
Abstract
Untargeted omics analyses aim to comprehensively characterize biomolecules within a biological system. Changes in the presence or quantity of these biomolecules can indicate important biological perturbations, such as those caused by disease. With current technological advancements, the entire genome can now be sequenced; however, in the burgeoning fields of lipidomics, only a subset of lipids can be identified. The recent emergence of high resolution tandem mass spectrometry (HR-MS/MS), in combination with ultra-high performance liquid chromatography, has resulted in an increased coverage of the lipidome. Nevertheless, identifications from MS/MS are generally limited by the number of precursors that can be selected for fragmentation during chromatographic elution. Therefore, we developed the software IE-Omics to automate iterative exclusion (IE), where selected precursors using data-dependent topN analyses are excluded in sequential injections. In each sequential injection, unique precursors are fragmented until HR-MS/MS spectra of all ions above a user-defined intensity threshold are acquired. IE-Omics was applied to lipidomic analyses in Red Cross plasma and substantia nigra tissue. Coverage of the lipidome was drastically improved using IE. When applying IE-Omics to Red Cross plasma and substantia nigra lipid extracts in positive ion mode, 69% and 40% more molecular identifications were obtained, respectively. In addition, applying IE-Omics to a lipidomics workflow increased the coverage of trace species, including odd-chained and short-chained diacylglycerides and oxidized lipid species. By increasing the coverage of the lipidome, applying IE to a lipidomics workflow increases the probability of finding biomarkers and provides additional information for determining etiology of disease.

ᅟ
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Lipids serve numerous functions in organisms, and the understanding of these roles has been shown to be important in the characterization of human disease, health, and nutrition. Lipids serve as membrane structure and functional components, modulators of immune system function [1], energy storage molecules, signaling molecules [2–4], and targets of oxidation [5]. Perturbations in these biological functions frequently correlate with shifts in lipid composition. Therefore, by examining the lipidome, scientists can identify new diagnostic markers that could increase our understanding of disease etiology, detection, and progression. Recently, biomedical-related international initiatives have inrcreased the awareness and methods for lipidomics (i.e., the measurement of all lipids within a biological substrate of interest using mass spectrometry) including Japan’s LipidBank founded in 1989 [6], Lipid Metabolites and Pathways Strategy (LIPID MAPS) in the USA founded in 2003 [7, 8], and the European Lipidomics Initiative: shaping the life sciences (ELIFE) founded in 2004 [9].
Comprehensive characterization of the lipidome in biological systems is challenging due to the diversity and quantity of lipid structures, wide range of biological concentrations, and subtle, but biologically important, differences in isomeric lipid structures. Lipid diversity stems from the numerous possible combinations of “backbones,” such as glycerol or sphingosine, with various fatty acids differing in carbon length, number, and positions of unsaturation. The number of lipids present in LIPID MAPS structural databases is over 40,000 [10], while over 300,000 possible lipid structures have been simulated in our in-house lipid identification workflow, LipidMatch, which takes into account oxidized species. Lipids have been shown to differ in concentrations of over seven orders of magnitude between classes; even within a lipid class, lipid concentrations can differ over three orders of magnitude [11–13]. Electrospray ionization mass spectrometry (ESI-MS) is the most widely employed ionization strategy for lipidomics [14] because its ability to ionize the diverse range of structures and concentrations.
However, isomeric species, lipid species containing different fatty acid constituents with the same total number of carbons and degrees of unsaturation [e.g., PC(16:0_20:1) and PC(18:0_18:1)], cannot be separated using ESI-MS alone. If this structural detail is desired, one solution is to employ liquid chromatography to separate the isomers for quantification based on polarity using reverse phase chromatography, in combination with tandem mass spectrometry (MS/MS) to identify the fatty acid constituents based on fragmentation patterns. However, this strategy often leads to a conundrum, as increasing the peak capacity in liquid chromatography to deconvolute more lipid features for quantification requires narrower chromatographic peaks, thus limiting the number of MS/MS scans that can be obtained across the chromatographic peaks [15]. Therefore, within lipid-rich retention time and m/z regimes, numerous ions of different mass-to-charge ratios will be ionized at the same elution time, but only a few can be selected for fragmentation in a single injection. For lipidomic experiments where the lipids of interest are unknown, heuristic rules have been developed to fragment ions. One approach is to select ions with the highest intensity for fragmentation, commonly referred to as data-dependent topN (ddMS2-topN). Owing to concentration bias, this strategy could miss important less abundant lipid species, such as diacylglycerides and phosphatidylinositols in plasma, which are both important signal molecule classes [16, 17].
A strategy that overcomes the drawback of traditional ddMS2-topN, in terms of the limited number of MS/MS spectra acquired at any given retention time, is to continuously repeat ddMS2-topN analysis on the same sample, excluding previously selected precursors ions in each sequential analysis. Theoretically, iterative repeat injections can be used to acquire MS/MS of all precursor ions above background signal, providing a substantial wealth of information for identification. A schematic of this technique, iterative exclusion (IE), is shown in Figure 1. Although still uncommon, this technique has been applied in the proteomics community. In 2009, Bendall et al. designed a proteomics software approach for their strategy termed iterative exclusion-mass spectrometry (IE-MS) [18], which excluded all ions in previous runs regardless of assignment. Using this technique, Bendall et al. identified 30% more unique proteins after five IE-MS acquisitions, compared with five repetitive traditional data-dependent scans. Rudomin et al. [19] were also able to identify 49% more unique proteins using this strategy. IE has been applied to LC/MS approaches with both LTQ-Orbitrap [20] and qTOF platforms [18] and has been shown to be advantageous for proteomic applications. Examples employing this approach include using IE to study post-translational modifications of proteins [21], discover previously unknown human embryonic growth promoters [18], identify genital track markers [20], characterize Matrigel marketed as a basement membrane matrix for stem cell growth [22], and track pH-induced protein changes [23]. In all these applications, IE enabled the ability to characterize a greater variety of proteins, including trace proteins.

