have been tested and employed both in laboratory and industrial scale. Among the various methods, organosolv pretreatment is gaining attention due to the fact that it produces lignin in its most unaltered form [43]. The above is crucial in the current situation as the industries reached a realization that an integrated bio-refinery, producing not only ethanol but also chemicals, is the economical approach [32]. This could make lignin, which traditionally is used as an energy source, a valuable byproduct, if produced with minimum modifications. Organosolv pretreatment is a process in which the biomass is delignified using organic solvents with or without the presence of catalysts [18,19,20,21]. The kinetics of the reactions (hemicellulose and lignin removal and furan formation) is enhanced by addition of catalysts like e.g. H2SO4. Apart from the fact that the organosolv pretreatment produces the least altered lignin, it has the advantage over other conventional pretreatments, such as ease of solvent recovery due to the low boiling point of solvent. This solvent recovery also leaves the lignin as a solid and carbohydrates, mainly from hemicellulose, as a syrup [43]. It also possesses some disadvantages such as high cost of organic solvents and expensive equipment development due to highly volatile nature of many organic solvents.
Organosolv pretreatment has been utilized for more than 100 years [42]. Pretreatment is the most important step in the biofuel production as it influences the hydrolysis and the ethanol yield and is the most energy demanding step in the whole process [34]. An optimization of the process parameters is hence inevitable for an economical operation of the plant. Among the process parameters, reaction time, temperature, solvent concentration, and catalyst concentration are the most important. An effort to express the combination of time and temperature as a single value has been done, since expressing those parameters separately became cumbersome and confusing. Several parameters have been introduced such as the severity factor and the H-factor [7]. The severity factor is used extensively for characterization of organosolv severity in pretreatment these days, but has the disadvantage of not including the heating time to the reaction temperature. This disadvantage can be overcome by the use of the H-factor, a parameter used more often in pulping processes. Versions of the severity factor can be found in the literature where an integration is used to accommodate the non-isothermal operation. R0 and ROH were calculated as the integral value in several works [4, 27, 29, 31]. It is worth noting that the severity factor concept was developed on the same basis as the H-factor concept; however, the H-factor is a direct integral of the Arrhenius equation whereas the severity factor often uses a Taylor series expansion of the exponential [1, 14]. The H-factor does not take into consideration the catalyst concentration and the concentration of the solvent used. Hence, it is very important to report these parameters when reporting the H-factor since the change in catalyst concentration or the solvent loading will affect the activation energy and thus the H-factor value. The extended severity factor enables the inclusion of catalyst effect to the overall severity of the reaction by introducing the pH term [7].
1.1 H-factor
The H-factor is a concept first introduced by K.E. Vroom in 1957 for the kraft pulping process and is today a well-established concept used for quantifying the extent of reaction during kraft pulping [2, 10, 41]. The H-factor is derived by assuming the reaction is first order and follows the Arrhenius equation. Then the reaction rate constant k is given by Eq. 1
And the H-factor can be written as
where A is not the same as the frequency parameter in the Arrhenius equation, but a constant introduced by assuming a relative reaction rate of unity at 100 ∘C
Lignocellulosic biomass is a highly complex heterogeneous system, and hence, a single value of kinetic parameters cannot define the whole system. A common assumption is to consider biomass as a “lump system” consisting of discrete subunits or “lumps” with individual kinetic parameters [1]. This model assumption allows us to assign individual H-factors for the components in biomass. Furthermore, by assuming a first-order reaction, we can write the general rate of reaction as,
by rearranging and integration, the equation becomes
By comparing Eq. 2 and Eq. 4, it can be written
Equation 5 suggests that a graph of the natural logarithm of the initial concentration of one species to the final concentration against H-factor should be a straight line with a slope m. This article is investigating the extent of this linear relationship. If found, such a relationship will help in tailoring the pretreatment process in such a way that it can result in desired output composition.
The concept of using a different activation energy than what Vroom used for the H-factor is not new. Herbert Sixta has mentioned the SF-factor for sulfite pulping and the P-factor for prehydrolysis in kraft pulping [35]. The prehydrolysis factor or P-factor concept was already been proposed by Brasch and Free in 1965 where it was assumed that the prehydrolysis rate triples for each 10 ∘C rise in temperature [3]. Lin tried to use the P-factor concept with a fixed activation energy value of 125.6 kJ/mol and got a good fit [35]. This value is a typical activation energy for glycosidic bond cleavage of carbohydrates in wood. The use of the P factor concept can be found in other literature such as Yaldez and Ecker 2000 and Testova 2011 [38]. To the extent of the authors’ knowledge, no such attempt to use the H-factor concept to individual compounds of wood has been done before and the authors would like to introduce the “O factor” concept, an organosolv pretreatment analogue to the H-factor concept.
