
Abstract
During the recent years, viral epidemic due to coronaviruses, such as SARS (Severe Acute Respiratory Syndrome), Middle East Respiratory Coronavirus Syndrome (MERS), and COVID-19 (coronavirus disese-19), has become a global problem. In addition to causing cardiovascular and respiratory lethal dysfunction, these viruses can cause neurodegeneration leading to neurological disorders. Review of the current scientific literature reveals the multiple neuropathies and neuronal dysfunction associated with these viruses. Here, we review the major findings of these studies and discuss the main neurological sequels and outcomes of coronavirus infections with SARS, MERS, and COVID-19. This article analyzes and discusses the main mechanisms of coronavirus-induced neurodegeneration according to the current experimental and clinical studies. Coronaviruses can damage the nerves directly through endovascular dysfunctions thereby affecting nerve structures and synaptic connections. Coronaviruses can also induce neural cell degeneration indirectly via mitochondrial dysfunction inducing oxidative stress, inflammation, and apoptosis. Thus, coronaviruses can cause neurological disorders by inducing neurovascular dysfunction affecting nerve structures and synaptic connections, and by inducing inflammation, oxidative stress, and apoptosis. While some of these mechanisms are similar to other RNA viruses, the neurotoxic mechanisms of COVID-19, MERS, and SARS-CoV viruses are unknown and need detailed clinical and experimental studies.
Similar content being viewed by others


Introduction
Coronavirus disease (COVID)-19 is a lethal syndrome characterized by severe acute respiratory syndrome induced by coronavirus 2 (SARS-CoV2) infections. COVID-19 has become a global pandemic that started in 2019 that became a public health emergency killing over 2 million people in the first year (Spinelli and Pellino 2020; Watkins 2020). Coronaviruses have an average diameter of 100 nm with a spherical or oval shape. The name of coronavirus refers to the large spikes of glycoproteins in the membrane on this virus causing a typical crown-like shape under electron microscopy (Ziebuhr 2004; Siddell et al. 1983; Satija and Lal 2007). These coronaviruses are single-stranded RNA viruses with a genome length of approximately 26–32 kb representing the largest genome of currently known RNA viruses (Satija and Lal 2007; Anand et al. 2002). These coronaviruses enter the host cell via ACE2 receptors, and its mechanism of replication is represented in Fig. 1. Coronaviruses induce multiple disorders including respiratory, cardiovascular, hepatocellular, and renal dysfunction (Wu et al. 2020; Xu et al. 2020; Madjid et al. 2020; Eckerle et al. 2013) (Fig. 2). However, some viruses such as SARS (severe acute respiratory syndrome), Middle East respiratory coronavirus syndrome (MERS), and coronavirus disese-19 (COVID-19) can become lethal with severe long-term respiratory and cardiovascular dysfunctions (Madjid et al. 2020; Xiong et al. 2020; Guo et al. 2019). One of the fatal consequences of coronavirus infection is a lasting neurodegeneration and neuropathy even in those patients surviving the infection (Nagashima et al. 1979; Bergmann et al. 2006; Yeh et al. 2004). Multiple studies show that coronaviruses are neurotropic as they can invade nervous tissues and infect the microglia or astrocytes in the CNS (Desforges et al. 2014a; Li et al. 2020a). Clinical studies show that COVID-19 patients can have seizures and loss of consciousness that can become fatal. Latest studies report that approximately 36% of COVID-19 patients exhibit neurological symptoms such as stroke, headache, impaired consciousness, and paresthesia (Wu et al. 2020; Bagheri et al. 2020). COVID-19 patients can also exhibit neurobehavioral symptoms such as anxiety and depression, as well as cognitive dysfunction especially in elderly patients, which are the most susceptible to the infection (Khan et al. 2020; Watanabe et al. 1983). Autopsies of COVID-19 patients show edema, neuronal degeneration, and viral encephalitis as common neurological consequences of the infection (Barton et al. 2020; Jacomy and Talbot 2003; Jacomy et al. 2006). This critical impact of COVID-19 on the nervous system and its social and clinical implications has fostered many investigators to analyze the molecular mechanisms mediating this neurotoxicity (Jacomy et al. 2006; Tsukamoto et al. 1990). The neurotropic potential of these viruses was first suggested by their concentrations in the cerebrospinal fluid, but their pathological mechanisms are still moot (Hung et al. 2003; Doenges et al. 2016). Here, we review the clinical and experimental studies to discuss the key mechanisms of coronavirus-induced neuropathies. We report the major findings, clinical management strategies, and therapies related to neuropathies induced by these coronaviruses. This information provides to clinicians better neurotoxicity and neuropathies induced by the coronaviruses.

The life cycle of coronavirus: At the beginning, S protein binds to the ACE2 receptor in host cells. Then, the conformation of S protein is changed and causes the virus envelope to merge with the cell membrane via the endosomal pathway. Next coronavirus releases its RNA into the host cell. Genome RNA is translated to viral replicas polyproteins pp1a and 1ab. That polyproteins disparted to small proteins by proteinases. Polymerase makes some of subgenomic mRNAs by discontinuous transcription and finally translated to viral proteins. Viral proteins and genome RNA are assembled to the ER and Golgi. After that, it transported via vesicles and released out of the cell. ACE2, angiotensin-converting enzyme 2; ER, endoplasmic reticulum; ERGIC, ER–Golgi intermediate compartment

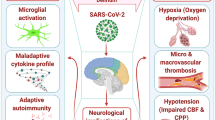
Multiple effects of infection with Coronaviruses Family, such as SARS (Severe Acute Respiratory Syndrome), Middle East Respiratory Coronavirus Syndrome (MERS) and COVID-19 (coronavirus disese-19), on body organs and systems
Effects of multiple main types of coronaviruses on nervous system
SARS-CoV neurological sequels
In 2002–2003, there was a global epidemic of severe acute respiratory syndrome (SARS), a zoonotic respiratory disease that started in Asia and spread worldwide (Ong et al. 2020). SARS patients typically have respiratory-related symptoms associated with fever, chills, dry cough, and difficult breathing, as well as neurological manifestations that were rarely described (Paybast et al. 2020; Zegarra-Valdivia et al. 2020). Neurological signs included axonopathic polyneuropathy, myopathy, encephalitis, and ischemic aortic stroke (Zegarra-Valdivia et al. 2020; Calcagno et al. 2020). Myopathic and neuropathic complications have also been reported in SARS patients, and clinicians should be aware of the potential neuromuscular complaints (Ding et al. 2004; Lau and Chan 2005). SARS-CoV infection can cause transient ischemic attack, partial epilepsy, and mild cognitive impairments (Umapathi et al. 2004; Lin et al. 2020). Some authors suggested that neurobehavioral symptoms such as agitation and mood-related behavior can be signs of SARS-CoV infection associated with cerebral edema and meningeal vasodilation (Iroegbu et al. 2020; Chevance et al. 2020).
Leukocytes, such as monocytes and lymphocytes, were also increased in the vessel wall of the brain of SARS-CoV patients, leading to ischemic and inflammatory changes in the nerves and in extreme cases demyelination of nerve fibers (Paniz-Mondolfi et al. 2020; Conde et al. 2020). Several studies showed the presence of these viral particles and genome in the cerebrospinal fluid (CSF) to confirm the direct effects of SARS-CoV on brain tissues (Paniz-Mondolfi et al. 2020; Conde et al. 2020).
MERS-CoV neurological sequels
The Middle East respiratory syndrome (MERS), caused by MERS-CoV, was first reported in Saudi Arabia in June 2012 and also originated from bats with camels reported as intermediate host (Alfaraj et al. 2019; Banik et al. 2015). MERS-CoV causes extreme respiratory infections leading to acute respiratory distress (ARDS) syndrome, which causes pneumonia, fever, myalgia, cough, dyspnea, septic shock, multiple organ failure, and death (Zumla et al. 2015; Group WM-CR 2013). Previous reported MERS-CoV is a neuroinvasive and neurotrophic virus that can trigger neurological effects such as loss of consciousness, paralysis, ischemic stroke, Guillain–Barre syndrome, and other neuropathies (Desforges et al. 2014a; Li et al. 2020a; Yashavantha Rao and Jayabaskaran 2020). MERS-CoV induces widespread, bilateral, hyper-intensive lesions in the white matter and subcortical areas of the basal ganglia, corpus callosum, frontal, temporal, and parietal lobes (Desforges et al. 2014a; Coupanec et al. 2015). The presence of MERS-CoV in the cerebrospinal fluid (CSF) has not been confirmed, but its neurotoxicity has been well accepted (Desforges et al. 2014a; Coupanec et al. 2015).
SARS-CoV-2 (COVID-19)
The genetic similarity between SARS-CoV-2 and SARS-CoV is 79.5%. Therefore, these two viruses are expected to have similar pathological mechanisms (Lai et al. 2020; Zhang et al. 2020). SARS-CoV-2 causes multiple symptoms, such as fever, mild pneumonia cough, and multiple organ dysfunctions with a mortality rate of 2–4% (Zheng et al. 2020; Velavan and Meyer 2020). COVID-19 induces neurological effects including headache, epilepsy, and impaired consciousness (Baig 2020; Ye et al. 2020). Anosmia and dysgeusia with sudden loss of odor or taste can occur in COVID-19 patients (Moein et al. 2020; Eliezer et al. 2020; Lechien et al. 2020). Recent genome sequencing analyses confirmed the presence of SARS-CoV-2 in cerebrospinal fluids associated with some cases of viral encephalitis caused by COVID-19. Clinical reports have shown that patients with extreme COVID-19 infection can have headaches, projectile vomiting, vision impairment, and limbs paralysis (Wu et al. 2020; Li et al. 2020b).
Mechanisms of coronavirus-induced neurological dysfunction
According to recent neurological studies, coronaviruses can induce neurotoxicity through (1) direct neuronal damage or (2) indirect mechanisms, such as pathological inflammation of the nerve, oxidative stress, and apoptosis (Fig. 3).

Mechanisms of coronavirus-induced neurological dysfunction via directly damaging the virus particle to the nerve or microvascular system and indirectly activating of pathological mechanisms such as inflammation of the nerve, oxidative stress and apoptosis was indicated. Each of these two pathways was effect on the neural system via downstream specific mechanisms which mentioned and described in diagram
Direct neuronal damage is mediated through the following pathways:
-
1.