Strategy for iterative exclusion (IE) based data-dependent topN analysis (IE-ddMS2-topN). Multiple injections of a sample are analyzed. The first injection (a) is used to create an exclusion list that is applied for the second injection (b). Hence, the next top N most abundant ions are selected and this process can be continued for N injections
Although numerous applications have shown the benefit of using IE across platforms for proteomics, most omics analyses do not take full advantage of IE. In part, this may be due to a lack of a simple software program capable of generating exclusion lists automatically as traditionally this is achieved manually. Furthermore, using available software, iterative acquisitions cost time and money, thus putting heavy emphasis on determining whether additional sample injections for IE are worth added instrument time. To this end, we have developed an R-script named “IE-omics” for generating exclusion lists from open-source formatted data easily converted from various vendor formats; as a demonstration, we have applied it to lipidomics. IE-Omics is advantageous over the IE-MS script in providing multiple user parameters in a relatively simple interface and providing the ability to directly import multiple vendor formats.
Recently, IE has been adapted to other omics fields, such as in lipidomics and metabolomics, as noted in Sandra et al. [24] and Edmands et al. [25], respectively. For lipidomics, IE type analyses have been used in direct infusion approaches by increasing the duration of dynamic exclusion to the length of the analysis. For example, Nazari and Muddiman [26] used gas-phase fractionation and dynamic exclusion to increase coverage of the lipidome, especially of low abundance species. Schwudke et al. [27] emulated precursor and neutral loss scanning using data-dependent analysis with a dynamic exclusion and inclusion list-based workflow for increasing the lipidome coverage. The success of IE type approaches for direct infusion supports the utility of IE for LC-MS/MS. While direct infusion allows rapid biomarker discovery, LC-MS/MS has been found to be more comprehensive [28].
To our knowledge, no research has shown the benefit of applying IE versus traditional approaches for lipidomics using LC-MS/MS. In addition, no omics studies have compared results across different matrices with varying amounts of features. Herein, we report the use of our user customizable R-script for IE to lipid extracts of both Red Cross plasma and substantia nigra brain tissue in both positive and negative polarity. The results show that due to the spectral density of numerous lipid species in a chromatographic run, especially in positive ion mode, a substantial benefit is obtained using IE for LC-MS/MS based lipidomics.
Experimental
Chemicals and Materials
Ammonium acetate and all analytical grade solvents (formic acid, chloroform, and methanol) were purchased from Fisher Scientific (Waltham, MA, USA). All mobile-phase solvents were Fisher Optima LC/MS grade (acetonitrile, isopropanol, and water). For Red Cross plasma, the following lipid standards were used: triglyceride [TG(15:0/15:0/15:0) and TG(17:0/17:0/17:0)] purchased from Sigma-Aldrich (St. Louis, MO, USA), and lysophosphatidylcholine [LPC(17:0) and LPC(19:0)], phosphatidylcholine [PC(17:0/17:0) and PC(19:0/19:0)], phosphatidylethanolamine [PE(15:0/15:0) and PE(17:0/17:0)], phosphatidylserine [PS(14:0/14:0) and PS(17:0/17:0)], and phosphatidylglycerol [PG(14:0/14:0) and PG(17:0/17:0)] purchased from Avanti Polar Lipids (Alabaster, AL, USA). For substantia nigra samples, the following standards were used: TG(15:0/15:0/15:0) from Sigma-Aldrich, and PC(19:0/19:0), DG(14:0/14:0), SM(d18:1/17:0), Cer(d18:1/17:0), 13C2-cholesterol, PE(15:0/15:0), LPC(19:0), PG(14:0/14:0), and PS(14:0/14:0) purchased from Avanti Polar Lipids. All lipid standards were diluted prior to analysis in 1:2 (v/v) chloroform:methanol and a working 100 mg/L standard mix was then prepared by diluting the stock solution with the same solvent mixture.
Sample Preparation
Pooled Red Cross human EDTA plasma was purchased from the American Red Cross National Testing Laboratories (Detroit, MI, USA) and samples were stored at –80 °C. All plasma aliquots (40 μL) were thawed on ice prior to extraction.
Substantia nigra samples were obtained from C57BL/6 mice. The Institutional Animal Care and Use Committee (IACUC # 20148382) at the University of Florida approved the use of all mice and procedures. The mice were housed with a 12 h light-12 h dark schedule and were provided food and water ad libitum. Five-month-old mice were anesthetized using isoflurane vapors. The mice were sacrificed and whole brains were harvested immediately from the skull and placed on a glass petri dish. A scalpel was used to carefully remove the substantia nigra region of the brain. Upon receipt, the tissue was placed in a freezer maintained at –80 °C for storage. A Bel-Art mortar (Bel-Art Scienceware, Wayne, NJ, USA) was used to pulverize the frozen tissue samples under liquid nitrogen condition. The frozen tissue powder (10–20 mg, in triplicate) was weighed in homogenization tubes containing zirconium beads (0.7 mm diameter; BioSpec Products, Bartlesville, OK, USA).