2 Materials and methods
2.1 Raw materials and chemicals
The wood chips used in this study were industrial Norway spruce (Picea abies) chips from Norske Skog Skogn, Norway. The chips were dried and fractionated to different particle sizes at RISE PFI. The − 8/ + 7 mm fraction was stored at room temperature until further use. Absolute ethanol was obtained from VWR chemicals. Sulphuric acid (ACS reagent, 95% to 98%) was received from Sigma-Aldrich.
HPLC analytical standards glucose (ACS reagent, ≥ 99.5%), xylose (ACS reagent, ≥ 99%), arabinose (ACS reagent, ≥ 99%), galactose (ACS reagent, ≥ 99%), mannose (ACS reagent, ≥ 99%), furfural (ACS reagent, 99%), and 5-HMF (ACS reagent, ≥ 99%) were obtained from Sigma-Aldrich. HPLC internal standard mannitol (ACS reagent, ≥ 98%) was obtained from Sigma-Aldrich. All the chemicals were used as received.
2.2 Moisture analysis
Moisture content of the sample was calculated gravimetrically. A pre-weighed amount of sample was dried in an oven at 105 ∘C overnight and then weighed again.
2.3 Reactor setup and organosolv pretreatment
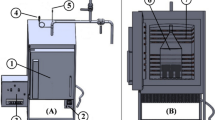
The reactor used in these experiments is a custom-made autoclave reactor system with 6 parallel autoclaves from TOP industries, France. Each autoclave has an internal volume of 1L and the system uses electrical heating. The operating pressure and temperature are 50 bar and 220 ∘C. The reactor cap is fitted with a themocouple which is extended to the inside to measure the internal temperature. The reactor is seated in an inclined position and is agitated throughout the reaction for proper mixing and even heat distribution. 50 g of wood chips (oven-dried weight) was used in all the experiments. A 63% (w/w) solution of ethanol in water was used as the solvent and 1% H2SO4 (oven-dried wood basis) was used as catalyst. A liquid to solid ratio of 7.5:1 was used. After mixing the solvent with the wood chips and the catalyst, the reactor was inserted into the heating jacket and let being agitated for 10 min to ensure proper impregnation of the wood chips. A fixed heating rate of 2 ∘C per minute was used to heat the system to the desired temperature unless stated differently. Upon completion of the reaction, the reactor was cooled in a water bath to stop the reaction. The effluent was separated from the solid residue by filtration and was stored at 4 ∘C until further use. The solid residue was washed first with solvent of same concentration as used for the reaction followed by about 10 L of warm water. The pretreated chips were then collected in a zip-lock bag, weighed, and stored at 4 ∘C.
Table 1 shows the reaction temperature and heating time used for each experiment. The validation experiments are marked with v.
2.4 Carbohydrate and lignin analysis of the biomass
The carbohydrate and lignin analysis of untreated and pretreated biomass was based on the NREL method for structural carbohydrate and lignin analysis in biomass [37]. The sample was air-dried to a moisture content below 10%. The sample was then powdered and 300 ± 10 mg of sample was transferred to a reaction tube. Three milliliters of 72% H2SO4 was added to the sample and mixed thoroughly with a Teflon rod. The tube was then placed in a Laboshake at 30 ∘C and incubated for 60 min with stirring the sample every 5 to 10 min. After incubation, the tube was removed and the contents of the tube were transferred to a 100-ml pyrex glass bottle and diluted with 84 ml distilled water to make the acid concentration be 4%. The sample was autoclaved at 121 ∘C for 1 h, cooled to room temperature and filtered through a pre-weighed filter paper. The liquid was collected, neutralized using Ca(OH)2, and analyzed using HPLC. The filter paper and the undissolved content were dried in an oven at 105 ∘C overnight and the Klason lignin was analyzed gravimetrically. The liquid fraction was analyzed for acid soluble lignin before neutralization using UV/Vis spectrometry at a wavelength of 205 nm. The composition of the pretreated biomass samples is given in Table 4 (Appendix).