Entering CNS through bloodstream and inducing endovascular dysfunction
-
a)
Invasion by infected circulating leukocytes:
Coronaviruses can enter the nervous system without infecting neurons, as they can infect leukocytes, which circulate in the bloodstream and can penetrate the parenchyma of the brain (Zhong et al. 2003; Nowacki and Charley 1993). This mechanism of neuronal penetration and infection is not completely understood (Nowacki and Charley 1993; Desforges et al. 2014b), but it is described in literature as the “Trojan horse” entry, as the pathogens concealed inside the leukocytes can cross the blood-brain-barrier (Paniz-Mondolfi et al. 2020; Talbot et al. 2011). Although this mechanism has not been confirmed yet, indirect evidence showing the involvement of monocytes/macrophages in the pathogenesis strongly supports their contribution to viral penetration into the brain (Paniz-Mondolfi et al. 2020; Mesel-Lemoine et al. 2012). Coronaviruses primarily target CD4+ T-lymphocytes and macrophages/microglia, which are specialized phagocytic and antigen-presenting cells in the CNS, and astrocytes expressing CD4 cell surface receptor (Pewe and Perlman 2002; Bergmann et al. 2004). The replication of coronaviruses is greatly affected by the immune responses with multiple cytokines and chemokines modulating its potential to enter the CNS (Brian and Baric 2005). Thus, CNS invasion may occur depending on the immune responses and the immunologic state (Desforges et al. 2014b, 2013). Infected monocyte/macrophage, which are involved in the coronaviruses pathogenicity, can cross the blood–brain-barrier and differentiate into perivascular microglia (Brian and Baric 2005). Replication of the virus in microglia and subsequent induction of inflammatory cytokines damages the CNS causing neurodegeneration and causing symptoms such as seizures and un-conciseness (Bergmann et al. 2006; Talbot et al. 2011; Parra et al. 1997). The role of coronavirus in peripheral nervous system has not been well studied, but axonopathic polyneuropathy has been reported in patients with SARS-CoV (Zegarra-Valdivia et al. 2020; Li et al. 2020c). Coronavirus can also infect oligodendrocytes (cell-producing myelin) and astrocytes, leading to fatal inflammatory disorders in the brain and degenerative symptoms such as seizures (Turner et al. 2004; Dandekar et al. 2005) (Fig. 3).
-
b)
Infection via brain microvascular endothelium
Viral particles in the circulatory system can reach and infect brain microvascular endothelial cells (BMVECs), the major constituent of blood-brain barrier. This phenomenon occurred in RNA viruses such as coronaviruses, but the details of this mechanism are not known (Argyris et al. 2003; Liu et al. 2002). BMVEC activation can cause brain inflammation and thus neurological disorders (Wu et al. 2020; Liu et al. 2002). BMVEC activation can cause brain inflammation and thus neurological disorders (Wu et al. 2020). For example, encephalitis and meningitis may be caused by BMVEC in a percentage of infected people with coronaviruses (Wu et al. 2020). Coronavirus CNS infections are frequently associated with immune dysfunction allowing viral replication and viremia (Bergmann et al. 2006, 2004). Certain coronavirus neuropathogenesis produced by the disruption of the blood-brain barrier may allow immune cells to enter the brain and cause neurodegeneration (Wu et al. 2020; Desforges et al. 2014b; Gallagher and Buchmeier 2001). This infection not only affects the integrity of tight junction proteins, in both epithelial and endothelial cells, but also induces inflammatory factors that can degrade the basement membrane (Gallagher and Buchmeier 2001; Miner and Diamond 2016; Sun and Guan 2020). As a result, leukocytes can cross the capillaries into the surrounding tissue and produce inflammatory cytokines (Chen et al. 2001). It has been proposed that BMVECs are the only type of cells that can mediate coronavirus entry into the brain (Chen et al. 2001; Cabirac et al. 1995). Coronavirus CNS infection is also associated with increased production of pro-inflammatory cytokines (IL1, TNFα, IL12, and IL18) and creatine, with cause microglia and astrocyte activation and neurological disorders (Cabirac et al. 1995; Conti et al. 2020). A major infection can cause fatal neurodegeneration in sensory neurons and induce abnormalities such as dysosmia and dysgeusia (Keyhan et al. 2019; Lao et al. 2020). In addition to BMVECs, coronaviruses can also affect astrocytes, pericytes, neurons, microglial cells, and even neural stem cells (Chen et al. 2001; Cabirac et al. 1995). During infections, astrocytes and microglial cells can produce copious quantities of inflammatory cytokines and induce fetal neurodevelopmental disorders (Dörries 2001; Edwards et al. 2000). Virus infection of BMVECs induces the expression of inflammatory cytokines and chemokines, disrupting the blood-brain barrier and causing inflammatory neurological disorders and encephalitis (Chen et al. 2001; Desforges et al. 2007). Although the invasion of the nervous system by coronaviruses via the bloodstream is rare, there is evidence that these viruses cause vascular endothelial system disturbances that are likely to disrupt the blood-brain barrier allowing coronaviruses to enter the CNS (Wu et al. 2020; Sun and Guan 2020). Other studies have indicated that these viruses replicate in the body in mononuclear macrophages and increase cytokine production, thus raising the risk of blood-brain barrier disorders and contribute to meningitis or encephalitis (Sun and Guan 2020). After the virus reaches the nervous system, viral proteins can cause neuronal damage (Bergmann et al. 2006; Sun and Guan 2020). Some studies suggest that viruses can infect sensory or motor nerve endings, inducing retro or anterograde neuronal transport through the motor proteins such as dynein and kinesins (Bergmann et al. 2006) (Fig. 3).
-
2.
Entering CNS through various nerve ending and disrupting nerve structures
-
a)
Invasion of sensory nerve endings
Recent studies indicate that coronaviruses may enter the peripheral nervous system (PNS) by binding to sensory and autonomic axon-terminal receptors that convey sensory and visceral information; this phenomenon has occurred in SARS-CoV inducing axonopathic polyneuropathy (Zegarra-Valdivia et al. 2020; Li et al. 2020c). Although the mechanism and route of coronaviruses entering the PNS is unknown, it appears to be mediated by cellular adhesion molecules, especially in SARS-CoV (Heerssen et al. 2004). Coronavirus particles reach sensory nerve endings via membrane fusion and can use dynein motors for retrograde transport to the neuronal cell body (81). Virus capsids will dock at the nuclear pore, and the viral RNA is instilled into the nucleus for an acute or quiescent, latent infection (Walker and Ghildyal 2017; Prone 2007). By infecting these sensory neurons, coronaviruses can penetrate into the brain tissue through the direct connections between the peripheral and central nervous system (Wu et al. 2020) (Fig. 3).
-
b)
Invasion of motor neurons at neuromuscular junctions (NMJs)
NMJs are specialized synapses between motor neurons and muscles to control movements and represent a gateway into the CNS for some viruses (Lai and Ip 2003; Turgay et al. 2015). Most motor neurons have their cell bodies in the spinal cord where they can connect with the brain’s motor centers (Turgay et al. 2015). There are no data showing coronaviruses infecting motor neurons, but preliminary results show acute flaccid paralysis (AFP) in human coronavirus patients (Turgay et al. 2015; Guidon and Amato 2020). However, about 1% of coronavirus infections result in motor neuron infection leading to some motor dysfunction and paralysis (Guidon and Amato 2020; Cokyaman et al. 2015). Coronavirus receptors can be expressed in axonal membranes (Dubé et al. 2018). Viral particles can enter motor neuronal axons in the NMJ by binding unknown receptors or neural cell adhesion molecules (NCAMs) (Bergmann et al. 2006; Bender and Weiss 2010). Trans-neuronal spread occurs exclusively between synoptically bound neurons and the muscle with the virus moving directly into the muscles and upstream neurons, which can lead to paralysis or neurodegeneration (Hirano et al. 2004; Cowley and Weiss 2010). The analyses of motor neurons at neuromuscular junctions may define their effects on the musculoskeletal system, which is likely to depend on the genetic status of the patient (91) (Cowley and Weiss 2010) (Fig. 3).
-
c)
Invasion via the olfactory epithelium and olfactory neurons
The olfactory system provides a special and direct gateway for coronaviruses to enter the CNS (Mori et al. 2005). The neurons that act as bipolar olfactory receptor have many branches of dendrites in the olfactory epithelium at the roof of the nasal-pharyngeal cavity where odors are detected (Mori et al. 2005; Riel et al. 2015). Dysosmia and dysgeusia are typical signs of viral infection strongly suggesting their causative effects in olfactory receptors (Keyhan et al. 2019; Lao et al. 2020). The olfactory nerve, which is part of the PNS, originates from the olfactory epithelium to the olfactory bulb in the CNS proper (Keyhan et al. 2019; Lao et al. 2020). The olfactory epithelium is well protected from most common infections by mucus and several pathogen recognition receptors (Koyuncu et al. 2013). Multiple studies suggested that coronaviruses may cause olfactory dysfunction through multiple mechanisms (Keyhan et al. 2019; Lao et al. 2020). Coronaviruses can enter the CNS through a trans-neuronal route and spread through neuroanatomic pathways to multiples brain regions (Desforges et al. 2014b). Coronaviruses tend to infect the CNS through the olfactory system after intranasal infection (Desforges et al. 2014b; Sun and Perlman 1995). For instance, human coronavirus (HCoV) can enter the CNS through the olfactory bulb, causing inflammation and demyelination (Desforges et al. 2014a; St-Jean et al. 2004). Then, the virus can infect the entire brain and CSF in less than 7 days and can cause demyelination as reported in several studies (Yeh et al. 2004; Murray et al. 1992). Neurons are highly polarized, with distinct dendrite and axonal processes (Murray et al. 1992). The maintenance and communication of distal axons with their cell bodies requires highly specialized signal transduction, intracellular sorting, trafficking, and membrane systems; coronaviruses as neurotrophic viruses are dependent on these cellular processes for migration into brain tissue (Dubé et al. 2018) (Fig. 3).