Both Red Cross plasma and frozen substantia nigra tissue powder were extracted using the Folch method (2:1, v/v, chloroform:methanol) [29]. Briefly, 5 μL of internal standard (IS) mix (100 mg/L) was spiked into the Red Cross blood plasma (40 μL) on ice (IS info in Chemicals and Materials). For Red Cross plasma, 160 μL of methanol and 320 μL of chloroform were added to all samples. Samples were incubated on ice for 30 min and centrifuged at 4 °C for 5 min at 15,000 rpm. To induce phase separation, 150 μL of water was added and samples were incubated on ice for an additional 10 min. The organic layer was removed and the aqueous layer was re-extracted with 250 μL of chloroform:methanol (2:1, v/v). The organic layers were combined, evaporated under nitrogen, and reconstituted in 100 μL of isopropanol (for lipidomics). For substantia nigra tissue, the ground tissue was homogenized for 120 s, with 100 μL of methanol and 200 μL of chloroform for every 15 mg of tissue. Five μL of IS mix (100 mg/L) was spiked into the chloroform:methanol (2:1, v/v) mixture before homogenization. Samples were incubated as for plasma, and water was added at a volume of one-fourth of the Folch solvent. Substantia nigra was re-extracted with 50 μL of methanol and 100 μL of chloroform for every 15 mg of tissue and dried down. Substantia nigra samples were reconstituted in 200 μL of isopropanol.
Data Acquisition
For both lipidomics analyses, a Dionex Ultimate 3000 RS UHLPC system (Thermo Scientific, San Jose, CA, USA) was employed. Ionization was performed with heated electrospray ionization probe (HESI II) and mass spectra acquired using a Q-Exactive Orbitrap (Thermo Scientific). Source parameters for lipidomics, in positive and negative polarity, are provided in Supplementary Table S-3. Samples were maintained at 4 °C in the autosampler. Two μL of sample was injected onto a Waters Acquity BEH C18 column (50 mm × 2.1 mm, 1.7 μm, Waters, Milford, MA, USA) maintained at 30 °C. For negative ion mode, 5 μL of sample was injected onto the column and analyzed with the same mass spectral parameters (Supplementary Table S-2). A gradient ramp (Supplementary Table S-1) was employed consisting of mobile phase C (60:40 acetonitrile:water, volume fraction) and mobile phase D (90:8:2 isopropanol:acetonitrile:water, volume fraction), both with 10 mmol/L ammonium formate and 0.1% formic acid.
Mass spectra were acquired in full scan mode using data-dependent top5 analysis (ddMS2-top5) in both positive and negative polarity with a mass resolution of 70,000. Full scan and ddMS2-top5 scan parameters are shown in Supplementary Table S-2. Before each analysis, the instrument was externally calibrated and at least three blanks were analyzed. Internal mass calibrants (lock masses) were used in positive ion mode and consisted of diisooctyl phthalate (m/z 391.2842) and polysiloxanes (m/z 371.1012 and 445.1200). No stable lock mass was observed to be used in negative ion mode. To compare IE with traditional ddMS2-top5 for lipidomics, a minimum of four sequential injections were analyzed by ddMS2-top5 with IE and four without IE, for both negative and positive polarity analysis of Red Cross and substantia nigra lipid extracts. For excluding ions previously selected for fragmentation and placed on an exclusion list, a 10 ppm exclusion tolerance was used.
Software Platform for IE
A software program “IE-Omics” was written using R [30] to directly intake a .ms2 file converted using MSConvert [31] and output an exclusion list (.csv) in a format that can be directly imported by Q-Exactive instruments. User-defined parameters include the retention time and m/z window for combining selected precursors to reduce the size of the exclusion list. A 0.02 m/z window and 0.3 min retention time window was used in this experiment. In this case, ions selected at m/z values of 400.02, 400.01, and 400.01, and respective retention times of 5.10, 5.15, and 5.30 min, would be combined in one row as m/z 400.02 excluded between 4.95 and 5.45 min. In addition, users can denote the number of times ions with the same m/z are selected before being considered background ions and excluded for the entire duration of the chromatographic run. In this experiment, a minimum of 15 instances of ions selected for fragmentation with the same m/z was used, excluding these ions from 0 to 18 min. The IE-Omics script can be found in the Supplementary Information and the most up-to-date version on the Southeast Center for Integrated Metabolomics (SECIM) webpage (http://secim.ufl.edu/secim-tools/).
Feature Detection and Identification
Lipids were identified using both an in-house workflow, LipidMatch, and LipidSearch (Thermo Scientific, San Jose, CA, USA) [32]. LipidMatch consists of R-scripts which identify lipids by matching MS/MS fragments indicative of class and fatty-acid constituents from experimental fragmentation to in-silico fragmentation libraries (covering over 250,000 lipid species across 56 lipid types). Only exact mass of the MS/MS fragments (not intensity) is used for matching. Fragment criteria used for identification of lipids by LipidMatch can be found in Table S-6. Before LipidMatch was applied, features were determined using MZmine 2.0 [33], with the batch mode file containing all the parameters. Both the MZmine batch file and LipidMatch software can be found at <http://secim.ufl.edu/secim-tools/> in the LipidMatch zip file. LipidMatch used the features exact mass determined by MZmine with a 10 ppm m/z window for precursor ion matching. Both for LipidMatch and LipidSearch, a 10 ppm m/z window was used for fragment matching. In LipidMatch, fragments were only considered confirmed if they were above 1000 intensity units and found in at least one scan within a 0.3 min window of the feature being identified; in LipidSearch, only lipids classified with grade A were kept. The 0.3-min retention time used for finding MS/MS scans and excluding precursors in sequential injections employing the IE-Omics script was close to the median of the full width at half maximum (FWHM) for all features (Supplementary Figure S-1). After lipid ions were annotated, redundant annotations, for example, different lipid ion adducts of the same molecular species, were combined separately for positive and negative analysis.
