
Abstract
The conformations of glycans are crucial for their biological functions. In-electrospray ionization (ESI) hydrogen/deuterium exchange-mass spectrometry (HDX-MS) is a promising technique for studying carbohydrate conformations since rapidly exchanging functional groups, e.g., hydroxyls, can be labeled on the timeframe of ESI. However, regular application of in-ESI HDX to characterize carbohydrates requires further analysis of the in-ESI HDX methodology. For instance, in this method, HDX occurs concurrently to the analyte transitioning from solution to gas-phase ions. Therefore, there is a possibility of sampling both gas-phase and solution-phase conformations of the analyte. Herein, we differentiate in-ESI HDX of metal-adducted carbohydrates from gas-phase HDX and illustrate that this method analyzes solvated species. We also systematically examine the effects of ESI parameters, including spray solvent composition, auxiliary gas flow rate, sheath gas flow rate, sample infusion rate, sample concentration, and spray voltage, and discuss their effects on in-ESI HDX. Further, we model the structural changes of a trisaccharide, melezitose, and its intramolecular and intermolecular hydrogen bonding in solvents with different compositions of methanol and water. These molecular dynamic simulations support our experimental results and illustrate how an individual ESI parameter can alter the conformations we sample by in-ESI HDX. In total, this work illustrates how the fundamental processes of ESI alter the magnitude of HDX for carbohydrates and suggest parameters that should be considered and/or optimized prior to performing experiments with this in-ESI HDX technique.

ᅟ
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Glycans play important roles in many biological processes, such as cell-cell recognition [1], host-pathogen interactions [2], and disease progression [3, 4]. Currently, changes in glycosylation are used as biomarkers for detection of cancer [5, 6], liver disease [7], and other human disorders [8, 9]. Glycans are branched carbohydrates that result from multiple enzymatic reactions, yielding mixtures of heterogeneous structures containing stereoisomers, regioisomers, and different branching patterns [10]. This structural diversity has made detailed characterization of glycans an important but challenging task. Currently, there are few methods for analyzing carbohydrate conformations, which are crucial for the biological functions of these molecules [11,12,13].
There are several techniques that are widely used to characterize the conformations of biological macromolecules, such as X-ray crystallography [14], nuclear magnetic resonance spectroscopy (NMR) [14, 15], ion mobility-mass spectrometry (IM-MS) [16, 17], and hydrogen/deuterium exchange-mass spectrometry (HDX-MS) [18,19,20]. X-ray crystallography, which samples the lowest energy structures of molecules in a crystal lattice, has seen limited use in elucidating glycan structures due to the flexible glycosidic linkages, preventing glycans from crystalizing in repeating unit patterns [14]. In comparison, IM-MS has seen increasing use in analyzing the averaged conformations of gas-phase glycans and carbohydrates [21,22,23,24,25,26]. However, the gas-phase conformations of carbohydrates have been shown to contain different intramolecular hydrogen bonding compared to solvated carbohydrates, and thus, different three-dimensional conformations [27]. HDX-MS and NMR are advantageous as they provide three-dimensional, dynamic conformations of biomolecules. However, a significant advantage of HDX-MS over current NMR approaches is the high sensitivity of MS, which allows for characterization of specific biomolecules in biologically relevant concentrations within complex mixtures [28]. Additionally, in NMR, an average of all the solution-phase conformations is evaluated [29], whereas in HDX-MS, individual conformational states can be measured [30]. Despite the advantages of HDX-MS, traditional, solution-phase HDX techniques are not applicable to carbohydrate analyses due to the rapid back exchange of deuterium labels on hydroxyls following reaction quenching in protic solvents [31].
HDX can also be performed in the gas-phase by introducing gaseous deuterating reagent into different regions in the mass spectrometer (e.g., the analyzer or trap) where deuterium-containing molecules exchange with gas-phase analyte ions [32,33,34,35,36,37]. Because these reactions are performed under vacuum, back exchange does not occur. Gas-phase HDX has been applied to isomer differentiation of carbohydrates following collisional-induced activation and formation of oxonium ions [38]. However, collisional activation increases the internal energy of the carbohydrate and can induce conformational changes, limiting the use of this method for characterizing the conformations of native carbohydrates and glycans.
HDX reactions can also be monitored by introducing deuterating reagent molecules to analytes during electrospray ionization (ESI) and/or ESI droplet evaporation, herein referred to as in-ESI HDX. In previous studies, D2O has been introduced into the source by adding D2O vapor through the sheath gas or nebulizing gas [39], introducing liquid D2O via a dual sprayer [40], or placing a droplet of D2O on a plate in the source that evaporates to form a D2O-saturated atmosphere [41]. In-ESI HDX has also been considered advantageous as it can be carried out “on the fly” by coupling to LC systems [40]. Since both the exchange reaction for hydroxyls and ESI droplet evaporation occurs on the microsecond to millisecond time scale [42], in-ESI HDX provides a suitable sampling window for analysis of these rapidly exchanging functional groups. Kostyukevich et al. have reported useful applications of in-ESI HDX, describing the effects of solvent and capillary desolvation temperature on carbohydrate conformations [43, 44]. Such in-ESI HDX approaches require little instrument modification, making this method simple to implement and compatible with many instrument platforms. However, to date, beyond these reported qualitative discussions on the influence of these parameters on in-ESI HDX, there has not been a comprehensive study on the impact of various experimental parameters on in-ESI HDX reactions of carbohydrates. Furthermore, when performing in-ESI HDX, Kostyukevich, et al. described the sampled carbohydrate structures as gas-phase conformations [43, 44], though more recent work by the Nikolaev and Kim groups suggest that in-ESI HDX occurs in solvent clusters [45,46,47]. Thus, further research should clarify the conformational state being sampled by these in-ESI HDX methods.
ESI is a complex process in which analyte molecules transition from solvated states to gas-phase ions and hence both gas- and solution-phase conformations could be sampled during in-ESI HDX. In general, the electrospray process begins when a potential difference exists between an analyte solution exiting a capillary and the MS source inlet, resulting in the formation of a Taylor cone and emission of charged droplets as a spray plume. As the droplets move toward the MS inlet, solvent evaporates, and the charge densities of the droplets increase. When the charge density on the droplet overcomes its surface tension, or at the Rayleigh limit, the droplet breaks into smaller, highly charged droplets. These processes of evaporation and droplet fission continue until highly charged nanodroplets form. Depending on the properties and sizes of molecules within these highly charged nanodroplets, ionization of analytes occurs by one of multiple presumed mechanisms [48]. For instance, ionization of low molecular weight species, including protonated drugs and metabolites, can be described by the ion evaporation model (IEM) [48,49,50]. In IEM, solvated, charged analytes are released from nanodroplets as the droplet shape becomes distorted due to the opposing effects of coulombic repulsion (the droplet trying to push excess charge away) and ion solvation (the solvent pulling the ion back into the droplet) [51]. These processes have been modeled by molecular dynamics and show that ejected ions are present in small clusters of solvent molecules [51].
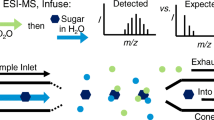
Due to the molecular weight of carbohydrates, we hypothesize that they will undergo ionization by the IEM. Thus, during in-ESI HDX, analytes are present in three different states, which potentially have different conformations that could be sampled. Specifically, analytes could exist as (1) solvated carbohydrates, representing solution-phase conformations; (2) carbohydrates and metal ions in small solvent clusters; and/or (3) gas-phase carbohydrate-metal ion adducts (Figure 1). Furthermore, as ESI parameters are modified, the distribution of carbohydrates in each state could be affected. Thus, to enhance the analytical utility of in-ESI HDX, it is crucial to understand the impact of experimental variations on both the HDX reaction outcome itself and analyte conformations. This work aims to characterize the effects of ESI conditions on in-ESI HDX of metal-carbohydrate adducts with the goal of future quantitative analyses. In this work, we examine the effects of spray solvent mixtures, auxiliary gas flow rate, sheath gas flow rate, sample infusion rate, sample concentration, and spray voltage on the magnitude of in-ESI HDX. Additionally, we interpret the effects of changing the source parameters within the framework of ESI fundamentals to further elucidate the mechanism of in-ESI HDX. Specifically, we differentiate in-ESI HDX from gas-phase HDX of metal-carbohydrate adducts. Finally, we correlate experimental data to simulations to determine how a single parameter, spray solvent composition, modifies the conformation of a model carbohydrate.