Indirect brain damage is mediated by the following molecular and cellular pathological pathways:
-
1)
Nerve inflammation
Neurotropic RNA viruses like coronaviruses are increasingly associated with central neurological disorders (Rejdak and Grieb 2020) and can cause immune-mediated CNS pathogenesis (Hosking and Lane 2010). This immune-mediated process has been well documented with coronaviruses (Bender and Weiss 2010). Mechanisms such as the “cytokine storm” caused by an over production of inflammatory factors after the viral infection can disrupt the blood-brain barrier (Bergmann et al. 2006; Dandekar and Perlman 2005). Local production of cytokines and chemokines in the CNS can also disrupt the blood-brain barrier and reduce tight junction’s stability (Weiss and Navas-Martin 2005; Weiss and Leibowitz 2011; Dörries et al. 1986). For instance, the expression of chemokine monocyte chemoattractant protein-1 (MCP-1, CCL2) is increased during coronavirus and other viral infections (Jiang et al. 2005; Law et al. 2005), and these proteins disrupt tight junction (Cheung et al. 2005). The effects of coronavirus infection on neuroinflammation depend on the host’s immune response; RNA replication interferes with multiple signaling pathways, disrupting the expression of cytokines, chemokines, prostaglandins, leukotrienes, and neurotrophins that regulate neuronal survival and function and apoptosis-related genes contributing to neurodegeneration (Cheung et al. 2005; Tirotta et al. 2013). One of the main processes is the so called cytokine storm, and it is reported as the leading critical cause of death in COVID-19 patients (Channappanavar et al. 2017). The “cytokine storm” is one of the key factors inducing neurodegeneration and neuronal dysfunction, due to the over production of inflammatory cytokines (IFNα, IL1β, IL6, IL12, IL18, IL33, TNFα, etc.) and chemokines (CCL2, CCL3, CCL5, CXCL8, CXCL9, CXCL10, etc.) to SARS-CoV and other coronavirus infections (Channappanavar et al. 2017; Tisoncik et al. 2012). Likewise, severe MERS-CoV infection also increased serum levels of IL6, IFNα, CCL5, CXCL8, and CXCL10 (Cheung et al. 2005; Thiel and Weber 2008). The cytokine storm triggers became more dangerous than the original infection and causes multiple organ failure and neurodegeneration contributing to neurological disorder (Thiel and Weber 2008). This process is also observed in experimental animal models, and coronaviruses infect both neuronal and non-neuronal cells (i.e. glial cells), inducing inflammatory responses leading to degenerative processes (Edwards et al. 2000; Whitman et al. 2009). A similar process has been reported with influenza infection, with inflammation leading to neurological and cognitive effects (Jang et al. 2009; Studahl 2003). Multiple studies show increased production of inflammatory cytokines and activation of microglia in coronavirus infection (Studahl 2003; Chiara et al. 2012). Elevated levels of multiple cytokines have been detected in SARS-CoV cerebrospinal fluids associated with acute encephalopathy (Wu et al. 2020; Bergmann et al. 2006). Although the molecular inflammatory mechanisms inducing neurological disorders are not known, primate and rodent models show that complex innate and adaptive immune responses are involved, including abnormal Th1/Th2 balance and defective T-cell proliferation during coronavirus infection (Zakhartchouk et al. 2005; Gupta et al. 2006). The inflammatory processes induced by coronaviruses can also affect the neuropathogenicity of other viruses and pathogens (Ng et al. 2003). These inflammatory responses have both an antiviral effect but also a detrimental effect in the nervous system (Chen et al. 2019). Thus, glia activation can induce both antiviral responses, while long-term glia activation may cause neuropathogenic processes (Chen et al. 2019). The innate immune responses induce the recruitment and activation of other peripheral immune cells via chemokines, modulating the blood-brain barrier and cell-cell interactions (Wu et al. 2020; Bergmann et al. 2006). Immune cell penetration is essential for viral infection in the CNS, but it may promote immune invasion resulting in neurological disorders (Bergmann et al. 2006) (Fig. 4). Taken together, these studies suggest that infection activates microglia to produce inflammatory cytokines. If unregulated, these cytokines can become detrimental and cause neurotoxicity and neurological dysfunction (Figs. 3, 4).
-
2)
Nerve oxidative stress
Viral infections trigger neurological disorders through a variety of pathological mechanisms such as mitochondrial dysfunction and oxidative stress (Shi et al. 2014; Liu et al. 2006). Coronaviruses also target mitochondria function and cause oxidative stress damage (Liu et al. 2006). Coronaviruses target both neuronal and glial cells and affect patients of all ages (Liu et al. 2006; Padhan et al. 2008). Viruses may undergo lytic replication in the cells and spread to other cells, until viral gene expression may be limited for viral latent infection (Lavi and Weiss 1989). Viral products and host mediators can cause tissue damage and, depending on the cell type and the nervous system area, this damage can lead to meningitis, neuritis, myelitis, encephalitis, vasculitis, or demyelination (Wu et al. 2020; Padhan et al. 2008). The molecular mechanisms by which coronavirus infections cause neurological dysfunction are not known (Wu et al. 2020). Multiple studies provide critical information about the potential of coronaviruses to activate the immune system, but it is unknown how this inflammatory response can become so detrimental in some patients and disrupt neural metabolism and cause neurological dysfunction or death (Wu et al. 2020; Poyiadji et al. 2020). The production of reactive oxygen species (ROS) and reactive nitrogen species (RNS) can become an important common pathway of tissue damage during viral infections (Ntyonga-Pono 2020; Hiffler 2020). ROS and RNS can be initiated within cells by redox reactions associated with normal physiological processes or enzymatic and non-enzymatic mechanisms associated with pathological processes induced by coronaviruses (Karadag et al. 2009). ROS and RNS are free radicals that have one or more unpaired electrons with a high ability to oxidize organelles, macromolecules, and key membrane and nucleic acid components (Karadag et al. 2009; Fransen et al. 2012). The most important examples of free radicals ROS and RNS include superoxide (O2−), hydroxyl (OH), nitric oxide (NO), and nitric dioxide (NO2). Examples of non-radical ROS and RNS include hydrogen peroxide (H2O2) and peroxynitrite (ONOO2) (Fransen et al. 2012; Seifried et al. 2007). Viral infections increase the production of ROS and RNS which confer their effects to O2- and NO (Valko et al. 2007), this free radical is the result of inflammatory processes (Groot-Mijnes et al. 2005). O2- and other ROS are produced by infiltration of NADPH oxidase-expressing phagocytic cells in viral inflamed tissues and by humoral reactions involving xanthine oxidase (XO) (Drummond et al. 2011). NO is mainly produced by the inducible synthase of nitric oxides (iNOS), expressed in multiple cell types including phagocytic cells (Qidwai and Jamal 2010; Lirk et al. 2002). NO and ROS, especially O2-, can form reactive nitrogen oxides such as peroxynitrite, which are potent protein oxidants, nucleic acids, and membrane-unsaturated lipids (Pohanka 2013). MDA is electrophilic species that can covalently modify and damage neural macromolecules (135). Coronavirus infection increase MDA levels in the brain (Namiduru et al. 2012; Engin and Bakir 2010). Some neurological disorders induced by coronaviruses are due to MDA, ROS, and NOS production (Desforges et al. 2014a; Savarin and Bergmann 2018). On the other hand, steady-state levels of oxidative damage occurred after the balance of tissue damage caused by ROS / RNS levels and antioxidant activity rates (Poljsak et al. 2013; Costantini and Verhulst 2009). Previous studies have shown that this balance is disrupted by viral infections such as coronaviruses (Delgado-Roche and Mesta 2020). Components of the antioxidant defense system include enzymes such as superoxide dismutase (SOD), glutathione peroxidase (GPx), glutathione reductase (GR), and catalase (CAT)(Delgado-Roche and Mesta 2020; Cheng 2020). Neurotrophic viral infections cause mitochondrial dysfunction in infected cells and reduce the activity of antioxidant enzymes (Shi et al. 2014; Yuan et al. 2006). Host functions and antioxidant defenses of neuronal cells are disrupted by coronaviruses and prevents normal cell and brain performance (Yuan et al. 2006). In addition, carbonylated proteins produced by coronavirus infection can cause DNA damage in brain tissues (Horner et al. 2015). In contrast, free radicals, such as ROS and NOS, damage arachidonic acid (AA) and docosahexaenoic acid (DHA) to produce inflammation affecting neuronal activity (Horner et al. 2015; Barnhart et al. 2013). This disturbance in AA affects the integrity of gray and white matter (Barnhart et al. 2013). Viral infection-induced oxidative damage is mediated by the generation of ROS and RNS, which can have a direct effect on the cells and trigger inflammation in the host (Valyi-Nagy and Dermody 2005). Other prominent molecules involved in viral oxidative stress induced is inducible iNOS, which have a prominent role in coronavirus infections and induces neurological dysfunction (Steer and Corbett 2003; Gomez et al. 2003). In most viral infections, regulated oxidative responses play beneficial roles by limiting viral replication through multiple mechanisms not well understood (Gomez et al. 2003). However, when unregulated these oxidative stress became detrimental to the host (Gomez et al. 2003; Li et al. 2009). For example, pharmacologic or genetic inhibition iNOS in mice is associated with reduced tissue damage during viral infection, so oxidative damage is also likely to contribute to coronavirus infections (Li et al. 2009). Coronavirus and other virus can induce ROS and RNS production in the CNS through an unknown mechanism (Delgado-Roche and Mesta 2020), but most likely mediated by viral components tohat can induce mitochondrial dysfunction and cytotoxic effects (Ntyonga-Pono 2020; Delgado-Roche and Mesta 2020). Glutathione is the main target of viral infections, it is an antioxidant cytosolic protein that plays a critical role in scavenging ROS and NOS (Cai et al. 2003). Glutathione (GSH) is a small thiol, the − SH group of its cysteine is extremely sensitive to oxidation, mainly by peroxide (Cai et al. 2003; Yang et al. 2017). However, when GSH is oxidized, GSH disulfide (GSSG) is formed and can be reduced by a specific enzyme, glutathione reductase (GSSG) (Yang et al. 2017). GSSG has detrimental effects and its concentration is increased during viral infections (Cai et al. 2003; Yang et al. 2017). Thus, coronavirus infections induce production of free radicals such as ROS and NOS, and reduce antioxidant capacity such as SOD, GPx, GR, and glutathione. These effects affect neural connectivity and survival leading to neuronal dysfunction and neurodegeneration. Although many of these mechanisms are not confirmed in coronavirus infections, they are expected to produce a pathophysiology similar to that reported other RNA viruses (Figs. 3, 5).