Results and Discussion
By only fragmenting ions not selected in the previous injections, applying IE increased the coverage of both analyte and background ions for lipids (Figures 2 and 3). As can be seen, for example for the background ion m/z 300.2253 in Figure 2, some ions selected in the first injection and placed on an exclusion list were unexpectedly selected in the second injection. The reason for these ions not being excluded is that the mass trigger used to select ions for fragmentation is stored with a m/z value with two decimal digits in the Thermo.raw file. Therefore, the m/z 300.23 was placed on an exclusion list, and using a 10 ppm exclusion tolerance ions from 300.2270 to 300.2330 were excluded. In the second injection, the ion was measured at 300.2253 and therefore was not excluded, and again was placed on an exclusion list at 300.23. This problem can be overcome by either changes in Thermo.raw data storage, in order to store the mass trigger for obtaining MS/MS past the third decimal point (which has been implemented in the Q-Exactive Plus and HF), or by the user increasing the exclusion tolerance, such as to 100 ppm. Since different ions with m/z values within 100 ppm will all be isolated and fragmented using a 1 Da isolation window, this solution would be sufficient. Either modification would ensure that ions isolated and fragmented once are never isolated and fragmented again, thereby decreasing the number of injections needed to fragment all ions of interest.

Selected precursor ions retention time and m/z for Red Cross plasma compared between the first injection (black dots) and second injection (red dots) with IE ddMS2 applied. The Y region is an unknown region with molecules separated by 14 Da corresponding to CH2 repeating units, likely representing polymer species. Three background ions are indicated with arrows, which were selected at m/z 391.28, 354.29, and 303.23 from highest to lowest mass, respectively

Selected precursor ions m/z and retention times for six repetitive injections using the traditional ddMS2 approach (a) and iterative based-exclusion ddMS2 (IE-ddMS2) (b) for Red Cross plasma lipid extracts analyzed in positive mode. The higher density of selected precursor ions in (b) shows selection of precursor ions using IE, which were not fragmented using the traditional ddMS2 approach
It is well-known that background ions compete with analytes of interest for selection and fragmentation. Therefore, by discerning the background ion patterns and automatically placing those ions on an exclusion list, analyte coverage can be increased. A background ion from the mobile phase, ESI source, or from column bleed (to name a few sources), can be discerned by a single m/z covering a large portion of the retention time region, as in m/z 300.2253 discussed above. An exclusion list for background ions can often be generated by running several blank injections, but this can be an inefficient process; when the column or mobile phase changes, a new list would need to be created. From this IE type of run, it can be seen that a large portion of ions selected and fragmented are background ions as depicted by a horizontal pattern across the chromatographic run (Figure 2). Therefore, this pattern can be readily used to exclude background ions in the IE-Omics software.
Using the default IE-Omics parameters, if the same m/z is selected more than 15 times, with each instance being at least 0.15 min apart, that m/z is annotated as background and placed on an exclusion list across the entire analysis time. For example, m/z 391.28 was excluded across the entire retention time in the second injection, after being selected in the first injection of Red Cross plasma 134 times (Figure 2). Annotated background ions can also be excluded in future experiments. After the first injection, 17 ions were automatically annotated as background ions by IE-Omics software in positive ion mode of Red Cross plasma. After six injections, 54 m/z values were annotated as background ions according to this algorithm.
In addition to generation of exclusion lists of background ions, IE also enhanced coverage of the lipidome. When comparing the second to first injection after applying IE, it can be seen that many additional unique precursors are selected in both the glycerophospholipid (GP) (about m/z 700–900 at 5–10 min) and triglyceride (TG) regions (about m/z 700–1100 at 11–16 min) (Figure 2). Figure 3 compares unique ions selected for fragmentation in positive mode analysis of Red Cross plasma lipid extracts in six sequential injections without IE applied (Figure 3a) and with IE applied (Figure 3b). The IE-omics approach shows that after six sequential injections, the number of unique ions fragments is substantially higher (Figure 3b). As discussed previously, both new background ion signatures and lipid ions (as can be seen in the GP and TG region) are selected using IE (Figure 3). An analogous figure for substantia nigra is shown in the Supplementary Information (Supplementary Figure S-2), with a zoom in of the GP region overlaid with unique molecular species annotated by LipidMatch (Supplementary Figure S-3).