Schematic of ESI process, illustrating the formation of charged droplets, ions in solvent clusters due to IEM, and gas-phase ions, all of which could be sampled during in-ESI HDX experiments
Experimental
Materials
Melezitose, sodium chloride (NaCl), and deuterated ammonia (ND3, 99 atom % D) were purchased from Sigma-Aldrich (St. Louis, MO). Deuterium oxide (D2O, 99.96% purity) was purchased from Cambridge Isotope Laboratories, Inc. (Tewksbury, MA). Magnesium chloride (MgCl2) was purchased from VWR International, Inc. (Radnor, PA). A hexapeptide (MFRYGG) was custom synthesized by Peptide 2.0, Inc. (Chantilly, VA). Potassium chloride (KCl) and all other materials were purchased from Fisher Scientific International, Inc. (Pittsburgh, PA). All chemicals were used without further purification. Nanopure water was acquired from a Purelab Flex 3 water purification system (Elga, Veolia Environment S. A., Paris, France).
Gas-Phase Hydrogen/Deuterium Exchange
Carbohydrates ionize poorly in positive-ion mode ESI; thus, NaCl was added to carbohydrate samples to generate sodium-adduct ions. For gas-phase hydrogen/deuterium exchange (HDX) experiments, solutions of melezitose and NaCl were prepared at a 2:1 M ratio in water. These samples were diluted in methanol (final solvent composition 99:1 (v/v) methanol:water) prior to ESI. Peptide (MFRYGG) was prepared in a solution of 49.95:49.95:0.1 (v/v/v) water:methanol:acetic acid at ~ 1 μM.
Gas-phase HDX-MS experiments were conducted on a modified Fourier transform-ion cyclotron resonance (FT-ICR) MS (formerly IonSpec Corp.—now a division of Agilent Technologies, Inc., Santa Clara, CA) equipped with an open-ended cylindrical Penning trap and a 9.4 T superconducting magnet (Cryomagnetics Inc., Oak Ridge, TN). An Analytica ESI source (Analytica of Branford Inc., Branford, CT) was utilized with an applied voltage of + 3 kV. For direct infusion of the sample, a Harvard PHD 2000 syringe pump (Harvard Apparatus, Holliston, MA) was utilized at a flow rate of 5 μl/min. Following ionization and trapping, the sodiated-melezitose ion (m/z 527.1714) or protonated peptide ion (MFRYGG, m/z 730.3341) was isolated in the ICR cell using a two-step process (first: scanned isolation of a 0.750 m/z area within a 100 m/z window centered around the ion of interest; second: a stored waveform inverse Fourier transform (SWIFT) ejection of the remaining ions outside the 100 m/z window of the first step). A high-speed pulse valve setup was used for ND3 (99 atom % D, Sigma-Aldrich, St. Louis, MO) introduction into the ICR cell. In short, a gas cylinder containing gaseous ND3 was used to charge an expansion volume to ~ 40 mTorr. A pulse valve (DC12 Miniature High Speed High Vacuum Dispense Valve; Parker Hannafin, Hollins, NH) was actuated by a high-voltage PWM signal generated from an Iota Drive (Parker Hannafin, Hollins, NH) at 100 Hz. This design allowed for easily adjustable and stable introduction of gas-phase deuterating reagents for reaction times in excess of 60 s. Gas pressures were recorded from readouts of a Granville-Phillips dual ion gauge controller (Helix Technology Corp., Longmont, CO) using series 274 Bayard-Alpert type ionization gauge tube outputs [34]. Sodiated-melezitose was exposed to ND3 for 300 to 900 s at stable pressures ranging from ~ 10−8–10−7 Torr, while the hexapeptide was exposed to ND3 for 5 s at a pressure of ~ 10−8 Torr.
In-ESI Hydrogen/Deuterium Exchange
Carbohydrates are most widely studied by MS as metal adducts; therefore, we analyzed sodiated, potassiated, and magnesiated metal-ion adducts for this study. Melezitose was combined with chloride salts of Mg2+, Na+, and K+ at a molar ratio of 2:1:1:1 in water. Samples were diluted into mixtures of methanol and water (containing final volumetric amounts of 1 to 100% water) with the final melezitose concentration varied from 62.5 to 500 μM while maintaining the same molar ratio to salts. Samples were infused at flow rates of 1 to 20 μl/min.
In-ESI HDX-MS experiments were conducted using an Ion Max source coupled to an Orbitrap Discovery MS (Thermo-Fisher Scientific, Waltham, MA). In-ESI HDX-MS experiments were performed by modifying a previously described protocol [41]. Briefly, a 200-μl droplet of D2O was placed on an indentation (approximately 1.5 cm diameter) in a gold anodized aluminum plate. The plate was positioned approximately 3 cm below the spray needle and the droplet was approximately 2 cm from the ESI skimmer cone (see supplementary material, Fig. S1). Placing the droplet on the plate indentation allowed for reproducible positioning of the droplet for consecutive runs and ensured that the droplet was the same distance from the heated cone to generate reproducible D2O-saturated environments in the source. To ensure a consistent source environment for our analyses, we averaged spectra collected between 4.0 and 4.2 min, following positioning of the D2O droplet in the source and initiation of data acquisition, at which point, the D2O vapor concentration was consistent (see supplementary material, Fig. S2). Experiments utilized a desolvation capillary temperature of 300 °C (see supplementary material, Fig. S3). To characterize the effects of other source parameters on in-ESI HDX, the following electrospray parameters were individually modified; 0 to 1 arbitrary units for auxiliary gas (N2), 0 to 70 arbitrary units for sheath gas (N2), and + 2.5 to + 5.5 kV for ESI spray voltage. Details describing the source parameters for each experiment are provided in figure legends. After each sample run, the source was opened for 2 min to remove residual solvent vapors [52]. Based on a hygrometer positioned near the MS instrument, the atmospheric humidity varied by approximately 5% throughout the course of a day.
Data Processing and Statistics
Unlike for the gas-phase HDX reactions, a single precursor ion could not be isolated for in-ESI HDX reactions. Hence, our calculations account for contributions from all naturally occurring carbon and oxygen isotopes. Equation (1) was employed to calculate weighted average masses (M values) of undeuterated and deuterated metal-adducted carbohydrates using experimental mass-to-charge values (m/z) and ion intensities (I).
As shown in Eq. (2), the average number of H/D exchanges for carbohydrate-metal adducts were determined by calculating the difference between the weighted average masses of deuterated (MDeuterated) and undeuterated (MUndeuterated) counterparts:
The number of H/D exchanges is presented as the average ± the standard deviation for a minimum of three replicates. Unless stated otherwise, reported data represents average values with error (and error bars) indicating standard deviation. All comparisons were made on data collected on the same day. Students’ t test at the 95% confidence interval was used for statistical analyses and group average comparisons.
The curve fitting application of MATLAB [53] (The MathWorks, Inc., Natick, MA) was used to fit the distribution of peaks in the mass spectra following HDX. A custom code was used to visualize the component curves that fit the H/D exchange peak distributions. By fitting the data to one or more Gaussian distributions, the number of distinctive populations or conformations of the analyte present in the sample could be determined.