2.5 Analysis of the effluent
The amount of organosolv effluent lignin (OEL) was calculated by precipitating the lignin by adding three volumes of water to one volume of effluent. The precipitated lignin was separated by filtration and dried overnight in an oven and the amount was calculated. The amount of 5-HMF and furfural was determined using HPLC. The NREL procedure for determination of sugars, byproducts, and degradation products in liquid fraction process samples [36] was used for the analysis of carbohydrates. The composition of the effluent samples is given in Table 4 (Appendix).
2.6 High-performance liquid chromatography
The samples were analyzed on a Shimadzu Providence HPLC system provided with RI and multichannel UV-VIS detectors. Separation of alcohols, acids and furans was done on an Agilent Hi-Plex H column (300 mm × 7.8 mm) using 0.005M H2SO4 as mobile phase. The flow rate was 0.6 ml/min and the column temperature was 60 ∘C. Separation of monosaccharides and furans was done on an Agilent Hi-Plex Pb column (300 mm × 7.8 mm) with an inline deashing column (Bio-Rad Micro-Guard) using DI water as mobile phase. The flow rate was 0.6 ml/min and the column temperature was 50 ∘C.
2.7 Model fitting
Model fitting for the main components such as lignin, xylan, mannan, and the furans was done by regression analysis. The range of activation energy values for each compound was chosen based on the literature. Once the range of values was decided, the physical meaning of activation energy was disregarded while performing regression analysis and the value which gave the best model fit was calculated using R2 analysis. Once this value was calculated, this was compared to activation energy values from the literature to make a physical meaning to the value.
3 Results and discussion
The composition of untreated biomass is given in Table 2. The yield of biomass after pretreatment was plotted against the H-factor, calculated using an activation energy of 134 kJ/mol as in the original H-factor expression [41]. The resulting graph is given in Fig. 1. A single relationship was observed for all the temperatures chosen, suggesting that the H-factor correlation is valid for organosolv pretreatment. Based on Eq. 5, a relationship between the individual components of biomass and the O-factor was investigated. Lignin was chosen first and the O-factor for all the reactions was calculated for a range of activation energy values from 70 to 100 kJ/mol, based on the literature on lignin activation energies in pretreatment [9, 15, 33, 40]. A regression analysis gave the best fit with a R2 value of 0.95 for an activation energy value of 96 kJ/mol. Figure 2a shows this relationship. The y coordinate is the natural logarithm of the ratio of the initial lignin content to that after pretreatment. The obtained value is comparable to the values reported in literature shown in Table 3. The activation energy value obtained is very similar to the activation energy reported for methanol and ethanol pulping, but is lower than for autocatalyzed ethanol pulping. Since catalyst concentration affects the activation energy, this difference is expected. It should be noted that the value 96 is not the activation energy but the model parameter. The comparison of the value to the reported activation energy values shows the validity of the model.

Biomass yield after pretreatment plotted against the H-factor. A second-order polynomial regression was used

O-factor dependence of lignin, xylan, and mannan. a Plot of lignin vs O-factor, where the O-factor was calculated using an EA value of 96 kJ/mol. L0 and L represents the percentage of lignin in the untreated biomass and the pretreated biomass respectively b Plot of xylan vs O-factor, where the O-factor was calculated using an EA value of 142 kJ/mol. X0 and X are the percentage of xylan in the untreated and the pretreated biomass respectively. c Plot of mannan vs O-factor, calculated using an EA value of 104 kJ/mol. M0 and M represents the percentage of mannan in the untreated and the pretreated biomass respectively
Figure 2a shows that it is possible to predict the lignin content of the pretreated Norway spruce from the O-factor value. It can be seen from the graph that similar O-factor values were achieved with different temperatures but resulted in similar delignification. Such a relationship can be useful in industry when designing the operation parameters. It provides flexibility in choosing the reaction temperature and time. The investigation was then extended to xylan, one of the main polysaccharides in hemicellulose. A regression analysis strategy similar to the one used for lignin was used. An activation energy range of 120 kJ/mol to 150 kJ/mol was used based on the literature [12, 13, 26]. A good fit was not obtained for xylan, but the best fit was at an activation energy of 142 kJ/mol with a R2 value of 0.81. Figure 2b shows this relationship. Even though a good fit was not obtained over the entire O-factor range, it is interesting to note the change in slope at an O-factor value of approximately 2000. It can be seen to have two straight lines with different slopes meeting at 2000. The concept that there exist at least two kinds of xylan in biomass with different reactivity to hydrolysis has been suggested by other authors [17, 23, 26]. The differences in the structure and properties of these xylans are not clear, as this is a hypothesis formulated based on the observed change in activation energy of xylan during pretreatment. It is worth noting the last data point in Fig. 2b which indicates that the xylan removal has reached a plateau around an O-factor value of 6000. This was also observed in another study [24]. Mannan, a major hemicellulose-based monosugar, gave an acceptable fit at an activation energy of 104 kJ/mol, shown in Fig. 2c. An analysis of minor sugars such as galactose or arabinose was not done since those sugars were not detected in some of the experiments.