Figure 4 displays the cumulative number of features with lipid identifications across injection number using LipidMatch from both positive and negative mode analysis of plasma and substantia nigra lipid extracts. In all cases, a greater number of features were identified as lipids using the IE approach compared with a traditional ddMS2-top5 approach. The application of IE was most advantageous in positive ion mode analysis of plasma and substantia nigra lipid extracts. For plasma extracts in positive mode, applying IE and using six sequential injections increased the coverage of features annotated with unique molecular species by 69% compared with the traditional ddMS2-topN approach across six sequential injections. A total of 728 unique lipid molecular species were identified with IE, compared to 431 without IE (Figure 4a). In negative mode analysis of Red Cross plasma, only 10% more identifications were obtained using IE (Figure 4b). In positive mode analysis of substantia nigra, after the five sequential injections, 40% more features were identified using IE compared with without IE (Figure 4c), whereas in negative mode analysis, 18% more identifications were obtained when applying IE (Figure 4d). Applying a different identification software, LipidSearch, provided the same general trend, with IE providing the most advantage in positive mode analysis of Red Cross plasma and substantia nigra (71% and 34% more identifications, respectively) and least advantage in negative mode analysis of Red Cross plasma and substantia nigra (18% and 4%, respectively) (Supplementary Figure S-4). Unique annotations of lipid molecular species with retention time information, exact m/z from full scan data, and average peak intensity compiled across all sequential injections for positive and negative polarity analysis of Red Cross plasma and substantia nigra can be found in Supplementary Tables S-5 and S-4, for LipidSearch and LipidMatch, respectively. Fragments observed for identification by LipidSearch are also included in Supplementary Table S-5, and fragmentation criteria for LipidMatch is included in Supplementary Table S-6.

Cumulative unique lipid molecular identifications using LipidMatch software across multiple data acquisitions are shown. IE-based data-dependent top5 (IE-ddMS2-top5) described in this paper is compared with traditional ddMS2 top5 for extracts of Red Cross plasma in positive mode (a) and negative mode (b), and extracts of substantia nigra in positive mode (c) and negative mode (d)
Based on these results, it is clear that the number of additional identifications obtained when applying IE depends on sample type and the polarity measured by the mass spectrometer. It is expected that if the mass spectrum is sparse, a traditional ddMS2 approach will likely select the majority of ions above an MS2 threshold limit. In the lipidomic analyses, where applying IE was less advantageous in negative ion mode, negative ion spectra showed fewer ions than positive ion spectra. For example, the number of features (which is inversely related to spectral sparseness), was drastically lower in negative mode than positive mode, with only 4258 features in Red Cross negative mode data versus 19,231 features in Red Cross positive mode data. Therefore, after applying exclusion lists generated by IE, fewer precursors remain above the threshold to be selected for fragmentation in negative ion mode. For example, in negative polarity analysis of plasma, MS/MS scans drastically declined from the first to the fifth sequential injection (from 2491 to 414 scans), showing depletion of precursors for selection, whereas in positive polarity there was less of a decline in MS/MS scans (from 2746 to 2581 scans) (Supplementary Figure S-5). Therefore, experimental design affecting spectral density will be a major factor in determining the additional benefit of IE. For example, increasing the chromatographic gradient time would increase separation of lipids while decreasing spectral density at a given time point, and, hence, potentially decrease the advantage of applying IE versus traditional ddMS2-topN approaches.
It should be noted that additional identifications using IE are only useful if they provide unique information. After excluding previously selected high abundance lipids for fragmentation, sequential injections should provide fragmentation of lower abundance species when applying IE. Often, less abundant or trace species serve as critical biomarkers, such as phosphatidylinositol (PI), which is an important signaling molecule class. Phosphatidylcholine (PC) concentrations in plasma, for example, are about 20-fold higher than concentrations of phosphatidylinositol (PI), phosphatidylserine (PS), and phosphatidic acid (PA) combined [34]. After applying IE in the second injection, peak heights of identified lipids were significantly lower than the initial injection for positive and negative polarity analysis of both plasma and substantia nigra lipid extracts (P-value < 0.05) (Figure 5). For plasma in positive ion mode, the average intensity of selected precursors seemed to continue to decrease using IE up to the fourth injection, although not significantly (Supplementary Figure S-6; P-value > 0.05). Exclusion of trace ions close to the threshold intensity for fragmentation in certain chromatographic regions, while high intensity species such as in the TG region where spectra are dense, continue to be selected, explains why the average intensity of ions does not continue to decrease after a certain number of sequential injections. This is supported by the fact that the number of precursors selected declines across sequential injections when applying IE, and therefore in certain regions ions are no longer being selected for fragmentation (Supplementary Figure S-5). For example, the TG region contained 4819 features above 5 × 104 in 4 minutes (11 to 15 min) in Red Cross plasma, while the lysophospholipid region contained 3859 features in 4 min (0.5 to 4.5 min).

Boxplots of log transformed peak heights (base 10) from MZmine for unique lipid molecules identified in the first ddMS2-top5 acquisition using LipidMatch (IE1) and after applying an exclusion list using the algorithm described in this paper (IE2). Comparisons are made for extracts of Red Cross plasma in positive mode (a) and negative mode (b), and extracts of substantia nigra in positive mode (c) and negative mode (d). All differences where highly significant with a P-value for a Student t-test less than 0.001
The reduced intensity of precursor ions selected after applying IE suggests lower MS/MS spectral quality. This is especially true for positive ion mode fragmentation of most glycerophospholipids, where fatty acyl indicative fragments are of low abundance. To determine the quality of MS/MS spectra across sequential injections, the percentage of lipids identified with grade A, calculated by (A/(A + B + C)), was determined using LipidSearch. These grades are based on the number of fragments identified that contain species-specific structural information, with lipid identifications graded A having the most structural information in MS/MS spectra. Following a similar trend to the selected ion signal, the sequential injections after applying IE had a general drop in percent A and, hence, decrease in MS/MS spectral quality (Supplementary Figure S-7). Injections in which IE was applied had significantly lower average percentages of A compared with sequential injections without IE, for negative and positive polarity analysis of substantia nigra tissue lipid extracts (P-value < 0.05) and for positive polarity analysis of Red Cross plasma (P-value < 0.005). No significant difference was observed for negative polarity analysis of Red Cross plasma. Therefore, unique identifications provided by IE of low abundance species often provide less structural information and are more tentative.