Molecular Dynamics Simulations
We have performed a series of molecular dynamics (MD) simulations to investigate the molecular conformations of melezitose under various solvation conditions at an atomic scale. All molecular dynamics simulations were performed using the GROMACS [54] program and the CHARMM36 all-atom forcefield [55] with parameters for carbohydrates. The optimized structures of melezitose and methanol were obtained from the ZINC 15 database [56]. One melezitose molecule, 30 sodium ions, and 30 chloride ions were placed in a cubic box of 64 nm3 and solvated with explicit SPC/E water [56, 57] molecules. Four additional solvated systems were made by randomly populating the simulation box with sufficient methanol and water molecules to produce volumetric water compositions of 75, 50, 25, and 1%. Each melezitose-solvent system was modeled three times from independent configurations. Systems were first minimized using the steepest descent algorithm followed by temperature equilibration at 300 K. We then performed 1 ns isothermal-isobaric equilibrations using v-rescale thermostat [58] for temperature regulation and isotropic pressure coupling using a Parrinello-Rahman barostat [59, 60] at 1 atm. The systems were periodic in all dimensions and the verlet [61] cut-off scheme was employed with electrostatic and van der Waals cut-offs of 1.2 nm. Following all equilibrations, 30 ns production runs were performed. All systems were found to be stable within 2 ns of each production run. The equations of motion were integrated using the leapfrog algorithm with 1 fs time steps, and the LINCS [62] algorithm was used to constrain bonds.
Results and Discussion
Carbohydrate-Metal Adducts Do Not H/D Exchange in the Gas Phase
We carried out gas-phase HDX experiments to test whether gas-phase conformations of carbohydrate-metal adducts could contribute to the exchange measured during in-ESI HDX. For sodiated-melezitose, the deuteration time and deuterating reagent pressures were increased to ensure sufficient collisions between the analyte ions and deuterating reagents in the FT-ICR cell of the instrument (see discussion in supplementary material under Scheme S1). Under our experimental conditions (t = 900 s, P~10−7 Torr), no observable HDX was detected for the melezitose-sodium adducts in the gas-phase (Figure 2a). In contrast, under similar experimental conditions (t = 5 s, P~10−8 Torr), MFRYGG, a hexapeptide with a total of 15 exchangeable hydrogens, had an average exchange of 6.4 labile H replaced with D (Figure 2b). These results suggest that metal adducts of melezitose, a model carbohydrate, do not undergo gas-phase HDX reactions.

Representative mass spectra before and after gas-phase HDX with ND3 of (a) sodium-adducted melezitose and (b) MFRYGG
There are several factors that affect gas-phase HDX, including differences in the gas-phase basicities of the reactants and the mechanism of exchange [63,64,65,66,67]. If the gas-phase basicity difference between the deuterating reagent and the analyte is less than or equal to 85 kJ/mol, then proton and deuteron exchange can occur via an approximately thermoneutral reaction [68]. The gas-phase basicity for melezitose (or its sodium-metal adduct) is unknown, but α-d-glucose has a known gas-phase basicity of 778.0 kJ/mol [69]. ND3 has a gas-phase basicity of 853.5 kJ/mol [63]. Thus, approximating the gas-phase basicity of melezitose to be similar to that of α-d-glucose, the difference in gas-phase basicities between melezitose and ND3 is 75.5 kJ/mol. For comparison, D2O has a gas-phase basicity of 696.6 kJ/mol [63]. Thus, the difference in basicities between α-d-glucose and D2O would be 81.4 kJ/mol. We did not use D2O as the deuterating agent for these gas-phase HDX reactions. However, the energetic difference associated with ND3 as the deuterating reagent makes that reaction more feasible. Since the difference in gas-phase basicities is less than 85 kJ/mol for both these deuterating reagents, it is likely that the mechanism, rather than the difference in gas-phase basicities, is limiting the formation of deuterated product [70].
Peptides undergo gas-phase HDX by several mechanisms, including the onium and relay mechanisms, depending on the analyte and the deuterating reagent (Scheme S1) [63]. For both mechanisms, the proximity of the protonated charge site to the potential exchange site is a crucial factor [63, 68]. For these experiments with carbohydrates, the charge carrier is the metal ion, which is immobilized in the adduct, likely preventing the formation of resonance structures to stabilize the transition state and minimizing HDX for these adducts in the gas-phase [63, 70]. Other gas-phase HDX mechanisms exist, including the flip-flop mechanism, which does not require charge migration (Scheme 1) [71]. However, regardless of the mechanism, exchange is not observed for sodiated-melezitose adducts upon reaction with ND3. Our observations are consistent with observations made by the Brodbelt [70] and Guttman [38] groups when examining gas-phase HDX of flavonoids and collisionally activated oxonium ions, respectively. Similarly, slow H/D exchange (if any) has been observed with other metal-adducted species, such as platinum complexes of diethylenetriamine [72] and alkali-metal complexes of polyamines [73]. Likewise, metal adduction to peptides has also been shown to result in slower rates of exchange in comparison to protonated peptides [74, 75]. Thus, these results suggest that the conformations sampled by in-ESI HDX represent carbohydrates in solvated states or solvent clusters as gas-phase HDX of these compounds is highly unfavorable.
Characterizing the Effects of Source Parameters on in-ESI HDX: Comparison of Spray Solvents
The H/D exchange data was analyzed to examine the effects of solvent on in-ESI HDX as the electrospray droplet composition changed. Increasing the percentage of water in the spray solvent from 1 to 10% resulted in an increase in the number of observed H/D exchanges from 2.4 ± 0.2 D to 4.2 ± 0.2 D. When the solvent contained 25% water, the number of H/D exchanges was 4.2 ± 0.1 D, consistent with the number of exchanges observed in a solvent with 10% water. Increasing the percentage of water to 50% resulted in a decrease in the number of H/D exchanges to 3.5 ± 0.2 D. The deuterium uptake continued to decrease eventually reaching 2.6 ± 0.1 D as the water percentage increased to 100% (Figure 3a). Representative mass spectra are presented in Figure 4.

The number of H/D exchanges for in-ESI HDX of melezitose is dependent on ESI parameters. The number of H/D exchanges varies as (a) the percentage of water in a methanolic spray solvent increases, (b) the sheath gas flow rate increases, (c) the sample concentration increases, and (d–f) the spray voltage increases in solvents containing (d) 99:1 (v/v) methanol:water, (e) 50:50 (v/v) methanol:water, and (f) 100% water. All experiments utilized a consistent set of ESI parameters, except for the parameter under investigation. ESI parameters: spray voltage + 3.5 kV, capillary temperature 300 °C, auxiliary gas 0 arb units, sheath gas 12 arb units, sample concentration 500 μM melezitose with 250 μM of each metal ion (Mg2+, Na+, K+), spray solvent 99:1 (v/v) methanol:water

Representative mass spectra following in-ESI HDX of sodiated-melezitose. Spectra illustrate differences in the magnitude of H/D exchange associated with different concentrations of water in the spray solvent. In the solvent containing 1% water and 99% methanol, two melezitose conformations with different rates of exchange are detected. ESI parameters spray voltage + 3.5 kV, sheath gas 12 arb units, auxiliary gas 0 arb units, capillary temperature 300 °C, sample 500 μM melezitose and 250 μM of each metal ion (Mg2+, Na+, and K+)
The observed number of deuteriums exchanged is governed by the forward and back exchange of the analyte. The forward exchange in solution is limited by the time it takes for ESI droplet desolvation to occur since carbohydrate-metal adducts do not exchange in the gas-phase (Figure 2). Water evaporates slower than methanol as it has a lower volatility and a higher surface tension than methanol, leading to an increase in the size of ESI droplets and a longer desolvation time for ESI droplets containing water compared to methanol [76,77,78,79]. Therefore, changing the solvent from approximately 99% methanol to 100% water would result in an increase in the desolvation time and hence an increased number of forward H/D exchanges. In competition with forward exchange, back exchange causes the loss of deuterium labels when a labeled sample comes in contact with a protonated solvent. Increasing the percentage of water in the spray solvent would result in more back exchange as H2O in the droplets can react with labeled carbohydrates and remove deuterium labels. H2O contains twice as many exchangeable protons as methanol, and therefore can result in more back exchange. Additionally, residually accruing H2O vapor in the source atmosphere (from evaporation of ESI droplets) has been shown to accumulate in the source, saturate the environment, and promote additional in-ESI HDX reactions, including back exchange [52]. Thus, the water and methanol evaporating from the ESI droplets could promote back exchange of hydroxyls. For up to 10% water in the spray solvent, the dominating factor appears to be the increase in number of H/D exchanges due to the increase in desolvation time. However, with more than 25% water in the spray solvent, the amount of back exchange seems to become the dominating factor, resulting in a net decrease in the number of H/D exchanges. Another important consideration is the change in the probability of the reaction resulting in a labeling event. For example, evaporated methanol from the solvent can undergo deuteration, resulting in CH3OD, which can then react with the carbohydrate analytes in the source. Additionally, water can be deuterated to give HDO, which would reduce the probability of a deuterating event by half in comparison to D2O. Thus, this interplay of solvent deuteration and forward and back exchange determines the total number of H/D exchanges observed.