The graphs show a relationship between O-factor and the composition of different species in biomass. Having said that, a further investigation is required to analyze how well the O-factor concept fits for organosolv pretreatment and in what range the assumptions hold true. A method similar to the one used by A.L. Macfarlane [25] was employed. The biomass was treated with same liquor and same catalyst concentration as before. The reaction temperature and the time to reach the temperature were kept constant and only the time at reaction temperature was varied between 15 and 90 min. Instead of taking the composition of untreated biomass as the initial concentration, the concentration in the biomass treated for 15 min was taken as the initial concentration and all the others were compared to this. In this manner there is the possibility to compare isothermal reactions. So if the assumptions from Arrhenius equation are applicable, a graph of ln(C0/C) vs time would result in a straight line. This experiment was done at three different temperatures, which are 170 ∘C, 180 ∘C, and 190 ∘C. The graphs for lignin are given in Fig. 3. There are deviations from a straight line at the lower and upper ends but the middle range values follow a straight line. Now this would give us an idea about the range in which the O-factor-lignin relationship is valid. It is worth noting that all the experiments were conducted within this O-factor-lignin relationship linearity range. Furthermore, the deviation at low and high O-factors can be understood by the concept of three stages of delignification: initial, bulk, and residual delignification [22]. As per this concept, the delignification takes place in three stages, a fast first stage called initial delignification, a second slower delignification stage called the bulk delignification, and finally the slowest residual delignification. Most of the lignin removal takes place in the bulk phase, and it can be assumed that the bulk delignification was dominating during the experiments.

Holding time vs ln(L0/L) for three reaction temperatures, where L0 is the percentage of lignin in the biomass after pretreatment for 15 min at the reaction temperature. a Plot of ln(L0/L) vs time at the reaction temperature of 170 ∘C. b Plot of ln(L0/L) vs time at the reaction temperature of 180 ∘C. c Plot of ln(L0/L) vs time at the reaction temperature of 190 ∘C
The yield of furans, the main degradation products in organosolv pretreatment, was plotted against the O-factor since there are no detectable furans in the untreated biomass. An activation energy of 150 kJ/mol was used to calculate the O-factor, a value on the higher side of the range of values reported in the literature [11, 12]. The kinetics of formation of furans is greatly affected by the acid concentration and since a fairly low acid concentration was used in this study it is reasonable that the activation energy value obtained is on the higher side. The furan formation would be higher if the acid concentration was higher and would be lower in case of an uncatalyzed reaction. The plots for furfural and 5-HMF are given in Fig. 4. Both the furans followed a single relationship with regard to O-factor suggesting that the concept of O-factor is useful in predicting the degradation product formation as well. This furan/O-factor relationship can be utilized to optimize furan formation such that this undesired byproduct can be put to profitable use.

Weight % yield of furans vs the O-factor at an activation energy of 150 kJ/mol. a Plot of yield vs O-factor for furfural. b Plot of yield vs O-factor for 5-HMF. Both the plots are obtained using a second-order polynomial regression
Since validation of the obtained model parameters using an independent set of experiments is desirable in any modeling work, the validity of this work was also checked using an independent set of experiments. The results are shown in Fig. 5. The validation data points fit well with the experimental data used for the model. The experimental values showed a decent correlation to the theoretical values with a maximum error of 15%. Considering the experimental errors and the slight variation in wood composition, the authors believe that the error margin is acceptable.

4 Conclusion
This study shows that the lignin content in Norway spruce after organosolv pretreatment can be predicted from the O-factor calculated with an activation energy of 96 kJ/mol. Such a straightforward relationship was not possible for xylan since there seems to be at least two kinds of xylan present with different reaction parameters. Mannan can be predicted using an activation energy of 104 kJ/mol. The furans also followed a single relationship suggesting the validity of the O-factor concept in predicting the formation of degradation products. This study concludes that the O-factor is a useful tool in organosolv pretreatment which can be useful for predicting the product composition. Even though the O-factor can be a useful tool in designing the reaction, it should be noted that this is not a general model. The O-factor depends on the catalyst concentration, biomass type, and the solvent property. The values will change if one or all of the above parameters are changed.