Diglycerides (DG) are often present at low abundance and have been noted as important signaling molecules. In plasma lipid extracts, lower abundance DG species were identified after sequential injections applying IE (Supplementary Figure S-8). In the initial injection, all DG species identified except one had peak heights of 106 or 107, whereas after the sixth injection applying IE, all species identified had peak heights of 105 (Table 1). All DGs at the level of carbons and double bonds in Table 1 have been confirmed previously in human plasma [13], except for DG(30:3), identified as DG(12:0_18:3). In addition, all fatty acid constituents contained in DGs have been confirmed in plasma using fatty acyl profiling [13] or have been found in DGs using derivatization [35]. The lower abundance DG species identified after applying IE contained both odd-chain (15:0, 17:1, and 17:2), and shorter chain (12:0, 14:0, and 14:1) species, which were not identified without IE (Table 1). These fatty acids are in lower abundance in human plasma [13] and odd-chain species could represent exogenous fatty acid species or those produced by gut microbiota [36]. In this case, these species represent additional biological information otherwise not obtained.
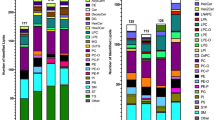
Coverage was improved for certain lipid classes using the IE approach. The majority of unique lipid molecules identified by IE in positive analysis of Red Cross plasma, but not by the traditional data-dependent approach, were mainly glycerolipids, specifically TGs, oxidized TGs, ether-linked TGs, and DGs (Figure 6a and c). These molecular species were of low intensity and were present in chromatographic regions where mass spectra were dense. Hence, using traditional approaches, these lower intensity ions generally never make it on the list of the top-5 most intense ions to be included for fragmentation. In addition, there was minimal identification of ether-linked TGs, oxidized lysophosphatidylcholines (OxLPC), and acyl-carnitines using the traditional ddMS2-top5 approach. Applying IE significantly increased the coverage of these lipid classes. Ether-linked TGs are a trace fraction of the total TGs in blood, for example, only comprising of 0.1% of chylomicrons in human blood plasma, where they have been noted to concentrate [37]. By applying IE, these low abundant ions (making up less than 0.1% of TG peak area signal) were selected for fragmentation and tentatively identified by exact mass of the precursor and exact masses of the neutral losses of the two non-ether fatty acyl constituents.

Distribution of lipids identified using LipidMatch by lipid class using IE-based data-dependent top5 (IE-ddMS2-top5) acquisitions in positive ion mode. The lipid class distribution of all identifications across sequential injections using the traditional ddMS2-top5 approach is shown for Red Cross plasma (a) and substantia nigra tissue lipid extracts (b). In addition, the distribution of additional unique lipid molecular identifications after applying IE across lipid classes are shown for Red Cross plasma (c) and substantia nigra lipid extracts (d)
In substantia nigra positive mode analysis (Figure 6b and d), IE improved the coverage of phosphatidylserine (PS), oxidized phosphatidylcholine (OxPC), phosphatidylglycerol (PG), and sulfatide species, which were minimally covered by the traditional approach. In negative ion mode analysis of substantia nigra tissue, using the traditional ddMS2-top5 approach, there was no coverage of sulfatides and minimal coverage of phosphatidic acid (PA). Applying IE significantly increased coverage of both of these species (Supplementary Figure S-9). These findings highlight that IE not only increases the total number of lipid identifications but increases identifications of trace lipid species of potential interest, which are minimally covered by traditional approaches.
Future developments will continue to increase the advantages of applying IE-Omics. Currently, the script is not integrated in Xcalibur software, and therefore exclusion lists are not generated in real time and must be uploaded into new method files before each iterative injection. In our lipidomics workflow, we suggest using three to four iterative injections on pooled samples, which can be used to identify features of a given sample group. Therefore, this method is sufficient for lipid identification in large quantitative studies to determine biomarkers where thousands of samples are required, as only a few additional injections are used for IE and, hence, there is minimal addition of acquisition time. Fewer injections may be required if the exclusion window is increased from 10 ppm, for example, to 100 ppm. By increasing the exclusion window, isobaric ions will only be selected in one injection, reducing the number of injections needed to select all ions above a certain threshold. In the future, fully automated exclusion list generation may be developed.
Conclusion
We have semi-automated the IE approach using a simple open source R script. The script uses open source formatted files that can be converted from various vendor formats and produces an exclusion list in the format required for importing into Thermo Scientific instruments (.csv). Features include smart exclusion list generation, which combines ions selected in similar m/z and retention time windows to generate a shorter exclusion list, and automatic annotation of background ions. After applying the software, IE-Omics, to lipidomic datasets in Red Cross plasma and substantia nigra brain tissue lipid extracts, IE was shown to be most advantageous in complex matrices with a high number of analyte species. Applying IE to lipidomics analyses in certain cases increased identifications by over 50%. The greatest advantage using IE was shown in positive ion mode and in Red Cross plasma versus substantia nigra lipid extracts, where spectra were most dense. In lipidomics, trace species, such as odd-chained and short-chained DGs, were identified only after applying the IE technique.