-
3)
Nerve cell death , apoptosis, and autophagy
The term of “apoptosis” was introduced based on the Greek word meaning “falling off” or “dropping off” in analogy to leaves falling off the trees or petals from flowers (Saikumar et al. 1999). There are two apoptotic pathways named: extrinsic or intrinsic apoptosis (Lockshin and Zakeri 2004; Love 2003). In the extrinsic pathway, ligands activate receptors, such as TRAIL and FAS, and thereby pro-caspase and caspase-8 and caspase-10, which activate caspase-3 and caspase-7, to trigger apoptosis. In the intrinsic pathway, mitochondrial damage, such as DNA damage or hypoxia, causes release of cytochrome C, which led to the production of apoptosome and therefore to the activation of caspasce-9, caspase-3, and caspase-7 and apoptosis. Some proteins such as BCL-2 and BCL-XL inhibit BAX, which stabilizes MOMP protein causing mitochondrial survival and preventing apoptosis. On the other hand, SMAC inhibits XIAP, which is a caspase-3 and caspase-7 inhibitor. Excessive apoptosis can contribute to the development or enhancement of neurodegenerative disorder (Sastre et al. 2000; Mattson 2000).
-
a)
Coronavirus induced apoptosis
Coronavirus-induced oxidative stress and inflammation can ultimately cause apoptosis (Maadidi et al. 2014; Orzalli and Kagan 2017). Cell death following viral infection is one of the most common causes of multi-organ dysfunction and death (Fung and Liu 2014; Yan et al. 2004). Mitochondrial dysfunction and the production of reactive oxygen species play an important role in the progression from cell dysfunction to apoptosis or autophagy (Liu et al. 2006). Cell death pathogenesis in coronavirus-mediated neural dysfunction is still not fully understood (Liu et al. 2006; Padhan et al. 2008). Although, it appears that dysfunction in lipid metabolism, protein synthesis, reduction in mitochondrial ß-oxidation, and alteration of the redox state contributes to coronavirus-induced neural dysfunction and death (Shi et al. 2014; Liu et al. 2006). Indirect evidence indicate that apoptosis could be a main mechanism for cell damage caused by MERS and SARS-CoV infections, but there is inadequate knowledge of the apoptotic pathways in COVID-19 (Liu et al. 2006; Padhan et al. 2008). Previous studies have shown that apoptosis in cardiorespiratory and neuronal cells is a significant pathological characteristic of MERS and SARS-Cov infections in both experimental and human subjects (Li et al. 2020a). Early studies confirmed the activation of caspases in the cardiorespiratory and neural systems during coronavirus infection (Li et al. 2020a, 2020d). Increased Bax as an apoptotic protein and decreased Bcl2 as an anti-apoptotic protein have been shown in MERS and SARS-Cov-infected cells (Rockx et al. 2020). These results show that coronaviruses can cause apoptosis and ultimately neuronal damage and death (Yan et al. 2004; Tan et al. 2004). Viral infection can cause cell damage by activating multiple apoptotic cascades that can cause DNA damage responsible for multiple cell death (Xu et al. 2011). The extent of apoptosis appears associated with viral burden and underlying disorders in infected patients (Xu et al. 2011). Experimental studies have shown that MERS and SARS-Cov induce cell death in both in vivo and in vitro (Yang et al. 2003; Gurwitz 2020). MERS and SARS-Cov infections stimulate both the intrinsic and extrinsic apoptotic pathways (Yang et al. 2003; Gurwitz 2020). The role of this mechanism in COVID-19 is still unknown at this time. Several studies indicated that MERS and SARS-Cov-induced apoptosis in neurons is due to mitochondrial dysfunction (Gurwitz 2020; Xie and Chen 2019). As described above, viral infection initiates mitochondrial dysfunction to produce ROS and induce apoptosis (Xie and Chen 2019). On the other hand, as the virus proliferates in the lung cells, it induces alveolar and interstitial inflammatory exudation, and edema. This mechanism contributes to alveolar gas exchange disorders that cause hypoxia in the CNS, increasing anaerobic metabolism in brain cell mitochondria, and apoptosis (Wu et al. 2020; Zanin et al. 2020). Accumulation of acid may cause cerebral vasodilation, swelling of brain cells, interstitial edema, obstruction of cerebral blood flow, and even headache due to ischemia and congestion (Zanin et al. 2020). If hypoxia persists, cerebral edema and circulation disorder will worsen (Felice et al. 2020). This hypoxia is the primary cause of apoptosis (Felice et al. 2020; Arabi et al. 2015). RNA mediated extrinsic apoptosis has been shown by the activation of Fas / Fas-ligand and TNF 1 / TNFα systems (Law PT-w. 2005). Given that brain cells also express Fas receptors, the death of neurons in infected patients can occur through detrimental inflammation as discussed above (Cowley and Weiss 2010; Law PT-w. 2005) (Figs. 3, 6).
The pathology of severe coronavirus infections is linked to systemic inflammation. These inflammatory reactions may be triggered by an extrinsic apoptotic pathway to cause neural death in infected patients (Dandekar and Perlman 2005; Memish et al. 2013). The persistence of coronavirus infections and its ability to infect macrophages, microglia, and astrocytes in the CNS are particularly important. These viruses can activate glial cells and induce inflammatory and apoptosis pathways. Inflammatory factors such as IL6, IL12, IL15, and TNFα are increased during coronavirus infections and all of these biomarkers can activate Fas receptor (Thiel and Weber 2008). On the other hand, clinical studies indicated that the mortality rate of COVID-19 in elderly and patients with underlying heart and respiratory conditions is significantly higher (Thiel and Weber 2008). The reduction of ACE2 expression appears to be one of the main reasons for this higher mortality rate (Burrell et al. 2012; Shenoy et al. 2010). ACE2 is a potent cardio-pulmonary protective protein that plays an important role preventing cardiovascular events and can also regulate the respiratory and nervous systems (Shenoy et al. 2010; Uhal et al. 2013; Cole-Jeffrey et al. 2015). Angiotensin-converting enzyme 2 (ACE2) produces angiotensin (Spinelli and Pellino 2020; Watkins 2020; Ziebuhr 2004; Siddell et al. 1983; Satija and Lal 2007; Anand et al. 2002; Wu et al. 2020) [Ang-(Spinelli and Pellino 2020; Watkins 2020; Ziebuhr 2004; Siddell et al. 1983; Satija and Lal 2007; Anand et al. 2002; Wu et al. 2020)] from angiotensin-2 (AngII). Previous studies have shown that the ACE2-Ang-(Spinelli and Pellino 2020; Watkins 2020; Ziebuhr 2004; Siddell et al. 1983; Satija and Lal 2007; Anand et al. 2002; Wu et al. 2020) axis plays a critical role regulating normal cardiovascular and respiratory physiology and it can be a therapeutic target in cardiopulmonary and neurological diseases (Shenoy et al. 2010; Dai et al. 2015). Some previous studies have shown that ACE2 can prevent cell death in cardiac and pulmonary systems and showed that this protein can inhibit apoptosis and autophagy-related signaling pathways and prevent cell death in various organs (Shenoy et al. 2010; Uhal et al. 2013, 2012; Cole-Jeffrey et al. 2015). Ang (Spinelli and Pellino 2020; Watkins 2020; Ziebuhr 2004; Siddell et al. 1983; Satija and Lal 2007; Anand et al. 2002; Wu et al. 2020) activates AT2R, the type-2 receptor for angiotensin. ACE-2/Ang(Spinelli and Pellino 2020; Watkins 2020; Ziebuhr 2004; Siddell et al. 1983; Satija and Lal 2007; Anand et al. 2002; Wu et al. 2020) /Mas causes Bcl-2/Beclin1 or Bcl-2/Bax complex stability and inhibits autophagy and apoptosis in the cardio-respiratory system, respectively (Dai et al. 2015; Meng et al. 2014). Ang (Spinelli and Pellino 2020; Watkins 2020; Ziebuhr 2004; Siddell et al. 1983; Satija and Lal 2007; Anand et al. 2002; Wu et al. 2020) can also inhibit phosphorylation by the c-Jun N-terminal kinas e(JNK). Apoptosis and autophagy are regulated by the complexes Bcl-2/Beclin1 and Bcl-2/Bax. Bcl-2 is an anti-apoptotic protein that interacts with Beclin1 (the key protein involved in autophagy) and Bax (the main protein involved in autophagy) when it is un-phosphorylated. Bcl-2 phosphorylation induced by autophagic or apoptotic pathways, such as the ACE-2-Mas-Ang pathway, in patients with cardio-respiratory diseases (Ferrario 2011; Uhal et al. 2011; Su et al. 2006). These pathway evokes JNK to phosphorylate and inactivate Bcl-2 by inducing its dissociation from Beclin1 or Bax, and thereby autophagia or apoptosis (Sadasivan et al. 2008; Ohishi et al. 2013). Some indirect evidence suggests that the lower expression and inactivation of ACE2-Mas-Ang (Spinelli and Pellino 2020; Watkins 2020; Ziebuhr 2004; Siddell et al. 1983; Satija and Lal 2007; Anand et al. 2002; Wu et al. 2020) in the cardiovascular system is one of the primary causes of increased mortality in high-risk patients with COVID-19. This mechanism causes the activation of JNC/Bcl2-Beclin1 or JNC/Bcl2-bax signaling pathway and induces autophagy or apoptosis (Sadasivan et al. 2008; Wang et al. 2011). These pathways affect neural connectivity and survival leading to neurodegeneration (Sadasivan et al. 2008; Wang et al. 2011). These pathways affect neural connectivity and survival leading to neurodegeneration (Sadasivan et al. 2008; Ohishi et al. 2013). Such degeneration contributes to neurological dysfunction, and although many of these mechanisms have not been well characterized in COVID-19, they are well documented in other similar RNA viruses, and predicted for coronavirus infections (Figs. 3, 7).