Comparison of Gas Flow Rates
Based on the previously discussed observation that HDX for melezitose-metal adducts does not occur in the gas-phase (Figure 2), we assumed that exchange in ESI droplets is limited to solution-assisted HDX for carbohydrate-metal adducts. Thus, source parameters that affect the droplet lifetime by altering the rate of desolvation are likely to alter the reaction time for HDX and thus the number of exchange events that can occur. Nitrogen gas is used in two ways in the Ion Max source to aid in droplet evaporation. The sheath gas aids in nebulization of the droplet spray and is inner coaxial with the spray emitter, while the auxiliary gas is outer coaxial with the spray emitter.
When utilizing the auxiliary gas, solvent evaporation can occur more rapidly during ESI [80]. As the auxiliary gas was varied from 0 to 1 arbitrary units, the number of H/D exchanges observed for sodiated-melezitose in 99% methanol decreased from 2.7 ± 0.3 D to 0.5 ± 0.2 D. Similar trends were observed for the potassium and magnesium adducts of melezitose. This significant decrease in the number of H/D exchanges with increased auxiliary gas flow is likely associated with the more rapid desolvation time. At higher auxiliary gas flow rates, analytes within the ESI solution transitioned to gas-phase at a more rapid rate, and thus the observed in-ESI HDX was minimal [80]. Though the increase in auxiliary gas flow rate decreases the partial pressure of D2O in the source atmosphere, we hypothesize that the HDX reaction is initiated by D2O vapor condensing in the ESI droplets [81] and initiating exchange. Thus, the mechanism of exchange is not altered by changing the auxiliary gas flow rate or the D2O partial pressure.
When the auxiliary gas flow rate was 1 arbitrary unit and the percentage of water in the methanolic spray solvent was varied from 1 to 25%, the number of H/D exchanges increased from 0.5 ± 0.2 D to 3.4 ± 0.3 D. As the percentage of water was increased to 50% in a methanolic spray solvent, the number of H/D exchanges remained consistent (3.6 ± 0.2 D) with the number of H/D exchanges measured in 25% water. However, when the solvent was altered to 100% water, the number of H/D exchanges decreased to 2.6 ± 0.1 D (see supplementary material, Fig. S4). Representative mass spectra are shown in the supplementary material, Fig. S5. Thus, at higher percentages of water in the spray solvent, the data are consistent with the trends observed without auxiliary gas flow (Figure 3a). The observation that increasing the percentage of water increases the number of H/D exchanges at auxiliary gas 1 indicates that the presence of the auxiliary gas does not hinder D2O vapor from interacting with the ESI droplets. With the increasing percentage of water in the spray solvent, the effect of the auxiliary gas in desolvation of the droplets likely becomes less prominent because water has a lower volatility than methanol and therefore, the slower desolvation results in more time for in-ESI HDX to occur in the droplets. Additionally, with an increasing percentage of water, the droplet size could also increase due to the increased surface tension of the solvent. This would also yield a longer time for in-ESI HDX. For the sample in 100% water, an increase in back exchange likely results in lower deuterium uptake compared to the other solvent mixtures.
For a sample in 99% methanol, increasing the sheath gas flow from 0 to 24 arbitrary units resulted in an increase in the number of H/D exchanges for sodiated-melezitose from 1.9 ± 0.2 D to 3.1 ± 0.5 D (Figure 3b and supplementary material, Fig. S6). Further increasing the sheath gas flow rate to 70 arbitrary units resulted in a decrease in the number of H/D exchanges to 1.4 ± 0.3 D. The sheath gas affects both the rate of desolvation and the size of the ESI droplets [82]. At low sheath gas flow rates, the droplets would be large and have a sufficient lifetime for exchange to occur. Increasing the sheath gas flow would result in smaller droplets with a greater surface area-to-volume ratio. We hypothesize that D2O vapor condenses on the surface of the droplets and diffuses through them, reacting with the analyte while it is solvated. The greater surface area-to-volume ratio of smaller droplets could result in more condensation and diffusion of D2O, enabling more HDX, and yielding a greater number of observed H/D exchanges [83]. However, at higher sheath gas flow rates, the continued increase in the rate of desolvation could decrease the size, and thus the lifetime, of the droplets, yielding fewer H/D exchange events observed for melezitose-metal adducts. Additionally, at high sheath gas flow rates, it is expected that the ion spray stability would decrease, leading to greater run-to-run variability as evidenced by the higher standard deviation for replicate measurements at sheath gas flow rates of 24 arbitrary units or higher.
Comparison of Direct Infusion Sample Flow Rates
Modifying the sample infusion flow rate was anticipated to alter the ESI droplet size [84] and therefore the droplet lifetime, which as we have already seen, can alter the time available for in-ESI HDX. Therefore, we wanted to optimize the infusion flow rate for our system. The number of H/D exchanges was measured to be 1.5 ± 0.5 D, 2.2 ± 0.1 D, and 2.5 ± 0.2 D for samples infused at 1, 5, and 20 μl/min, respectively. A flow rate of 1 μl/min resulted in higher standard deviation compared to the faster flow rates, likely due to poor spray stability and ion transmission (with the other ESI parameters kept fixed, including spray voltage + 3.5 kV, sheath gas 12 arb units, auxiliary gas 0 arb units, capillary temperature 300 °C, and sample concentration 500 μM melezitose and 250 μM of each metal ion (Na+, K+, and Mg2+) in 99:1 (v/v) methanol:water). The number of H/D exchanges measured when infusing at 5 or 20 μl/min was similar, indicating that the carbohydrate analytes underwent in-ESI HDX for similar lengths of time.
Comparing HDX for Different Sample Concentrations
Concentration is an important factor in any reaction since limiting reactants result in incomplete reactions. We examined the relationship between sample concentration and deuterium uptake. As the concentration of melezitose was increased from 62.5 to 500 μM, while maintaining a constant molar ratio of melezitose to each metal ion, the number of H/D exchanges for sodiated-melezitose decreased from 5.8 ± 0.3 D to 2.8 ± 0.1 D (Figure 3c). The samples with concentrations of 62.5 and 125 μM melezitose exhibited similar levels of H/D exchange with 5.8 ± 0.3 D and 5.6 ± 0.2 D, respectively. However, the extent of exchange for the samples with 125 μM melezitose was statistically greater than that observed for samples with either 250 μM melezitose (3.5 ± 0.2 D) or 500 μM melezitose (2.8 ± 0.1 D). Similar results were observed for all three metal-ion adducts examined in this study. Representative mass spectra are shown in supplementary material, Fig. S7. Although multimers, such as dimers and trimers of melezitose adducted to a single metal ion, were observed in the undeuterated controls, changes in the intensity of these multimers were not observed when increasing the melezitose and metal ion concentrations. Therefore, the changes in the number of H/D exchanges are unlikely to be the result of structural differences due to multimer adduct formation.