Future data acquisition strategies, for example only including precursor ions for fragmentation that match lipid masses and identifying polymer patterns for exclusion, could prove advantageous. In most cases, however, such as in negative mode, after only using a few sequential injections, all ions above the threshold limit for fragmentation were selected, and therefore new data-acquisition methods would not provide additional advantage in terms of MS/MS spectral coverage. New data-acquisition methods might be able to reduce the number of injections needed for coverage of the majority of lipid ions and notify the user when additional injections are no longer required. Applying IE expands the scope of the lipidome covered, both increasing the total number and diversity of lipids identified.
References
de Pablo, M.A., Puertollano, M.A., de Cienfuegos, G.: Biological and clinical significance of lipids as modulators of immune system functions. Clin. Diagn. Lab. Immunol. 9, 945–950 (2002)
Edwards, P.A., Ericsson, J.: Sterols and isoprenoids: signaling molecules derived from the cholesterol biosynthetic pathway. Annu. Rev. Biochem. 68, 157–185 (1999)
Martin, T.F.J.: Phosphoinositide lipids as signaling molecules: common themes for signal transduction, cytoskeletal regulation, and membrane trafficking. Annu. Rev. Cell Dev. Biol. 14, 231–264 (1998)
Simons, K., Toomre, D.: Lipid rafts and signal transduction. Nat. Rev. Mol. Cell Biol. 1, 31–39 (2000)
Sies, H.: Oxidative stress: oxidants and antioxidants. Exp. Physiol. 82, 291–295 (1997)
Watanabe, K., Yasugi, E., Oshima, M.: How to search the glycolipid data in “LIPID BANK for the Web” – the newly developed lipid database in Japan. Trends Glycosci. Glycotechnol. 12, 175–184 (2000)
Dove, A.: Greasing the Wheels of Lipidomics, available at: http://www.sciencemag.org/site/products/lst_20150213.xhtml. Accessed 25 Dec 2016 (2015)
Schmelzer, K., Fahy, E., Subramaniam, S., Dennis, E.A.: The Lipid Maps Initiative in Lipidomics. Methods Enzymol. 432, 171–183 (2007)
Spener, F.: European Commission funds lipidomics project. Eur. J. Lipid Sci. Technol. 107, 1–2 (2005)
Sud, M., Fahy, E., Cotter, D., Brown, A., Dennis, E.A., Glass, C.K., Merrill, A.H., Murphy, R.C., Raetz, C.R.H., Russell, D.W., Subramaniam, S.: LMSD: LIPID MAPS Structure database. Nucleic Acids Res. 35, D527–D532 (2007)
Lintonen, T.P.I., Baker, P.R.S., Suoniemi, M., Ubhi, B.K., Koistinen, K.M., Duchoslav, E., Campbell, J.L., Ekroos, K.: Differential mobility spectrometry-driven shotgun lipidomics. Anal. Chem. 86, 9662–9669 (2014)
Han, X., Gross, R.W.: Global analyses of cellular lipidomes directly from crude extracts of biological samples by ESI mass spectrometry a bridge to lipidomics. J. Lipid Res. 44, 1071–1079 (2003)
Quehenberger, O., Armando, A.M., Brown, A.H., Milne, S.B., Myers, D.S., Merrill, A.H., Bandyopadhyay, S., Jones, K.N., Kelly, S., Shaner, R.L., Sullards, C.M., Wang, E., Murphy, R.C., Barkley, R.M., Leiker, T.J., Raetz, C.R.H., Guan, Z., Laird, G.M., Six, D.A., Russell, D.W., McDonald, J.G., Subramaniam, S., Fahy, E., Dennis, E.A.: Lipidomics reveals a remarkable diversity of lipids in human plasma. J. Lipid Res. 51, 3299–3305 (2010)
Knittelfelder, O.L., Weberhofer, B.P., Eichmann, T.O., Kohlwein, S.D., Rechberger, G.N.: A versatile ultra-high performance LC-MS method for lipid profiling. J. Chromatogr. B 951/952, 119–128 (2014)
Andrews, G.L., Dean, R.A., Hawkridge, A.M., Muddiman, D.C.: Improving proteome coverage on a LTQ-Orbitrap using design of experiments. J. Am. Soc. Mass Spectrom. 22, 773–783 (2011)
Hirasawa, K., Nishizuka, Y.: Phosphatidylinositol turnover in receptor mechanism and signal transduction. Annu. Rev. Pharmacol. Toxicol. 25, 147–170 (1985)
Tang, X., Edwards, E.M., Holmes, B.B., Falck, J.R., Campbell, W.B.: Role of phospholipase C and diacylglyceride lipase pathway in arachidonic acid release and acetylcholine-induced vascular relaxation in rabbit aorta. Am. J. Physiol. - Heart Circ. Physiol. 290, H37–H45 (2006)
Bendall, S.C., Hughes, C., Campbell, J.L., Stewart, M.H., Pittock, P., Liu, S., Bonneil, E., Thibault, P., Bhatia, M., Lajoie, G.A.: An enhanced mass spectrometry approach reveals human embryonic stem cell growth factors in culture. Mol. Cell. Proteom. 8, 421–432 (2009)
Rudomin, E.L., Carr, S.A., Jaffe, J.D.: Directed sample interrogation utilizing an accurate mass exclusion-based data-dependent acquisition strategy (AMEx). J. Proteome Res. 8, 3154–3160 (2009)
Rolland, A.D., Lavigne, R., Dauly, C., Calvel, P., Kervarrec, C., Freour, T., Evrard, B., Rioux-Leclercq, N., Auger, J., Pineau, C.: Identification of genital tract markers in the human seminal plasma using an integrative genomics approach. Hum. Reprod. 28, 199–209 (2013)