Coronaviruses induced neural neuro-inflammation was mediated via production of some cytokine and chemokine such as Interleukin (IL)-1,6,12,18,36 and Tumor Necrosis Factor alpha(TNF-α) and Interferon alpha(IFN-α) and Tumor Growth Factor beta(TGF-β) and some proteins such as chemokine chemoattractant protein-1 (CCL2) such as activation, monocyte chemoattractant protein-1(MCP-1). All this biomarkers cause initiation of inflammatory signaling in astrocyte and neuron which led to activation of extrinsic pathway of apoptosis in neuronal cell and lead neurodegeneration

Coronaviruses induced neural oxidative stress which was mediated via some parameters such as activation of NADH oxidase, induction of blood barriers barrier damage, induction of damage to neural structure, induction of hypoxia, induction of disturbances in neurotrophic factor, and activation of nitric oxide synthase enzyme (NOS).This parameters causes mitochondrial dysfunction which lead to production of Reactive oxygens specie(ROS) and Nitrogen Oxygen Species (NOS) and cause disturbances in glutathione pathway and antioxidant enzymes such as superoxide dismutase(SOD), Glutathione peroxidase(GPx) and Glutathione Reductase(GR), also causes occurrences of apoptosis and secretion of lactate dehydrogenases(LDH) and cytochrome C enzyme which all this event causes neurodegeneration

Coronaviruses causes apoptosis in both intrinsic and in-extrinsic pathways. In the extrinsic pathway of apoptosis, death ligand such as TRAIL and FAS causes activation of death receptor, which lead to the activation of pro-caspase and caspase 8 and 10, which consequently activated caspase 3 and 7 and lead to apoptosis in the intrinsic pathway of apoptosis. This family viruses causes mitochondrial damage via activation of NADH oxidase, induction of blood barriers barrier damage, induction of damage to neural structure, induction of hypoxia, induction of disturbances in neurotrophic factor, and activation of nitric oxide synthase enzyme (NOS).Which lead to release of cytochrome C and production of apoptosome and therefore causes activation of caspasce-9, caspase 3 and 7 and, finally, apoptosis. Some proteins such as BCL-2 cause inhibition of Bax, which has led to the stability and mitochondrial survival and inhibited the process of apoptosis. FADD: Fas-associated protein with death domain, TRAIL: TNF-related apoptosis-inducing ligand,, Bcl-2: B-cell lymphoma 2, mitochondrial outer membrane permeabilization

The angiotensin-converting enzyme 2(ACE2) induces angiotensin (1–7) [Ang-(1–7)] development of angiotensin-2(AngII). Ang(1–7) causes AT2R, which is the angiotensin receptor type-2.On the other hand, it has been shown that ACE-2/Ang(1–7)/Mas causes Bcl-2/Beclin1 or Bcl-2/Bax complex stability and inhibits autophagy and apoptosis in the cardio-respiratory system respectively. Ang(1–7) can inhibit phosphorylation by the c-Jun N-terminal kinase(JNK). Apoptosis and autophagy are regulated by the complexes Bcl-2/Beclin1 and Bcl-2/Bax. Bcl-2 is an anti-apoptotic protein that interacts with Beclin1 (the key protein involved in autophagy) and Bax(the main protein involved in autophagy) in an unphosphorylated form. Induction of Bcl-2 phosphorylation after indications of autophagy or apoptosis such as dysfunction in the signaling pathway of ACE-2/Mas / Ang (1–7), which occurred in patient with underlying cardio-respiratory disease, activating JNK which led to phosphorylation (inactivation) of Bcl-2 and dissociation of this protein from Beclin1 or Bax and thus causes autophagy or apoptosis in respiratory and cardiac tissue of patients and increased their mortality rate. Some indirect evidence indicates that one of the key causes of increased mortality rates in high-risk patients with COVID-19 virus infection is lower expression and inactivation of ACE-2/Mas / Ang (1–7) in the cardiovascular system, resulting in activation of JNC / Bcl-2-Beclin1 or JNC / Bcl-2-bax signaling pathway and initiation of autophagy or apoptosis and thus death signal
Conclusion
Taken together, the studies on the neurological effects of COVID-19, MERS, and SARS-CoV indicate that these lethal RNA viruses can cause direct damage to the nervous system by entering the CNS through the bloodstream and nerve endings. Viral infection directly damages the nerve structures and causes endovascular dysfunctions. Coronaviruses can also trigger detrimental inflammation, oxidative stress, and apoptosis, and all these mechanisms can induce neurotoxicity, neurodegeneration, and neurological dysfunction. While some of these mechanisms were reported in similar RNA viruses, the mechanisms inducing neurological dysfunction in COVID-19, MERS, and SARS-CoV viruses are not well known at this time and need further evaluation both in clinical settings and experimental animal models.
References
Alfaraj SH, Al-Tawfiq JA, Memish ZA (2019) Middle East Respiratory Syndrome Coronavirus (MERS-CoV) infection during pregnancy: Report of two cases & review of the literature
Anand K, Palm GJ, Mesters JR, Siddell SG, Ziebuhr J, Hilgenfeld R (2002) Structure of coronavirus main proteinase reveals combination of a chymotrypsin fold with an extra α-helical domain. EMBO J 21(13):3213–3224
Arabi Y, Harthi A, Hussein J, Bouchama A, Johani S, Hajeer A et al (2015) Severe neurologic syndrome associated with Middle East respiratory syndrome corona virus (MERS-CoV). Infection 43(4):495–501
Argyris EG, Acheampong E, Nunnari G, Mukhtar M, Williams KJ, Pomerantz RJ (2003) Human immunodeficiency virus type 1 enters primary human brain microvascular endothelial cells by a mechanism involving cell surface proteoglycans independent of lipid rafts. J Virol 77(22):12140–12151
Bagheri SHR, Asghari AM, Farhadi M, Shamshiri AR, Kabir A, Kamrava SK et al (2020) Coincidence of COVID-19 epidemic and olfactory dysfunction outbreak. Medrxiv
Baig AM (2020) Neurological manifestations in COVID-19 caused by SARS-CoV-2. CNS Neurosci Ther 26(5):499
Banik G, Khandaker G, Rashid H (2015) Middle East respiratory syndrome coronavirus “MERS-CoV”: current knowledge gaps. Paediatr Respir Rev 16(3):197–202
Barnhart MD, Moon SL, Emch AW, Wilusz CJ, Wilusz J (2013) Changes in cellular mRNA stability, splicing, and polyadenylation through HuR protein sequestration by a cytoplasmic RNA virus. Cell Rep 5(4):909–917
Barton LM, Duval EJ, Stroberg E, Ghosh S, Mukhopadhyay S (2020) Covid-19 autopsies, oklahoma, usa. Am J Clin Pathol 153(6):725–733
Bender SJ, Weiss SR (2010) Pathogenesis of murine coronavirus in the central nervous system. J Neuroimmune Pharmacol 5(3):336–354
Bergmann CC, Parra B, Hinton DR, Ramakrishna C, Dowdell KC, Stohlman SA (2004) Perforin and gamma interferon-mediated control of coronavirus central nervous system infection by CD8 T cells in the absence of CD4 T cells. J Virol 78(4):1739–1750
Bergmann CC, Lane TE, Stohlman SA (2006) Coronavirus infection of the central nervous system: host–virus stand-off. Nat Rev Microbiol 4(2):121–132
Brian D, Baric R (2005) Coronavirus genome structure and replication. Springer, Coronavirus replication and reverse genetics, pp 1–30
Burrell LM, Burchill L, Dean RG, Griggs K, Patel SK, Velkoska E (2012) Chronic kidney disease: cardiac and renal angiotensin-converting enzyme (ACE) 2 expression in rats after subtotal nephrectomy and the effect of ACE inhibition. Exp Physiol 97(4):477–485
Cabirac G, Murray R, McLaughlin L, Skolnick D, Hogue B, Dorovini-Zis K et al (1995) In vitro interaction of coronaviruses with primate and human brain microvascular endothelial cells. Springer, Corona-and Related Viruses, pp 79–88
Cai J, Chen Y, Seth S, Furukawa S, Compans RW, Jones DP (2003) Inhibition of influenza infection by glutathione. Free Radical Biol Med 34(7):928–936
Calcagno N, Colombo E, Maranzano A, Pasquini J, Keller Sarmiento IJ, Trogu F et al (2020) Rising evidence for neurological involvement in COVID-19 pandemic. Neurol Sci 1–3
Channappanavar R, Perlman S (2017) Pathogenic human coronavirus infections: causes and consequences of cytokine storm and immunopathology. Seminars in immunopathology. Springer
Chen BP, Kuziel WA, Lane TE (2001) Lack of CCR2 results in increased mortality and impaired leukocyte activation and trafficking following infection of the central nervous system with a neurotropic coronavirus. J Immunol 167(8):4585–4592
Chen G, Wu D, Guo W, Cao Y, Huang D, Wang H et al (2020) Clinical and immunological features of severe and moderate coronavirus disease 2019. The J Clin Investig 130(5)
Cheng RZ (2020) Can early and high intravenous dose of vitamin C prevent and treat coronavirus disease 2019 (COVID-19)? Medicine in Drug Discovery 5:100028
Cheung CY, Poon LL, Ng IH, Luk W, Sia S-F, Wu MH et al (2005) Cytokine responses in severe acute respiratory syndrome coronavirus-infected macrophages in vitro: possible relevance to pathogenesis. J Virol 79(12):7819–7826
Chevance A, Gourion D, Hoertel N, Llorca P, Thomas P, Bocher R et al (2020) Ensuring mental health care during the SARS-CoV-2 epidemic in France: a narrative review. L'encephale