Availability of data and material
Not applicable
Code availability
Not applicable
References
Abatzoglou N, Chornet E, Belkacemi K, Overend R P (1992) Phenomenological kinetics of complex systems: the development of a generalized severity parameter and its application to lignocellulosics fractionation. Chem Eng Sci 47(5):1109–1122
Biermann CJ (1996) Handbook of pulping and papermaking. Elsevier
Brasch D, Free K (1965) Prehydrolysis-kraft pulping of pinus radiata grown in new zealand. Tappi 48(4):245–248
Brosse N, Sannigrahi P, Ragauskas A (2009) Pretreatment of miscanthus x giganteus using the ethanol organosolv process for ethanol production. Ind Eng Chem Res 48(18):8328–8334
Carroll A, Somerville C (2009) Cellulosic biofuels. Ann Rev Plant Biol 60:165–182
Chen H (2014) Chemical composition and structure of natural lignocellulose. In: Biotechnology of lignocellulose. Springer, pp 25–71
Chum H L, Johnson D K, Black S K (1990) Organosolv pretreatment for enzymic hydrolysis of poplars. 2. catalyst effects and the combined severity parameter. Ind Eng Chemi Res 29(2):156–162
Esteghlalian A, Hashimoto A G, Fenske J J, Penner M H (1997) Modeling and optimization of the dilute-sulfuric-acid pretreatment of corn stover, poplar and switchgrass. Bioresour Technol 59 (2-3):129–136
Faass G, Roberts R, Muzzy J (1989) Buffered solvent pulping
Fardim P (2007) Chemical pulping Part 1, fibre chemistry and Technology, vol 6. Paper Engineers’ Association/Paperi ja Puu Oy; Helsinki
Garrote G, Domıínguez H, Parajo JC (2001a) Kinetic modelling of corncob autohydrolysis. Process Biochem 36(6):571–578
Garrote G, Domínguez H, Parajó JC (2001b) Manufacture of xylose-based fermentation media from corncobs by posthydrolysis of autohydrolysis liquors. Appl Biochem Biotechnol 95(3):195–207
Garrote G, Domínguez H, Parajó JC (2002a) Autohydrolysis of corncob: study of non-isothermal operation for xylooligosaccharide production. J Food Eng 52(3):211–218
Garrote G, Domínguez H, Parajó JC (2002b) Interpretation of deacetylation and hemicellulose hydrolysis during hydrothermal treatments on the basis of the severity factor. Process Biochem 37(10):1067–1073
Gilarranz MA, Rodríguez F, Santos A, Oliet M, García-Ochoa F, Tijero J (1999) Kinetics of eucalyptus globulus delignification in a methanol- water medium. Ind Eng Chem Res 38(9):3324–3332
Hardenberg HO, Morey S (1992) Samuel Morey and his atmospheric engine. SAE
Jacobsen SE, Wyman CE (2000) Cellulose and hemicellulose hydrolysis models for application to current and novel pretreatment processes. In: Twenty-first symposium on biotechnology for fuels and chemicals. Springer, pp 81–96
Kleinert T, v Tayenthal K (1931) ÜBer neuere versuche zur trennung von cellulose und inkrusten verschiedener hölzer. Angew Chem 44(39):788–791
Kleinert T N (1971) Organosolv pulping and recovery process. US Patent 3,585,104
Kleinert T N (1974) Organosolv pulping with aqueous alcohol. Tappi 57(8):99–102
Kleinert T N (1975) Ethanol-water delignification of wood–rate constants and activation energy. Tappi 58(8):170–171
Kleppe P J (1970) Kraft pulping. Tappi 53(1):35–47
Kobayashi T, Sakai Y (1956) Hydrolysis rate of pentosan of hardwood in dilute sulfuric acid. J Agricultural Chem Soc Jpn 20(1):1–7
Liu J, Gong Z, Yang G, Chen L, Huang L, Zhou Y, Luo X (2018) Novel kinetic models of xylan dissolution and degradation during ethanol based auto-catalyzed organosolv pretreatment of bamboo. Polymers 10(10):1149
Macfarlane A, Farid M, Chen J (2009) Kinetics of delignification using a batch reactor with recycle. Chem Eng Process Process Intens 48(4):864–870
Maloney M T, Chapman T W, Baker A J (1985) Dilute acid hydrolysis of paper birch: Kinetics studies of xylan and acetyl-group hydrolysis. Biotechnol Bioeng 27(3):355–361