Seo, J., Jeong, J., Kim, Y.M., Hwang, N., Paek, E., Lee, K.-J.: Strategy for comprehensive identification of post-translational modifications in cellular proteins, including low abundant modifications: application to glyceraldehyde-3-phosphate dehydrogenase. J. Proteome Res. 7, 587–602 (2008)
Hughes, C.S., Postovit, L.M., Lajoie, G.A.: Matrigel: a complex protein mixture required for optimal growth of cell culture. Proteomics 10, 1886–1890 (2010)
Vahidi, S., Stocks, B.B., Liaghati-Mobarhan, Y., Konermann, L.: Mapping pH-induced protein structural changes under equilibrium conditions by pulsed oxidative labeling and mass spectrometry. Anal. Chem. 84, 9124–9130 (2012)
Sandra, K., t'Kindt, R., Jorge, L. Sandra, P.: The art and practice of lipidomics. In: Lämmerhofer, M., Weckwerth, W. (eds.) Metabolomics in practice: successful strategies to generate and analyze metabolic Data, p. 137–173. John Wiley and Sons, Hoboken, NJ (2013)
Edmands, W.M., Ferrari, P., Rothwell, J.A., Rinaldi, S., Slimani, N., Barupal, D.K., Biessy, C., Jenab, M., Clavel-Chapelon, F., Fagherazzi, G., Boutron-Ruault, M.-C., Katzke, V.A., Kühn, T., Boeing, H., Trichopoulou, A., Lagiou, P., Trichopoulos, D., Palli, D., Grioni, S., Tumino, R., Vineis, P., Mattiello, A., Romieu, I., Scalbert, A.: Polyphenol metabolome in human urine and its association with intake of polyphenol-rich foods across European countries. Am. J. Clin. Nutr. 102, 905–913 (2015)
Nazari, M., Muddiman, D.C.: Enhanced lipidome coverage in shotgun analyses by using gas-phase fractionation. J. Am. Soc. Mass Spectrom. 27, 1735–1744 (2016)
Schwudke, D., Oegema, J., Burton, L., Entchev, E., Hannich, J.T., Ejsing, C.S., Kurzchalia, T., Shevchenko, A.: Lipid profiling by multiple precursor and neutral loss scanning driven by the data-dependent acquisition. Anal. Chem. 78, 585–595 (2006)
Lin, L., Yu, Q., Yan, X., Hang, W., Zheng, J., Xing, J., Huang, B.: Direct infusion mass spectrometry or liquid chromatography mass spectrometry for human metabonomics? A serum metabonomic study of kidney cancer. Analyst 135, 2970–2978 (2010)
Folch, J., Lees, M., Sloane Stanley, G.H.: A simple method for the isolation and purification of total lipides from animal tissues. J. Biol. Chem. 226, 497–509 (1957)
R Development Core Team: R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna (2016)
Kessner, D., Chambers, M., Burke, R., Agus, D., Mallick, P.: ProteoWizard: open source software for rapid proteomics tools development. Bioinformatics Oxf. Engl. 24, 2534–2536 (2008)
Taguchi, R., Ishikawa, M.: Precise and global identification of phospholipid molecular species by an Orbitrap mass spectrometer and automated search engine LIPID Search. J. Chromatogr. A 1217, 4229–4239 (2010)
Pluskal, T., Castillo, S., Villar-Briones, A., Orešič, M.: MZmine 2: modular framework for processing, visualizing, and analyzing mass spectrometry-based molecular profile data. BMC Bioinformatics 11, 395 (2010)
Turner, J.D., Rouser, G.: Precise quantitative determination of human blood lipids by thin-layer and triethylaminoethylcellulose column chromatography. Anal. Biochem. 38, 437–445 (1970)
Wang, M., Hayakawa, J., Yang, K., Han, X.: Characterization and quantification of diacylglycerol species in biological extracts after one-step derivatization: a shotgun lipidomics approach. Anal. Chem. 86, 2146–2155 (2014)
Jenkins, B., West, J.A., Koulman, A.: A review of odd-chain fatty acid metabolism and the role of pentadecanoic acid (C15:0) and heptadecanoic acid (C17:0) in health and disease. Molecules 20, 2425–2444 (2015)
Gilbertson, J.R., Karnovsky, M.L.: Nonphosphatide fatty acyl esters of alkenyl and alkyl ethers of glycerol. J. Biol. Chem. 238, 893–897 (1963)
Acknowledgements
This research was done in collaboration between Core 1 and Core 3 of the Southeast Center for Metabolomics (SECIM) <http://secim.ufl.edu/> (NIH grant #U24 DK097209). The authors thank Jason Cochran for writing R code to compile the fragments required for identification in Supplementary Table S-6, and for helping to expand the in-silico libraries used for lipid identification.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Disclaimer
Certain commercial equipment, instruments, or materials are identified in this paper to specify adequately the experimental procedure. Such identification does not imply recommendation or endorsement by the National Institute of Standards and Technology, nor does it imply that the materials or equipment identified are necessarily the best for the purpose.
Rights and permissions
About this article

Cite this article
Koelmel, J.P., Kroeger, N.M., Gill, E.L. et al. Expanding Lipidome Coverage Using LC-MS/MS Data-Dependent Acquisition with Automated Exclusion List Generation. J. Am. Soc. Mass Spectrom. 28, 908–917 (2017). https://doi.org/10.1007/s13361-017-1608-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13361-017-1608-0