Cokyaman T, Emine T, Ozlem K, Muhammet SP, Haydar AT (2015) A rare cause of acute flaccid paralysis: Human coronaviruses. J Pediatr Neurosci 10(3)
Cole-Jeffrey CT, Liu M, Katovich MJ, Raizada MK, Shenoy V (2015) ACE2 and microbiota: emerging targets for cardiopulmonary disease therapy. J Cardiovasc Pharmacol 66(6):540
Conde G, Pájaro LDQ, Marzola IDQ, Villegas YR, Salazar LRM (2020) Neurotropism of SARS-CoV 2: Mechanisms and manifestations. J Neurol Sci
Conti P, Ronconi G, Caraffa A, Gallenga C, Ross R, Frydas I et al (2020) Induction of pro-inflammatory cytokines (IL-1 and IL-6) and lung inflammation by Coronavirus-19 (COVI-19 or SARS-CoV-2): anti-inflammatory strategies. J Biol Regul Homeost Agents 34(2):1
Costantini D, Verhulst S (2009) Does high antioxidant capacity indicate low oxidative stress? Funct Ecol 23(3):506–509
Cowley TJ, Weiss SR (2010) Murine coronavirus neuropathogenesis: determinants of virulence. J Neurovirol 16(6):427–434
Dai H, Jiang L, Xiao Z, Guang X (2015) ACE2–angiotensin-(1–7)–Mas axis might be a promising therapeutic target for pulmonary arterial hypertension. Nat Rev Cardiol 12(6):374
Dandekar AA, Perlman S (2005) Immunopathogenesis of coronavirus infections: implications for SARS. Nat Rev Immunol 5(12):917–927
Dandekar AA, O’Malley K, Perlman S (2005) Important roles for gamma interferon and NKG2D in γδ T-cell-induced demyelination in T-cell receptor β-deficient mice infected with a coronavirus. J Virol 79(15):9388–9396
De Chiara G, Marcocci ME, Sgarbanti R, Civitelli L, Ripoli C, Piacentini R et al (2012) Infectious agents and neurodegeneration. Mol Neurobiol 46(3):614–638
De Felice FG, Tovar-Moll F, Moll J, Munoz DP, Ferreira ST (2020) Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2) and the Central Nervous System. Trends Neurosci
de Groot-Mijnes JD, Van Dun JM, Van Der Most RG, De Groot RJ (2005) Natural history of a recurrent feline coronavirus infection and the role of cellular immunity in survival and disease. J Virol 79(2):1036–1044
Delgado-Roche L, Mesta F (2020) Oxidative Stress as Key Player in Severe Acute Respiratory Syndrome Coronavirus (SARS-CoV) infection. Arch Med Res
Desforges M, Favreau DJ, Brison É, Desjardins J, Meessen-Pinard M, Jacomy H et al (2013) Human Coronaviruses: Respiratory pathogens revisited as infectious neuroinvasive, neurotropic, and neurovirulent agents
Desforges M, Miletti TC, Gagnon M, Talbot PJ (2007) Activation of human monocytes after infection by human coronavirus 229E. Virus Res 130(1–2):228–240
Desforges M, Le Coupanec A, Brison É, Meessen-Pinard M, Talbot PJ (2014a) Neuroinvasive and neurotropic human respiratory coronaviruses: potential neurovirulent agents in humans. Springer, Infectious Diseases and Nanomedicine I, pp 75–96
Desforges M, Le Coupanec A, Stodola JK, Meessen-Pinard M, Talbot PJ (2014) Human coronaviruses: viral and cellular factors involved in neuroinvasiveness and neuropathogenesis. Virus Res 194:145–158
Ding Y, He L, Zhang Q, Huang Z, Che X, Hou J et al (2004) Organ distribution of severe acute respiratory syndrome (SARS) associated coronavirus (SARS-CoV) in SARS patients: implications for pathogenesis and virus transmission pathways. The Journal of Pathology: A Journal of the Pathological Society of Great Britain and Ireland 203(2):622–630
Doenges SJ, Weber K, Dorsch R, Fux R, Fischer A, Matiasek LA et al (2016) Detection of feline coronavirus in cerebrospinal fluid for diagnosis of feline infectious peritonitis in cats with and without neurological signs. J Feline Med Surg 18(2):104–109
Dörries R (2001) The role of T-cell-mediated mechanisms in virus infections of the nervous system. The Mechanisms of Neuronal Damage in Virus Infections of the Nervous System: Springer 219–45
Dörries R, Watanabe R, Wege H, Ter Meulen V (1986) Murine coronavirus-induced encephalomyelitides in rats: analysis of immunoglobulins and virus-specific antibodies in serum and cerebrospinal fluid. J Neuroimmunol 12(2):131–142
Drummond GR, Selemidis S, Griendling KK, Sobey CG (2011) Combating oxidative stress in vascular disease: NADPH oxidases as therapeutic targets. Nat Rev Drug Discovery 10(6):453–471
Dubé M, Le Coupanec A, Wong AH, Rini JM, Desforges M, Talbot PJ (2018) Axonal transport enables neuron-to-neuron propagation of human coronavirus OC43. J Virol 92(17):e00404-e418
Eckerle I, Müller MA, Kallies S, Gotthardt DN, Drosten C (2013) In-vitro renal epithelial cell infection reveals a viral kidney tropism as a potential mechanism for acute renal failure during Middle East Respiratory Syndrome (MERS) Coronavirus infection. Virology journal 10(1):359
Edwards JA, Denis F, Talbot PJ (2000) Activation of glial cells by human coronavirus OC43 infection. J Neuroimmunol 108(1–2):73–81
El Maadidi S, Faletti L, Berg B, Wenzl C, Wieland K, Chen ZJ et al (2014) A novel mitochondrial MAVS/Caspase-8 platform links RNA virus–induced innate antiviral signaling to Bax/Bak-independent apoptosis. J Immunol 192(3):1171–1183
Eliezer M, Hautefort C, Hamel A-L, Verillaud B, Herman P, Houdart E et al (2020) Sudden and complete olfactory loss function as a possible symptom of COVID-19. JAMA otolaryngology–head & neck surgery
Engin A, Bakir S (2010) Malondialdehyde, vitamin E, and anti-oxidant enzyme activity levels in patients with crimean-congo hemorrhagic fever. African journal of microbiology research 4(22):2402–2409
Ferrario CM (2011) ACE 2: More of Ang 1–7 or less Ang II? Curr Opin Nephrol Hypertens 20(1):1
Fransen M, Nordgren M, Wang B, Apanasets O (2012) Role of peroxisomes in ROS/RNS-metabolism: implications for human disease. Biochimica et Biophysica Acta (BBA)-Mol Basis Dis 1822(9):1363–73
Fung TS, Liu DX (2014) Coronavirus infection, ER stress, apoptosis and innate immunity. Front Microbiol 5:296
Gallagher TM, Buchmeier MJ (2001) Coronavirus spike proteins in viral entry and pathogenesis. Virology 279(2):371–374
Gomez RM, Yep A, Schattner M, Berría MI (2003) Junin virus-induced astrocytosis is impaired by iNOS inhibition. J Med Virol 69(1):145–149
Group WM-CR (2013) State of knowledge and data gaps of Middle East respiratory syndrome coronavirus (MERS-CoV) in humans. PLoS currents 5
Guidon AC (2020) Amato AA. COVID-19 and neuromuscular disorders. Neurology
Guo T, Fan Y, Chen M, Wu X, Zhang L, He T et al (2020) Cardiovascular implications of fatal outcomes of patients with coronavirus disease 2019 (COVID-19). JAMA Cardiol
Gupta V, Tabiin TM, Sun K, Chandrasekaran A, Anwar A, Yang K et al (2006) SARS coronavirus nucleocapsid immunodominant T-cell epitope cluster is common to both exogenous recombinant and endogenous DNA-encoded immunogens. Virology 347(1):127–139
Gurwitz D (2020) Angiotensin receptor blockers as tentative SARS‐CoV‐2 therapeutics. Drug Dev Res
Heerssen HM, Pazyra MF, Segal RA (2004) Dynein motors transport activated Trks to promote survival of target-dependent neurons. Nat Neurosci 7(6):596–604
Hiffler L (2020) Selenium and RNA viruses interactions: Potential implications for SARS-Cov-2 infection (Covid-19). Available at SSRN 3594240
Hirano N, Nomura R, Tawara T, Tohyama K (2004) Neurotropism of swine haemagglutinating encephalomyelitis virus (coronavirus) in mice depending upon host age and route of infection. J Comp Pathol 130(1):58–65
Horner SM, Wilkins C, Badil S, Iskarpatyoti J, Gale Jr M (2015) Proteomic analysis of mitochondrial-associated ER membranes (MAM) during RNA virus infection reveals dynamic changes in protein and organelle trafficking. PloS One 10(3)
Hosking MP, Lane TE (2010) The pathogenesis of murine coronavirus infection of the central nervous system. Crit Reviews™ Immunol 30(2)
Hung EC, Chim SS, Chan PK, Tong YK, Ng EK, Chiu RW et al (2003) Detection of SARS coronavirus RNA in the cerebrospinal fluid of a patient with severe acute respiratory syndrome. Clin Chem 49(12):2108–2109
Iroegbu JD, Ifenatuoha CW, Ijomone OM (2020) Potential neurological impact of coronaviruses: implications for the novel SARS-CoV-2. Neurol Sci 1
Jacomy H, Talbot PJ (2003) Vacuolating encephalitis in mice infected by human coronavirus OC43. Virology 315(1):20–33
Jacomy H, Fragoso G, Almazan G, Mushynski WE, Talbot PJ (2006) Human coronavirus OC43 infection induces chronic encephalitis leading to disabilities in BALB/C mice. Virology 349(2):335–346
Jang H, Boltz D, Sturm-Ramirez K, Shepherd KR, Jiao Y, Webster R et al (2009) Highly pathogenic H5N1 influenza virus can enter the central nervous system and induce neuroinflammation and neurodegeneration. Proc Natl Acad Sci 106(33):14063–14068
Jiang Y, Xu J, Zhou C, Wu Z, Zhong S, Liu J et al (2005) Characterization of cytokine/chemokine profiles of severe acute respiratory syndrome. Am J Respir Crit Care Med 171(8):850–857
Karadag A, Ozcelik B, Saner S (2009) Review of methods to determine antioxidant capacities. Food Anal Methods 2(1):41–60
Keyhan SO, Fallahi HR, Cheshmi B (2020) Dysosmia and dysgeusia due to the 2019 Novel Coronavirus; a hypothesis that needs further investigation. Springer Open
Khan S, Siddique R, Li H, Ali A, Shereen MA, Bashir N et al (2020) Impact of coronavirus outbreak on psychological health. J Glob Health 10(1)