There are two possible explanations for an increase in concentration resulting in a decrease in the number of H/D exchanges. Firstly, D2O vapor may be limiting in samples with higher concentrations, reducing the average number of H/D exchanges for each molecule. Secondly, changes in concentration would lead to changes in the distribution of melezitose molecules in ESI droplets and their lifetime as solvated species. Melezitose is a small molecule that is expected to be ionized and released into the gas-phase during electrospray via the IEM [48, 82]. Upon initial formation of ESI droplets, the molecular distribution is assumed to be similar to the bulk solution; thus, a higher analyte concentration would result in a larger number of melezitose molecules per droplet and thus more molecules would be distributed near the droplet surface. A higher concentration of analyte near the surface will likely result in a higher probability of the analytes being released into the gas-phase at early time points by IEM and would shorten the time available for these molecules to undergo in-ESI HDX. In contrast, lower concentrations will likely result in fewer melezitose molecules near the surface of the droplet since carbohydrates are expected to hydrogen bond to both methanol and water, lessen the number of melezitose molecules released into the gas-phase at early time points, and thus lengthen the time available for HDX since melezitose may stay solvated for a longer duration.
Comparison of Spray Voltages
During in-ESI HDX, small changes in electrospray conditions can have significant effects on HDX due to the fast exchanging nature of hydroxyl functional groups. Thus, we examined the effect of spray voltage on H/D exchange. Water requires a spray voltage of approximately + 4 kV [77] to form a stable Taylor cone due to the high surface tension of the liquid. Alternatively, methanol has lower surface tension and requires a spray voltage of approximately + 2.2 kV to generate a stable Taylor cone [77]. However, for the 99% methanol, 50% water and 50% methanol, and 100% water solvents, the average number of H/D exchanges was similar for spray voltages between + 2.5 and + 5.5 kV (Figure 3d–f). Representative mass spectra are presented in the supplementary material as Figs. S8, S9, and S10. Thus, in this range, voltage had little effect on the magnitude of HDX. Yet, when comparing the different voltages, the magnitude of HDX was consistent with the percentage of water in the solvent.
Comparison of Melezitose Conformations in Different Spray Solvents
The Nikolaev group reported that solvent composition affects the number of carbohydrate conformations observed during in-ESI HDX [43, 44]. Their previous work focused on examining the number of distributions of peaks following in-ESI HDX for carbohydrates in a single solvent (water, acetonitrile, methanol, or ethanol) or a 50% mixture of water and methanol [44]. They observed that two conformations with different exchange rates were present when samples were sprayed in methanol, ethanol, or a 50% mixture of water and methanol. Additionally, the population that exchanged more rapidly, and thus had a higher amount of H/D exchange, was more abundant at higher temperature. However, samples in water or acetonitrile resulted in a single carbohydrate conformation [44]. In biological systems, glycans function in aqueous environments, though glycans and carbohydrates are often sprayed in mixtures of water and organic solvent for MS analyses. Thus, in an effort to characterize the biologically relevant conformations of carbohydrates, we wanted to examine how changing the percentage of methanol in a water-based solvent (a) modified the distribution of H/D exchange peaks, (b) altered the magnitude of H/D exchange, and (c) changed the carbohydrate structure.
Following in-ESI HDX, the distribution of H/D exchange peaks for each solvent mixture was fit to one or more Gaussian distributions to determine the number of sample populations with different exchange rates, and thus the number of conformers present (Figure 4 and supplementary material, Fig. S11). For samples containing, 10, 25, 50, 75, and 100% H2O, the distribution of peaks was fit to a single Gaussian distribution (R2 of 0.9981, 0.9996, 0.9982, 0.9996, and 0.9946, respectively), indicating that a single carbohydrate conformation was present. The sample in 1% water had a broad set of peaks that best fit to two Gaussian distributions (R2 of 0.9996 for fitting to two Gaussian distributions and R2 of 0.9605 for fitting a single Gaussian distribution); thus, in 99% methanol two conformations, representing two differently exchanging populations, were observed. This data shows some differences compared to that reported by the Nikolaev group. In 100% methanol, they observed a single, slow exchanging conformation based on the single distribution of peaks for a dextran trisaccharide at capillary temperatures of 300 and 200 °C. However, at 400 °C, both a slow exchanging and a fast exchanging conformation were observed [44]. In comparison, we used a spray solvent with 99% methanol and varied the capillary temperature from 250 to 350 °C (see supplementary material, Fig. S3). We observed an overall increase in the number of H/D exchanges with increasing temperature (2.7 ± 0.3 D to 3.5 ± 0.6 D). Temperatures higher than 350 °C were not used for our experiments as consecutive measurements in triplicate resulted in caramel formation on the sweep cone. Additionally, in the 99:1 (v/v) methanol:water solvent, we observed two conformations at all capillary temperatures (250, 300,and 350 °C), as the data was fit by two Gaussian distributions (R2 of 0.9997, 0.9991, and 0.9977, respectively).
MD Simulation of Carbohydrate Conformations in Varying Solvent Compositions
Since spray solvents that yield good ion signal often contain organic solvents [85], it is also crucial to observe how solvent mixtures affect the observed conformations of analytes. We performed MD simulations to examine how increasing the concentration of methanol in an aqueous solvent affected the conformation of melezitose.
We analyzed the MD trajectories, investigating the extent with which intramolecular hydrogen bonds affected the melezitose structure. A geometric criterion was specified for the occurrence of hydrogen bonds, where a hydrogen bond was considered to exist between a donor-hydrogen-acceptor group if the donor-acceptor distance was less than or equal to 0.35 nm and donor-hydrogen-acceptor angle was less than or equal to 30°. The number of intramolecular hydrogen bonds per timeframe (NHB) within the melezitose molecule increases from 1.21 hydrogen bonds in the presence of 100% water to an average of 2.15 hydrogen bonds in the presence of 99:1 (v/v) methanol:water. This increasing intramolecular hydrogen bond trend occurs as the number of hydrogen bonds per timeframe between melezitose and water decreases (18.5 to 0.67 hydrogen bonds) and that between melezitose and methanol increases (1.70 to 9.49 hydrogen bonds) (Table 1). The capacity for melezitose to form more intramolecular hydrogen bonds in the presence of methanol, as opposed to water, is due to the fact that while the hydrogen bonding with methanol increases with an increase in methanol composition, that rate of increase is less than the rate of decrease of hydrogen bonds with water.
To characterize how the change in the number of intramolecular hydrogen bonds affects the conformation of melezitose, we analyzed the solvation shell of the melezitose molecule by calculating the radial distribution functions (RDF) of melezitose with water and methanol. The radial distribution function (gAB) between two particles A and B is defined by Eq. (3).
gAB(r) is the particle density of type B around particle A at a distance of r, with 〈ρB〉local as the particle density of type B averaged over all spheres around particle A. By integrating the RDF until a specific distance, we obtained the coordination number (Cnum) of water or methanol within that distance (Table 1). Normalized RDFs for melezitose in different solvents are presented in the supplementary material, Fig. S12.
The first minimum in the RDF traces delineate the distance within which the closest water or methanol molecule lies. On average, the first minimum for the first water shell was at r = 0.38 nm while that of methanol was at r = 0.44 nm, owing to the smaller dimension of the water molecule versus that of methanol. Interestingly, we find that for melezitose in water, we obtained 1.51 ± 0.02 water molecules within the first minimum in pure water systems, while for systems with 99:1 methanol:water, we obtain 0.020 ± 0.001 water and 1.28 ± 0.05 methanol molecules at that corresponding distance (Table 1). The distance of the first minimum corresponds to a region near the center of mass of the melezitose molecule where only one water or methanol molecule could fit. We present the second coordination numbers to represent the numbers of solvent molecules where melezitose is fully solvated (Table 1). The coordination numbers of melezitose to water have a decreasing trend as you decrease the percent composition of water. With fewer water molecules within the solvation shell of melezitose as the percentage of water in the solvent decreases, there is a reduction in the number of intermolecular hydrogen bonds between melezitose and water and an increase in the number of intramolecular hydrogen bonds within melezitose. In Figure 5, we present a visual representation of the solvation of melezitose in 100% water and 1% water systems.