Martínez J, Reguant J, Salvadó J, Farriol X (1997) Soda-anthraquinone pulping of a softwood mixture: Applying a pseudo-kinetic severity parameter. Bioresour Technol 60(2):161–167
Mohr A, Raman S (2013) Lessons from first generation biofuels and implications for the sustainability appraisal of second generation biofuels. Energy Policy 63:114–122
Montane D, Farriol X, Salvado J, Jollez P, Chornet E (1998) Fractionation of wheat straw by steam-explosion pretreatment and alkali delignification. cellulose pulp and byproducts from hemicellulose and lignin. J Wood Chem Technol 18(2): 171–191
Oliet M, Rodríguez F, Santos A, Gilarranz MA, García-Ochoa F, Tijero J (2000) Organosolv delignification of eucalyptus g lobulus: Kinetic study of autocatalyzed ethanol pulping. Ind Eng Chem Res 39(1):34–39
Overend R P, Chornet E (1987) Fractionation of lignocellulosics by steam-aqueous pretreatments. Philosophical Transactions of the Royal Society of London Series A. Math Phys Sci 321(1561): 523–536
Rosales-Calderon O, Arantes V (2019) A review on commercial-scale high-value products that can be produced alongside cellulosic ethanol. Biotechnol Biofuels 12(1):240
Santos A, Rodríguez F, Gilarranz MA, Moreno D, García-Ochoa F (1997) Kinetic modeling of kraft delignification of eucalyptus globulus. Ind Eng Chem Res 36(10):4114–4125
da Silva ARG, Errico M, Rong BG (2018) Techno-economic analysis of organosolv pretreatment process from lignocellulosic biomass. Clean Techn Environ Policy 20(7):1401–1412
Sixta H (2006) Handbook of pulp. Wiley-vch
Sluiter A, Hames B, Ruiz R, Scarlata C, Sluiter J, Templeton D, et al. (2006) Determination of sugars, byproducts and degradation products in liquid fraction process samples. National Renewable Energy Laboratory, Golden
Sluiter A, Hames B, Ruiz R, Scarlata C, Sluiter J, Templeton D, Crocker D, et al. (2008) Determination of structural carbohydrates and lignin in biomass. Labor Anal Proced 1617:1–16
Testova L, Chong SL, Tenkanen M, Sixta H (2011) Autohydrolysis of birch wood: 11th ewlp, hamburg, germany, august 16–19, 2010. Holzforschung 65(4):535–542
Thomas V, Kwong A (2001) Ethanol as a lead replacement: phasing out leaded gasoline in africa. Energy Policy 29(13):1133–1143
Vázquez G, Antorrena G, González J (1995) Kinetics of acid-catalysed delignification of eucalyptus globulus wood by acetic acid. Wood Sci Technol 29(4):267–275
Vroom K (1957) The “h” factor: a means of expressing cooking times and temperatures as a single variable. PPMC 58:228–231
Zhang K, Pei Z, Wang D (2016) Organic solvent pretreatment of lignocellulosic biomass for biofuels and biochemicals: a review. Bioresour Technol 199:21–33
Zhao X, Cheng K, Liu D (2009) Organosolv pretreatment of lignocellulosic biomass for enzymatic hydrolysis. Appl Microbiol Biotechnol 82(5):815
Acknowledgements
This project was funded by the Department of Chemical Engineering, NTNU, Trondheim. The authors would like to acknowledge RISE PFI for technical assistance and providing the laboratory facilities. This work is carried out as part of the Norwegian national research infrastructure project NorBioLab (“Norwegian Biorefinery Laboratory”) and Norwegian Centre for Sustainable Bio-based Fuels and Energy (Bio4Fuels). We gratefully acknowledge The Research Council of Norway for financial support.
Funding
Open access funding provided by NTNU Norwegian University of Science and Technology (incl St. Olavs Hospital - Trondheim University Hospital).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Appendix
Appendix
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Joseph, P., Opedal, M.T. & Moe, S.T. The O-factor: using the H-factor concept to predict the outcome of organosolv pretreatment. Biomass Conv. Bioref. 13, 6727–6736 (2023). https://doi.org/10.1007/s13399-021-01667-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13399-021-01667-8