Koyuncu OO, Hogue IB, Enquist LW (2013) Virus infections in the nervous system. Cell Host Microbe 13(4):379–393
Lai CC, Shih TP, Ko WC, Tang HJ, Hsueh PR (2020) Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) and corona virus disease-2019 (COVID-19): the epidemic and the challenges. Int J Antimicrobial agents 105924
Lai K-O, Ip NY (2003) Central synapse and neuromuscular junction: same players, different roles. Trends Genet 19(7):395–402
Lao WP, Imam SA, Nguyen SA (2020) Anosmia, hyposmia, and dysgeusia as indicators for positive SARS-CoV-2 infection. World J Otorhinolaryngol Head Neck Surg
Lau PY, Chan CW (2005) SARS (Severe Acute Respiratory Syndrome): reflective practice of a nurse manager. J Clin Nurs 14(1):28–34
Lavi E, Weiss SR (1989) Coronaviruses. Springer, Clinical and Molecular Aspects of Neurotropic Virus Infection, pp 101–139
Law HK, Cheung CY, Ng HY, Sia SF, Chan YO, Luk W et al (2005) Chemokine up-regulation in sars-coronavirus–infected, monocyte-derived human dendritic cells. Blood 106(7):2366–2374
Law PTW (2005) Complete genome sequencing of severe acute respiratory syndrome-associated coronavirus (SARS-CoV) and the functional characterization of the 3a protein: The Chinese University of Hong Kong (Hong Kong)
Le Coupanec A, Desforges M, Meessen-Pinard M, Dubé M, Day R, Seidah NG et al (2015) Cleavage of a neuroinvasive human respiratory virus spike glycoprotein by proprotein convertases modulates neurovirulence and virus spread within the central nervous system. PLoS Pathogens 11(11)
Lechien JR, Chiesa-Estomba CM, De Siati DR, Horoi M, Le Bon SD, Rodriguez A, et al (2020) Olfactory and gustatory dysfunctions as a clinical presentation of mild-to-moderate forms of the coronavirus disease (COVID-19): a multicenter European study. Eur Arch Oto-Rhino-Laryngol 1–11
Li W, Yang F, Liu Y, Gong R, Liu L, Feng Y et al (2009) Negative feedback regulation of IL-32 production by iNOS activation in response to dsRNA or influenza virus infection. Eur J Immunol 39(4):1019–1024
Li Y, Bai WZ (2020) Hashikawa T. Response to Commentary on:“The neuroinvasive potential of SARS‐CoV‐2 may play a role in the respiratory failure of COVID‐19 patients”. J Med Virol
Li YC, Bai WZ, Hashikawa T (2020) The neuroinvasive potential of SARS‐CoV2 may play a role in the respiratory failure of COVID‐19 patients. J Med Virol
Li Z, Huang Y, Guo X (2020) The brain, another potential target organ, needs early protection from SARS-CoV-2 neuroinvasion. Sci China Life Sci 63(5):771–773
Li Z, Liu T, Yang N, Han D, Mi X, Li Y et al (2020) Neurological manifestations of patients with COVID-19: potential routes of SARS-CoV-2 neuroinvasion from the periphery to the brain. Front Med 1–9
Lin L, Lu L, Cao W, Li T (2020) Hypothesis for potential pathogenesis of SARS-CoV-2 infection–a review of immune changes in patients with viral pneumonia. Emerging microbes & infections 9(1):727–732
Lirk P, Hoffmann G, Rieder J (2002) Inducible nitric oxide synthase-time for reappraisal. Current Drug Targets-Inflammation & Allergy 1(1):89–108
Liu NQ, Lossinsky AS, Popik W, Li X, Gujuluva C, Kriederman B et al (2002) Human immunodeficiency virus type 1 enters brain microvascular endothelia by macropinocytosis dependent on lipid rafts and the mitogen-activated protein kinase signaling pathway. J Virol 76(13):6689–6700
Liu Y, Pu Y, Zhang X (2006) Role of the mitochondrial signaling pathway in murine coronavirus-induced oligodendrocyte apoptosis. J Virol 80(1):395–403
Lockshin RA, Zakeri Z (2004) Apoptosis, autophagy, and more. Int J Biochem Cell Biol 36(12):2405–2419
Love S (2003) Apoptosis and brain ischaemia. Prog Neuropsychopharmacol Biol Psychiatry 27(2):267–282
Madjid M, Safavi-Naeini P, Solomon SD, Vardeny O (2020) Potential effects of coronaviruses on the cardiovascular system: a review. JAMA Cardiol
Mattson MP (2000) Apoptosis in neurodegenerative disorders. Nat Rev Mol Cell Biol 1(2):120–130
Memish ZA, Zumla AI, Al-Hakeem RF, Al-Rabeeah AA, Stephens GM (2013) Family cluster of Middle East respiratory syndrome coronavirus infections. N Engl J Med 368(26):2487–2494
Meng Y, Yu C-H, Li W, Li T, Luo W, Huang S et al (2014) Angiotensin-converting enzyme 2/angiotensin-(1–7)/Mas axis protects against lung fibrosis by inhibiting the MAPK/NF-κB pathway. Am J Respir Cell Mol Biol 50(4):723–736
Mesel-Lemoine M, Millet J, Vidalain P-O, Law H, Vabret A, Lorin V et al (2012) A human coronavirus responsible for the common cold massively kills dendritic cells but not monocytes. J Virol 86(14):7577–7587
Miner JJ, Diamond MS (2016) Mechanisms of restriction of viral neuroinvasion at the blood–brain barrier. Curr Opin Immunol 38:18–23
Moein ST, Hashemian SM, Mansourafshar B, Khorram‐Tousi A, Tabarsi P, Doty RL (2020) Smell dysfunction: a biomarker for COVID‐19. International Forum of Allergy & Rhinology: Wiley Online Library
Mori I, Nishiyama Y, Yokochi T, Kimura Y (2005) Olfactory transmission of neurotropic viruses. J Neurovirol 11(2):129–137
Murray RS, Cai G-Y, Hoel K, Zhang J-Y, Soike KF, Cabirac GF (1992) Coronavirus infects and causes demyelination in primate central nervous system. Virology 188(1):274–284
Nagashima K, Wege H, Meyermann R, Ter Meulen V (1979) Demyelinating encephalomyelitis induced by a long-term corona virus infection in rats. Acta Neuropathol 45(3):205–213
Namiduru ES, Namiduru M, Tarakçioğlu M, Tanriverdi M (2012) Levels of malondialdehyde, myeloperoxidase and nitrotyrosine in patients with chronic viral hepatitis B and C. Adv Clin Exp Med 21(1):47–53
Ng M, Tan S, See E, Ooi E, Ling A (2003) Early events of SARS coronavirus infection in vero cells. J Med Virol 71(3):323–331
Nowacki W, Charley B (1993) Enrichment of coronavirus-induced interferon-producing blood leukocytes increases the interferon yield per cell: a study with pig leukocytes. Res Immunol 144(2):111–120
Ntyonga-Pono M-P (2020) COVID-19 infection and oxidative stress: an under-explored approach for prevention and treatment?
Ohishi M, Yamamoto K, Rakugi H (2013) Angiotensin (1–7) and other angiotensin peptides. Curr Pharm Des 19(17):3060–3064
Ong SWX, Tan YK, Chia PY, Lee TH, Ng OT, Wong MSY et al (2020) Air, surface environmental, and personal protective equipment contamination by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) from a symptomatic patient. JAMA 323(16):1610–1612
Orzalli MH, Kagan JC (2017) Apoptosis and necroptosis as host defense strategies to prevent viral infection. Trends Cell Biol 27(11):800–809
Padhan K, Minakshi R, Towheed MAB, Jameel S (2008) Severe acute respiratory syndrome coronavirus 3a protein activates the mitochondrial death pathway through p38 MAP kinase activation. J Gen Virol 89(8):1960–1969
Paniz‐Mondolfi A, Bryce C, Grimes Z, Gordon RE, Reidy J, Lednicky J et al (2020) Central Nervous System Involvement by Severe Acute Respiratory Syndrome Coronavirus‐2 (SARS‐CoV‐2). J Med Virol
Parra B, Hinton DR, Lin MT, Cua DJ, Stohlman SA (1997) Kinetics of cytokine mRNA expression in the central nervous system following lethal and nonlethal coronavirus-induced acute encephalomyelitis. Virology 233(2):260–270
Paybast S, Emami A, Koosha M, Baghalha F (2020) Novel Coronavirus Disease (COVID-19) and Central Nervous System Complications: What Neurologist Need to Know. Acta Neurol Taiwan 29(1):24–31
Pewe L, Perlman S (2002) Cutting edge: CD8 T cell-mediated demyelination is IFN-γ dependent in mice infected with a neurotropic coronavirus. J Immunol 168(4):1547–1551
Pohanka M (2013) Role of oxidative stress in infectious diseases. A review Folia microbiologica 58(6):503–513
Poljsak B, Šuput D, Milisav I (2013) Achieving the balance between ROS and antioxidants: when to use the synthetic antioxidants. Oxid Med Cell Longev
Poyiadji N, Shahin G, Noujaim D, Stone M, Patel S, Griffith B (2020) COVID-19–associated acute hemorrhagic necrotizing encephalopathy: CT and MRI features. Radiology 201187
Prone RRIE (2007) Virus replication strategies. Fields’. Virology 1:119
Qidwai T, Jamal F (2010) Inducible nitric oxide synthase (iNOS) gene polymorphism and disease prevalence. Scand J Immunol 72(5):375–387
Rejdak K, Grieb P (2020) Adamantanes might be protective from COVID-19 in patients with neurological diseases: multiple sclerosis, parkinsonism and cognitive impairment. Mult Scler Relat Disord 102163
Rockx B, Kuiken T, Herfst S, Bestebroer T, Lamers MM, Munnink BBO, et al (2020) Comparative pathogenesis of COVID-19, MERS, and SARS in a nonhuman primate model. Science
Sadasivan S, Dunn WA Jr, Hayes RL, Wang KK (2008) Changes in autophagy proteins in a rat model of controlled cortical impact induced brain injury. Biochem Biophys Res Commun 373(4):478–481
Saikumar P, Dong Z, Mikhailov V, Denton M, Weinberg JM, Venkatachalam MA (1999) Apoptosis: definition, mechanisms, and relevance to disease. Am J Med 107(5):489–506
Sastre J, Pallardó FV, Viña J (2000) Mitochondrial oxidative stress plays a key role in aging and apoptosis. IUBMB Life 49(5):427–435
Satija N, Lal SK (2007) The molecular biology of SARS coronavirus. Ann N Y Acad Sci 1102(1):26