Snapshots of melezitose structures in (a) pure water and (b) 99:1 (v/v) methanol:water solvent systems. Structures illustrate intramolecular hydrogen bonds within melezitose (blue) and hydrogen bonds between melezitose and solvent (water or methanol in purple). Water molecules are yellow, methanol molecules are gray, and the carbon, oxygen, and hydrogen atoms of melezitose are cyan, red, and white, respectively
To observe differences in conformations of melezitose, we report the distributions of the rotation of dihedral angles about the glycosidic bonds (φ and ψ) as well as rotation about the CCOH dihedrals (ω). We did not observe meaningful differences in φ and ψ with a change in solvent (see supplementary material, Fig. S15); however, we did observe the rotameric transitions in CCOH dihedrals corresponding to the rotation of the hydroxyl groups, presented in Figure 6. The variations in those curves are more prominent for the groups that contributed the most to hydrogen bonding (see supplementary material, Fig. S13). When melezitose hydroxyl groups are not bound to water, there is less restriction on the conformation of the melezitose molecule which allows an increase in intermolecular interactions. This indicates that differences in conformation of melezitose arise because of intramolecular hydrogen bonding within melezitose, which is associated with the nature of the solvent.

Normalized probability distributions of CCOH dihedrals involving melezitose hydroxyl groups in solvents of decreasing water composition
Conclusions
In this work, we have shown that the carbohydrate-metal adduct conformations characterized by in-ESI HDX are representative of solution-phase conformations. However, these species cannot currently be distinguished as either analytes that are fully solvated in droplets or analytes residing in solvent clusters following IEM (Figure 1). We have also highlighted how ESI parameters and processes (droplet formation, solvent evaporation, and ionization) affect H/D exchange of carbohydrate-metal adducts. This fundamental knowledge regarding the relationship between ESI and HDX provides a foundation for future applications of this methodology. We have not provided a set of optimized conditions since researchers interested in carbohydrate conformations may have different goals in performing in-ESI HDX experiments, and thus, may require the use of different conditions. However, this work suggests parameters that should be considered when analyzing carbohydrates or other analytes during rapid HDX reactions in ESI droplets, including spray solvent composition, sample concentration, and gas flow rates. These findings will play a significant role in the continued development and application of in-ESI HDX to carbohydrate conformational analyses.
References
Bucior, I., Burger, M.M.: Carbohydrate–carbohydrate interactions in cell recognition. Curr. Opin. Struct. Biol. 14, 631–637 (2004)
Le Pendu, J., Nyström, K., Ruvoën-Clouet, N.: Host–pathogen co-evolution and glycan interactions. Curr. Opin. Virol. 7, 88–94 (2014)
Freeze, H.H., Kinoshita, T., Varki, A.: Glycans in acquired human diseases. In: Varki, a., Cummings, R.D., Esko, J.D., (Eds) Essentials of Glycobiology (3rd Ed.). Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY, Chapter 46. (2017)
Dube, D.H., Bertozzi, C.R.: Glycans in cancer and inflammation—potential for therapeutics and diagnostics. Nat. Rev. Drug Discov. 4, 477–488 (2005)
Qin, R., Zhao, J., Qin, W., Zhang, Z., Zhao, R., Han, J., Yang, Y., Li, L., Wang, X., Ren, S., Sun, Y., Gu, J.: Discovery of non-invasive glycan biomarkers for detection and surveillance of gastric cancer. J. Cancer. 8, 1908–1916 (2017)
Adamczyk, B., Tharmalingam, T., Rudd, P.M.: Glycans as cancer biomarkers. Biochim. Biophys. Acta. 1820, 1347–1353 (2012)
Ocho, M., Togayachi, A., Iio, E., Kaji, H., Kuno, A., Sogabe, M., Korenaga, M., Gotoh, M., Tanaka, Y., Ikehara, Y., Mizokami, M., Narimatsu, H.: Application of a Glycoproteomics-based biomarker development method: alteration in glycan structure on colony stimulating factor 1 receptor as a possible glycobiomarker candidate for evaluation of liver cirrhosis. J. Proteome Res. 13, 1428–1437 (2014)
Akinkuolie, A.O., Buring, J.E., Ridker, P.M., Mora, S.: A novel protein glycan biomarker and future cardiovascular disease events. J. Am. Heart Assoc. 3, e001221 (2014)
An, H.J., Kronewitter, S.R., de Leoz, M.L.A., Lebrilla, C.B.: Glycomics and disease markers. Curr. Opin. Chem. Biol. 13, 601–607 (2009)
Bertozzi, C.R., Rabuka, D.: Structural basis of glycan diversity. In: Varki, A., Cummings, R.D., Esko, J.D., Freeze, H.H., Stanley, P., Bertozzi, C.R., Hart, G.W., Etzler, M.E. (eds.) Essentials of Glycobiology (2nd Ed.). Cold Spring Harbor Laboratory Press, p. 2. Chapter, Cold Spring Harbor (2009)
Kizuka, Y., Kitazume, S., Fujinawa, R., Saito, T., Iwata, N., Saido, T.C., Nakano, M., Yamaguchi, Y., Hashimoto, Y., Staufenbiel, M., Hatsuta, H., Murayama, S., Manya, H., Endo, T., Taniguchi, N.: An aberrant sugar modification of BACE1 blocks its lysosomal targeting in Alzheimer disease. EMBO Mol Med. 7, 175 (2015)
Nagae, M., Kanagawa, M., Morita-Matsumoto, K., Hanashima, S., Kizuka, Y., Taniguchi, N., Yamaguchi, Y.: Atomic visualization of a flipped-back conformation of bisected glycans bound to specific lectins. Sci. Rep. 6, 22973 (2016)
Nagae, M., Yamaguchi, Y.: Function and 3D structure of the N-glycans on glycoproteins. Int. J. Mol. Sci. 13, 8398–8429 (2012)
Wormald, M.R., Petrescu, A.J., Pao, Y.-L., Glithero, A., Elliott, T., Dwek, R.A.: Conformational studies of oligosaccharides and glycopeptides: complementarity of NMR, X-ray crystallography, and molecular modelling. Chem. Rev. 102, 371–386 (2002)
Thomas, W.A.: Nuclear magnetic resonance spectroscopy in conformational analysis. Annu. Rev. NMR Spectrosc., academic press, Cambridge. MA. 1, 43–89 (1968)
Bohrer, B.C., Merenbloom, S.I., Koeniger, S.L., Hilderbrand, A.E., Clemmer, D.E.: Biomolecule analysis by ion mobility spectrometry. Annu. Rev. Anal. Chem. 1, 293–327 (2008)
Verbeck, G., Ruotolo, B., Sawyer, H., Gillig, K., Russell, D.: A fundamental introduction to ion mobility mass spectrometry applied to the analysis of biomolecules. J. Biomol. Tech. 13, 56–61 (2002)
Englander, S.W.: Hydrogen exchange and mass spectrometry: a historical perspective. J. Am. Soc. Mass Spectrom. 17, 1481–1489 (2006)
Engen, J.R.: Analysis of protein conformation and dynamics by hydrogen/deuterium exchange MS. Anal. Chem. 81, 7870–7875 (2009)
Gallagher, E.S., Hudgens, J.W.: Mapping protein–ligand interactions with proteolytic fragmentation, hydrogen/deuterium exchange-mass spectrometry in:Kelman, Z (eds.)methods in enzymology pg 357-404, academic press, Cambridge (MA). In: 566 (2016)