Savarin C, Bergmann CC (2018) Fine tuning the cytokine storm by IFN and IL-10 following neurotropic coronavirus encephalomyelitis. Front Immunol 9:3022
Seifried HE, Anderson DE, Fisher EI, Milner JA (2007) A review of the interaction among dietary antioxidants and reactive oxygen species. J Nutr Biochem 18(9):567–579
Shenoy V, Ferreira AJ, Qi Y, Fraga-Silva RA, Díez-Freire C, Dooies A et al (2010) The angiotensin-converting enzyme 2/angiogenesis-(1–7)/Mas axis confers cardiopulmonary protection against lung fibrosis and pulmonary hypertension. Am J Respir Crit Care Med 182(8):1065–1072
Shi C-S, Qi H-Y, Boularan C, Huang N-N, Abu-Asab M, Shelhamer JH et al (2014) SARS-coronavirus open reading frame-9b suppresses innate immunity by targeting mitochondria and the MAVS/TRAF3/TRAF6 signalosome. J Immunol 193(6):3080–3089
Siddell S, Wege H, Ter Meulen V (1983) The biology of coronaviruses. J Gen Virol 64(4):761–776
Spinelli A, Pellino G (2020) COVID‐19 pandemic: perspectives on an unfolding crisis. Br J Surg
Steer SA, Corbett JA (2003) The role and regulation of COX-2 during viral infection. Viral Immunol 16(4):447–460
St-Jean JR, Jacomy H, Desforges M, Vabret A, Freymuth F, Talbot PJ (2004) Human respiratory coronavirus OC43: genetic stability and neuroinvasion. J Virol 78(16):8824–8834
Studahl M (2003) Influenza virus and CNS manifestations. J Clin Virol 28(3):225–232
Su Z, Zimpelmann J, Burns K (2006) Angiotensin-(1–7) inhibits angiotensin II-stimulated phosphorylation of MAP kinases in proximal tubular cells. Kidney Int 69(12):2212–2218
Sun N, Perlman S (1995) Spread of a neurotropic coronavirus to spinal cord white matter via neurons and astrocytes. J Virol 69(2):633–641
Sun T, Guan J (2020) Novel coronavirus and the central nervous system. Eur J Neurol
Talbot PJ, Desforges M, Brison E, Jacomy H, Tkachev S (2011) Coronaviruses as encephalitis-inducing infectious agents. Non-flavirus Encephalitis In-Tech 185–202
Tan Y-J, Fielding BC, Goh P-Y, Shen S, Tan TH, Lim SG et al (2004) Overexpression of 7a, a protein specifically encoded by the severe acute respiratory syndrome coronavirus, induces apoptosis via a caspase-dependent pathway. J Virol 78(24):14043–14047
Thiel V, Weber F (2008) Interferon and cytokine responses to SARS-coronavirus infection. Cytokine Growth Factor Rev 19(2):121–132
Tirotta E, Duncker P, Oak J, Klaus S, Tsukamoto MR, Gov L et al (2013) Epstein-Barr virus-induced gene 3 negatively regulates neuroinflammation and T cell activation following coronavirus-induced encephalomyelitis. J Neuroimmunol 254(1–2):110–116
Tisoncik JR, Korth MJ, Simmons CP, Farrar J, Martin TR, Katze MG (2012) Into the eye of the cytokine storm. Microbiol Mol Biol Rev 76(1):16–32
Tsukamoto T, Hirano N, Iwasaki Y, Haga S, Terunuma H, Yamamoto T (1990) Vacuolar degeneration in mice infected with a coronavirus JHM‐CC strain. Neurology 40(6):904
Turgay C, Emine T, Ozlem K, Muhammet SP, Haydar AT (2015) A rare cause of acute flaccid paralysis: human coronaviruses. J Pediatr Neurosci 10(3):280
Turner BC, Hemmila EM, Beauchemin N, Holmes KV (2004) Receptor-dependent coronavirus infection of dendritic cells. J Virol 78(10):5486–5490
Uhal BD, Li X, Xue A, Gao X, Abdul-Hafez A (2011) Regulation of alveolar epithelial cell survival by the ACE-2/angiotensin 1–7/Mas axis. American Journal of Physiology-Lung Cellular and Molecular Physiology 301(3):L269–L274
Uhal BD, Li X, Piasecki CC, Molina-Molina M (2012) Angiotensin signalling in pulmonary fibrosis. Int J Biochem Cell Biol 44(3):465–468
Uhal BD, Dang M, Dang V, Llatos R, Cano E, Abdul-Hafez A et al (2013) Cell cycle dependence of ACE-2 explains downregulation in idiopathic pulmonary fibrosis. Eur Respir J 42(1):198–210
Umapathi T, Kor AC, Venketasubramanian N, Lim CT, Pang BC, Yeo TT et al (2004) Large artery ischaemic stroke in severe acute respiratory syndrome (SARS). J Neurol 251(10):1227–1231
Valko M, Leibfritz D, Moncol J, Cronin MT, Mazur M, Telser J (2007) Free radicals and antioxidants in normal physiological functions and human disease. Int J Biochem Cell Biol 39(1):44–84
Valyi-Nagy T (2005) Dermody TS. Role of oxidative damage in the pathogenesis of viral infections of the nervous system. Histol Histopathol
van Riel D, Verdijk R, Kuiken T (2015) The olfactory nerve: a shortcut for influenza and other viral diseases into the central nervous system. J Pathol 235(2):277–287
Velavan TP, Meyer CG (2020) The COVID-19 epidemic. Tropical Med Int Health 25(3):278
Walker EJ, Ghildyal R (2017) Viral Interactions with the Nucleus. Front Microbiol 8:951
Wang Y, Ma T, Li M, Sun X, Wang Y, Gu S (2011) Regulated hypoxia/reperfusion-dependent modulation of ERK1/2, cPLA2, and Bcl-2/Bax: a potential mechanism of neuroprotective effect of penehyclidine hydrochloride. Int J Neurosci 121(8):442–449
Watanabe R, Wege H, Ter Meulen V (1983) Adoptive transfer of EAE-like lesions from rats with coronavirus-induced demyelinating encephalomyelitis. Nature 305(5930):150–153
Watkins J. Preventing a covid-19 pandemic. British Medical Journal Publishing Group; 2020.
Weiss SR, Leibowitz JL. Coronavirus pathogenesis. Advances in virus research. 81: Elsevier; 2011. p. 85–164.
Weiss SR, Navas-Martin S (2005) Coronavirus pathogenesis and the emerging pathogen severe acute respiratory syndrome coronavirus. Microbiol Mol Biol Rev 69(4):635–664
Whitman L, Zhou H, Perlman S, Lane TE (2009) IFN-γ-mediated suppression of coronavirus replication in glial-committed progenitor cells. Virology 384(1):209–215
Wu Y, Xu X, Chen Z, Duan J, Hashimoto K, Yang L, et al. Nervous system involvement after infection with COVID-19 and other coronaviruses. Brain, Behavior, and Immunity. 2020.
Xie M, Chen Q (2020) Insight into 2019 novel coronavirus—an updated intrim review and lessons from SARS-CoV and MERS-CoV. Int J Infect Dis
Xiong T-Y, Redwood S, Prendergast B, Chen M (2020) Coronaviruses and the cardiovascular system: acute and long-term implications. Eur Heart J
Xu LH, Huang M, Fang SG, Liu DX (2011) Coronavirus infection induces DNA replication stress partly through interaction of its nonstructural protein 13 with the p125 subunit of DNA polymerase δ. J Biol Chem 286(45):39546–39559
Xu L, Liu J, Lu M, Yang D, Zheng X (2020) Liver injury during highly pathogenic human coronavirus infections. Liver Int 40(5):998–1004
Yan H, Xiao G, Zhang J, Hu Y, Yuan F, Cole DK et al (2004) SARS coronavirus induces apoptosis in Vero E6 cells. J Med Virol 73(3):323–331
Yang Y, Peng F, Wang R, Guan K, Jiang T, Xu G, The deadly coronaviruses: The, et al (2003) SARS pandemic and the 2020 novel coronavirus epidemic in China. J Autoimmun 2020:102434
Yang C-W, Lee Y-Z, Hsu H-Y, Shih C, Chao Y-S, Chang H-Y et al (2017) Targeting coronaviral replication and cellular JAK2 mediated dominant NF-κB activation for comprehensive and ultimate inhibition of coronaviral activity. Sci Rep 7(1):1–13
Yashavantha Rao H, Jayabaskaran C (2020) The emergence of a novel coronavirus (SARS‐CoV‐2) disease and their neuroinvasive propensity may affect in COVID‐19 patients. J Med Virol
Ye M, Ren Y, Lv T (2020) Encephalitis as a clinical manifestation of COVID-19. Brain, Behav Immun
Yeh EA, Collins A, Cohen ME, Duffner PK, Faden H (2004) Detection of coronavirus in the central nervous system of a child with acute disseminated encephalomyelitis. Pediatrics 113(1):e73–e76
Yuan X, Shan Y, Yao Z, Li J, Zhao Z, Chen J et al (2006) Mitochondrial location of severe acute respiratory syndrome coronavirus 3b protein. Mol Cells (Springer Science & Business Media BV) 21(2)
Zakhartchouk AN, Viswanathan S, Mahony JB, Gauldie J, Babiuk LA (2005) Severe acute respiratory syndrome coronavirus nucleocapsid protein expressed by an adenovirus vector is phosphorylated and immunogenic in mice. J Gen Virol 86(1):211–215
Zanin L, Saraceno G, Panciani PP, Renisi G, Signorini L, Migliorati K et al (2020) SARS-CoV-2 can induce brain and spine demyelinating lesions. Acta Neurochirurgica 1–4
Zegarra-Valdivia J, Vilca BNC, Tairo T, Munive V, Lastarria C (2020) NEUROLOGICAL COMPONENT IN CORONAVIRUSES INDUCED DISEASE: SYSTEMATIC REVIEW OF SARS‐CoV, MERS‐CoV, AND SARS‐CoV‐2
Zhang T, Wu Q, Zhang Z (2020) Probable pangolin origin of SARS-CoV-2 associated with the COVID-19 outbreak. Curr Biol
Zheng Y-Y, Ma Y-T, Zhang J-Y, Xie X (2020) COVID-19 and the cardiovascular system. Nat Rev Cardiol 17(5):259–260
Zhong Y, Gao X, Wang S, Xie Z, Ma Y, Fang W et al (2003) Pathologic study of circulating blood leukocytes in severe acute respiratory syndrome. Zhonghua Yi Xue Za Zhi 83(24):2137–2141
Ziebuhr J (2004) Molecular biology of severe acute respiratory syndrome coronavirus. Curr Opin Microbiol 7(4):412–419
Zumla A, Hui DS, Perlman S (2015) Middle East respiratory syndrome. The Lancet 386(9997):995–1007
Author information
Authors and Affiliations
Corresponding authors
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Gholami, M., Safari, S., Ulloa, L. et al. Neuropathies and neurological dysfunction induced by coronaviruses. J. Neurovirol. 27, 380–396 (2021). https://doi.org/10.1007/s13365-021-00977-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13365-021-00977-x