Huang, Y., Dodds, E.D.: Ion mobility studies of carbohydrates as group I adducts: isomer specific collisional cross section dependence on metal ion radius. Anal. Chem. 85, 9728–9735 (2013)
Huang, Y., Dodds, E.D.: Discrimination of isomeric carbohydrates as the electron transfer products of group II cation adducts by ion mobility spectrometry and tandem mass spectrometry. Anal. Chem. 87, 5664–5668 (2015)
Clowers, B.H., Dwivedi, P., Steiner, W.E., Hill, H.H., Bendiak, B.: Separation of sodiated isobaric disaccharides and trisaccharides using electrospray ionization-atmospheric pressure ion mobility-time of flight mass spectrometry. J. Am. Soc. Mass Spectrom. 16, 660–669 (2005)
Fenn, L.S., McLean, J.A.: Structural resolution of carbohydrate positional and structural isomers based on gas-phase ion mobility-mass spectrometry. Phys. Chem. Chem. Phys. 13, 2196–2205 (2011)
Li, H., Giles, K., Bendiak, B., Kaplan, K., Siems, W.F., Hill, H.H.: Resolving structural isomers of monosaccharide methyl glycosides using drift tube and traveling wave ion mobility mass spectrometry. Anal. Chem. 84, 3231–3239 (2012)
Pettit, M.E., Brantley, M.R., Donnarumma, F., Murray, K.K., Solouki, T.: Broadband ion mobility deconvolution for rapid analysis of complex mixtures. Analyst. 143, 2574–2586 (2018)
Kirschner, K.N., Woods, R.J.: Solvent interactions determine carbohydrate conformation. Proc. Natl. Acad. Sci. 98, 10541–10545 (2001)
Kaltashov, I.A., Bobst, C.E., Abzalimov, R.R., Berkowitz, S.A., Houde, D.: Conformation and dynamics of biopharmaceuticals: transition of mass spectrometry-based tools from academe to industry. J. Am. Soc. Mass Spectrom. 21, 323–337 (2010)
Kleckner, I.R., Foster, M.P.: An introduction to NMR-based approaches for measuring protein dynamics. Biochim. Biophys. Acta. 1814, 942–968 (2011)
Lindner, R., Heintz, U., Winkler, A.: Applications of hydrogen deuterium exchange (HDX) for the characterization of conformational dynamics in light-activated photoreceptors. Front. Mol. Biosci. 2, 33 (2015)
Guttman, M., Scian, M., Lee, K.K.: Tracking hydrogen/deuterium exchange at glycan sites in glycoproteins by mass spectrometry. Anal. Chem. 83, 7492–7499 (2011)
Mao, D., Douglas, D.J.: H/D exchange of gas phase bradykinin ions in a linear quadrupole ion trap. J. Am. Soc. Mass Spectrom. 14, 85–94 (2003)
Green-Church, K.B., Limbach, P.A., Freitas, M.A., Marshall, A.G.: Gas-phase hydrogen/deuterium exchange of positively charged mononucleotides by use of Fourier-transform ion cyclotron resonance mass spectrometry. J. Am. Soc. Mass Spectrom. 12, 268–277 (2001)
Miladi, M., Olaitan, A.D., Zekavat, B., Solouki, T.: Competing noncovalent host-guest interactions and H/D exchange: reactions of benzyloxycarbonyl-proline glycine dipeptide variants with ND3. J. Am. Soc. Mass Spectrom. 26, 1938–1949 (2015)
Zekavat, B., Miladi, M., Al-Fdeilat, A.H., Somogyi, A., Solouki, T.: Evidence for sequence scrambling and divergent H/D exchange reactions of doubly-charged isobaric b-type fragment ions. J. Am. Soc. Mass Spectrom. 25, 226–236 (2014)
Nagy, K., Redeuil, K., Rezzi, S.: Online hydrogen/deuterium exchange performed in the ion mobility cell of a hybrid mass spectrometer. Anal. Chem. 81, 9365–9371 (2009)
Mistarz, U.H., Brown, J.M., Haselmann, K.F., Rand, K.D.: Probing the binding interfaces of protein complexes using gas-phase H/D exchange mass spectrometry. Structure. 24, 310–318 (2016)
Uppal, S.S., Beasley, S.E., Scian, M., Guttman, M.: Gas-phase hydrogen/deuterium exchange for distinguishing isomeric carbohydrate ions. Anal. Chem. 89, 4737–4742 (2017)
Takáts, Z., Schlosser, G., Vékey, K.: Hydrogen/deuterium exchange of electrosprayed ions in the atmospheric interface of a commercial triple–quadrupole mass spectrometer. Int. J. Mass Spectrom. 228, 729–741 (2003)
Wolff, J.C., Laures Alice, M.F.: ‘On-the-fly’ hydrogen/deuterium exchange liquid chromatography/mass spectrometry using a dual-sprayer atmospheric pressure ionisation source. Rapid Commun. Mass Spectrom. 20, 3769–3779 (2006)
Kostyukevich, Y., Kononikhin, A., Popov, I., Nikolaev, E.: Simple atmospheric hydrogen/deuterium exchange method for enumeration of labile hydrogens by electrospray ionization mass spectrometry. Anal. Chem. 85, 5330–5334 (2013)
Tittebrandt, S., Edelson-Averbukh, M., Spengler, B., Lehmann, W.D.: ESI hydrogen/deuterium exchange can count chemical forms of heteroatom-bound hydrogen. Angew. Chem. Int. Ed. 52, 8973–8975 (2013)
Kostyukevich, Y., Kononikhin, A., Popov, I., Nikolaev, E.: In-ESI source hydrogen/deuterium exchange of carbohydrate ions. Anal. Chem. 86, 2595–2600 (2014)
Kostyukevich, Y., Kononikhin, A., Popov, I., Nikolaev, E.: Conformations of cationized linear oligosaccharides revealed by FTMS combined with in-ESI H/D exchange. J. Mass Spectrom. 50, 1150–1156 (2015)
Zherebker, A., Kostyukevich, Y., Kononikhin, A., Roznyatovsky, V.A., Popov, I., Grishin, Y.K., Perminova, I.V., Nikolaev, E.: High desolvation temperature facilitates in ESI-source H/D exchange at non-labile sites of hydroxybenzoic acids and aromatic amino acids. Analyst. 141, 2426–2434 (2016)
Acter, T., Lee, S., Cho, E., Jung, M.-J., Kim, S.: Design and validation of in-source atmospheric pressure photoionization hydrogen/deuterium exchange mass spectrometry with continuous feeding of D2O. J. Am. Soc. Mass Spectrom. 29, 85–94 (2018)
Ahmed, A., Choi, C., Kim, S.: Mechanistic study on lowering the sensitivity of positive atmospheric pressure photoionization mass spectrometric analyses: size-dependent reactivity of solvent clusters. Rapid Commun. Mass Spectrom. 29, 2095–2101 (2015)
Konermann, L., Ahadi, E., Rodriguez, A.D., Vahidi, S.: Unraveling the mechanism of electrospray ionization. Anal. Chem. 85, 2–9 (2013)
Iribarne, J.V., Thomson, B.A.: On the evaporation of small ions from charged droplets. J. Chem. Phys. 64, 2287–2294 (1976)
Znamenskiy, V., Marginean, I., Vertes, A.: Solvated ion evaporation from charged water Nanodroplets. J. Phys. Chem. A. 107, 7406–7412 (2003)
Ahadi, E., Konermann, L.: Ejection of solvated ions from Electrosprayed methanol/water Nanodroplets studied by molecular dynamics simulations. J. Am. Chem. Soc. 133, 9354–9363 (2011)
Kim, H.J., Liyanage, O.T., Mulenos, M.R., Gallagher, E.S.: Mass spectral detection of forward- and reverse-hydrogen/deuterium exchange resulting from residual solvent vapors in electrospray sources. J. Am. Soc. Mass Spectrom. 29, 2030–2040 (2018)
MATLAB: And statistics toolbox release 2016a, the MathWorks, Inc., Natick, Massachusetts. United States.
Hess, B., Kutzner, C., van der Spoel, D., Lindahl, E.: GROMACS 4: algorithms for highly efficient, load-balanced, and scalable molecular simulation. J. Chem. Theory Comput. 4, 435–447 (2008)
Guvench, O., Mallajosyula, S.S., Raman, E.P., Hatcher, E., Vanommeslaeghe, K., Foster, T.J., Jamison, F.W., MacKerell, A.D.: CHARMM additive all-atom force field for carbohydrate derivatives and its utility in polysaccharide and carbohydrate–protein modeling. J. Chem. Theory Comput. 7, 3162–3180 (2011)
Sterling, T., Irwin, J.J.: ZINC 15—ligand discovery for everyone. J. Chem. Inf. Model. 55, 2324–2337 (2015)
Berendsen, H.J.C., Grigera, J.R., Straatsma, T.P.: The missing term in effective pair potentials. J. Phys. Chem. 91, 6269–6271 (1987)
Bussi, G., Donadio, D., Parrinello, M.: Canonical sampling through velocity rescaling. J. Chem. Phys. 126, 014101 (2007)
Parrinello, M., Rahman, A.: Polymorphic transitions in single crystals: a new molecular dynamics method. J. Appl. Phys. 52, 7182–7190 (1981)
Nosé, S., Klein, M.L.: Constant pressure molecular dynamics for molecular systems. Mol. Phys. 50, 1055–1076 (1983)
Verlet, L.: Computer "experiments" on classical fluids. I. Thermodynamical Properties of Lennard-Jones Molecules. Phys. Rev. 159, 98–103 (1967)
Hess, B.: P-LINCS: a parallel linear constraint solver for molecular simulation. J. Chem. Theory Comput. 4, 116–122 (2008)
Campbell, S., Rodgers, M.T., Marzluff, E.M., Beauchamp, J.L.: Deuterium exchange reactions as a probe of biomolecule structure. fundamental studies of gas phase H/D exchange reactions of protonated glycine oligomers with D2O, CD3OD, CD3CO2D, and ND3. J. Am. Chem. Soc. 117, 12840–12854 (1995)
Ausloos, P., Lias, S.G.: Thermoneutral isotope-exchange reactions of cations in the gas phase. J. Am. Chem. Soc. 103, 3641–3647 (1981)
Gard, E., Green, M.K., Bregar, J., Lebrilla, C.B.: Gas-phase hydrogen/deuterium exchange as a molecular probe for the interaction of methanol and protonated peptides. J. Am. Soc. Mass Spectrom. 5, 623–631 (1994)
Wyttenbach, T., Bowers, M.T.: Gas phase conformations of biological molecules: the hydrogen/deuterium exchange mechanism. J. Am. Soc. Mass Spectrom. 10, 9–14 (1999)
Evans, S.E., Lueck, N., Marzluff, E.M.: Gas phase hydrogen/deuterium exchange of proteins in an ion trap mass spectrometer. Int. J. Mass Spectrom. 222, 175–187 (2003)
Winkler, H.D.F., Dzyuba, E.V., Sklorz, J.A.W., Beyeh, N.K., Rissanen, K., Schalley, C.A.: Gas-phase H/D-exchange reactions on resorcinarene and pyrogallarene capsules: proton transport through a one-dimensional Grotthuss mechanism. Chem. Sci. 2, 615–624 (2011)
Hunter, E.P.L., Lias, S.G.: Evaluated gas phase Basicities and proton affinities of molecules: an update. J. Phys. Chem. Ref. Data. 27, 413–656 (1998)
Zhang, J., Brodbelt, J.S.: Gas-phase hydrogen/deuterium exchange and conformations of deprotonated flavonoids and gas-phase acidities of flavonoids. J. Am. Chem. Soc. 126, 5906–5919 (2004)
Tian, Z., Reed, D.R., Kass, S.R.: H/D exchange pathways: Flip-flop and relay processes. Int. J. Mass Spectrom. 377, 130–138 (2015)
Reid, G.E., O’Hair, R.A.J., Styles, M.L., McFadyen, W.D., Simpson, R.J.: Gas phase ion–molecule reactions in a modified ion trap: H/D exchange of non-covalent complexes and coordinatively unsaturated platinum complexes. Rapid Commun. Mass Spectrom. 12, 1701–1708 (1999)
Reyzer, M.L., Brodbelt, J.S.: Gas-phase H/D exchange reactions of polyamine complexes: (M + H)+, (M + alkali metal+), and (M + 2H)2+. J. Am. Soc. Mass Spectrom. 11, 711–721 (2000)
Kaltashov, I.A., Vladimir, D.M., Cotter, R.J.: Gas phase hydrogen/deuterium exchange reactions of peptide ions in a quadrupole ion trap mass spectrometer. Proteins: structure. Function, and Bioinformatics. 28, 53–58 (1997)
Solouki, T., Fort, R.C., Alomary, A., Fattahi, A.: Gas phase hydrogen deuterium exchange reactions of a model peptide: FT-ICR and computational analyses of metal induced conformational mutations. J. Am. Soc. Mass Spectrom. 12, 1272–1285 (2001)
Kostiainen, R., Kauppila, T.J.: Effect of eluent on the ionization process in liquid chromatography–mass spectrometry. J. Chromatogr. A. 1216, 685–699 (2009)
Kebarle, P., Verkerk Udo, H.: Electrospray: from ions in solution to ions in the gas phase, what we know now. Mass Spectrom. Rev. 28, 898–917 (2009)
Zhou, S., Cook, K.D.: Probing solvent fractionation in electrospray droplets with laser-induced fluorescence of a Solvatochromic dye. Anal. Chem. 72, 963–969 (2000)
Lu, H.M., Jiang, Q.: Size-dependent surface tension and Tolman's length of droplets. Langmuir. 21, 779–781 (2005)
Kashiwagi, T., Yamada, N., Hirayama, K., Suzuki, C., Kashiwagi, Y., Tsuchiya, F., Arata, Y., Kunishima, N., Morikawa, K.: An electrospray-ionization mass spectrometry analysis of the pH-dependent dissociation and denaturation processes of a heterodimeric protein. J. Am. Soc. Mass Spectrom. 11, 54–61 (2000)
Wang, R., Zenobi, R.: Evolution of the solvent polarity in an electrospray plume. J. Am. Soc. Mass Spectrom. 21, 378–385 (2010)
Banerjee, S., Mazumdar, S.: Electrospray ionization mass spectrometry: a technique to access the information beyond the molecular weight of the Analyte. Int. J. Anal. Chem. 2012, 282574 (2012)
Li, Z., Li, L.: Chemical-vapor-assisted electrospray ionization for increasing Analyte signals in electrospray ionization mass spectrometry. Anal. Chem. 86, 331–335 (2014)
Schmidt, A., Karas, M., Dülcks, T.: Effect of different solution flow rates on analyte ion signals in nano-ESI MS, or: when does ESI turn into nano-ESI? J. Am. Soc. Mass Spectrom. 14, 492–500 (2003)
Thacker, J.B., Schug, K.A.: Effects of solvent parameters on the electrospray ionization tandem mass spectrometry response of glucose. Rapid Commun. Mass Spectrom. 32, 1191–1198 (2018)
Acknowledgements
O.T.L. and E.S.G. were supported by The Welch Foundation under award number AA-1899. M.R.B. and T.S. were supported by NSF IDBR award number 1455668. E.I.C. and K.L.S. were supported by the Chemical Sciences, Geosciences, and Biosciences Division, Office of Basic Energy Sciences, Office of Science, U.S. Department of Energy under Award Number DE-SC0019327. We thank the Baylor Mass Spectrometry Center (MSC) for providing instruments and resources used to collect the data presented here. We thank Baylor University’s High Performance and Research Computing Services (HPRCS) for technical support and access to the cluster (Kodiak). We thank H. Jamie Kim and Ian G. M. Anthony for assistance with solution-phase HDX experiments and data presentation software, respectively.
Author information
Authors and Affiliations
Corresponding author
Electronic Supplementary Material
ESM 1
(DOCX 11.6 mb)
Rights and permissions
About this article

Cite this article
Liyanage, O.T., Brantley, M.R., Calixte, E.I. et al. Characterization of Electrospray Ionization (ESI) Parameters on In-ESI Hydrogen/Deuterium Exchange of Carbohydrate-Metal Ion Adducts. J. Am. Soc. Mass Spectrom. 30, 235–247 (2019). https://doi.org/10.1007/s13361-018-2080-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13361-018-2080-1