
Abstract
Annotation of peptide sequence from tandem mass spectra constitutes the central step of mass spectrometry-based proteomics. Peptide mass spectra are obtained upon gas-phase fragmentation. Identification of the protein from a set of experimental peptide spectral matches is usually referred as protein inference. Occurrence and intensity of these fragment ions in the MS/MS spectra are dependent on many factors such as amino acid composition, peptide basicity, activation mode, protease, etc. Particularly, chemical derivatizations of peptides were known to alter their fragmentation. In this study, the influence of acetylation, guanidinylation, and their combination on peptide fragmentation was assessed initially on a lipase (LipA) from Bacillus subtilis followed by a bovine six protein mix digest. The dual modification resulted in improved fragment ion occurrence and intensity changes, and this resulted in the equivalent representation of b- and y-type fragment ions in an ion trap MS/MS spectrum. The improved representation has allowed us to accurately annotate the peptide sequences de novo. Dual labeling has significantly reduced the false positive protein identifications in standard bovine six peptide digest. Our study suggests that the combinatorial labeling of peptides is a useful method to validate protein identifications for high confidence protein inference.

ᅟ
Similar content being viewed by others


Introduction
Protein sequence coverage obtained in a proteomic analysis depends on multiple variables, including peptide size, peptide hydrophobicity, aromatic amino acid content, charged side chains, ability to form stable secondary structures, etc. On the other hand, protein identification is also dependent on workflow parameters such as ion selectivity, limit of detection, limit of quantification, dynamic range, data density, repeatability, and reproducibility [1]. For regular proteomic workflows, using database search for protein identification, a small stretch of contiguous b- or y-ions would be enough for mapping theoretical and experimental spectra. However, studies focused on de novo sequence assignments for peptide/protein mapping need to explore and optimize strategies that can improve peptide fragmentation in terms of ion occurrence as well as intensity. Peptide properties such as peptide basicity, location, and number of acidic and basic residues and activation mode influence the peptide fragmentation and thereby act as key players that determine the appearance of fragment ions [2–12].
Basic processes that govern peptide fragmentation in mass spectrometry are comprehensively explained by mobile proton model proposed by Gaskell and Wysocki [13–17]. Sequence-specific cleavages have been identified by careful analysis of CID fragmentation data [7, 9, 11, 18–24]. Peptide fragmentation can be tuned by addition/altering the charge localization by selective modification of peptide termini, especially internal residues such as K, R, S, T, C, etc. [25, 26]. Although many of the chemical modifications are known since 1970 [27], their utility was limited because of the incompatibility of these reactions to MS-based workflows, need for higher amounts of sample, interferences due to by-products, possible side reactions, and increased data analysis time. However, development of improved chemical labeling procedures has enabled researchers to integrate the chemical tags into traditional proteomic workflows [28]. Chemical tags have found three major applications in proteomics: (1) As affinity tags to reduce the problem of dynamic range; (2) as differential isotope labeled chemical tags in quantitative proteomics; (3) as chemical labels to engineer peptide fragmentation [26, 29, 30].
Peptide fragmentation in a typical CID results in the formation of b- and y-type ions. Representation of both b-type and y-type ions is favorable for confident protein sequence assignment. Moreover, in proteomic studies involving novel biological systems lacking cognate database, de novo peptide annotation followed by homology search is the most frequently adopted methodology for protein annotation [31–34]. In such a scenario, chemical labels that can optimize peptide fragmentation would provide comprehensive sequence information [35–37]. Chemical labeling strategies have provided an alternate platform for improving the confidence of protein identifications. The major contribution of chemical tags to the mainstream proteomic studies is in peptide quantitation, e.g., ITRAQ [26]. But one of the most popular applications of chemical tags is to direct the peptide fragmentation towards specific ion type (i.e., b-type and y-type ions). Based on their chemical nature, these tags can enhance or suppress the appearance of a specific type of fragment ion. For instance, sulphonation of peptides by 4-sulphophenyl isothiocyanate/3-sulpho propionic acid NHS ester results in a sulfate group at the peptide amino terminus, the negative charge of which neutralizes the protonated b-ion and, thus, the net charge on b-ion becomes zero; only y-ion series could be observed in the tandem mass spectra [38–40]. Whereas derivatization with 2,4,6-trimethylpyrillium tetrafluoroborate [41]/ 2,4,6- trimethyl pyridinium [41] or 4-amidino benzoic acid [42] or acetylation would result in dominant N-terminal ion series (i.e., b-type ions) [41, 43, 44].
However, b-type ions are under-represented in ion trap mass spectra [10, 45–52]. Among many chemical labeling approaches, acetylation and guanidinylation are simple to perform and are known to affect the peptide fragmentation. Acetylation is employed to study gas-phase peptide fragmentation and results in a mass shift of 42.01 Da, which helps us in differentiating lysine and glutamine [44, 49]. It has been shown that acetylation of N-terminus results in (1) peptide fragmentation at low energies [4, 8], (2) prevents gas phase cyclization of b-ions [44, 49, 53], (3) increases b-ion relative intensity [43, 54], and (4) b-ion occurrence [43]. Guanidinylation was initially used as a tool to improve the stability of the proteins [55]. Guanidinylation is specific to epsilon amine, it causes a mass shift of 42.02 Da, and results in an increased precursor ion intensity [56]. Guanidinylation improves peptide fragmentation via charge remote and charge directed mechanisms, which in turn results in an increased sequence coverage [36]. Although a combination of acetylation and guanidinylation has been utilized for quantitative labeling, the impact of dual labeling on peptide fragmentation efficiency and influence of each modification was not investigated systematically [57].
A combination of acetylation and guanidinylation would play a role at two stages of mass spectrometry. Guanidinylation is expected to improve ionization due to increased basicity rendered by guanidine group. On the other hand, acetylation leads to an improved b-ion relative intensity in the tandem mass spectra, which is otherwise dominated by y-type ions. Thus, a combination of acetylation and guanidinylation could yield improved peptide fragmentation. To evaluate the specific statistical advantages of dual labeling method, we have tested this workflow on a lipase from Bacillus subtilis. This lipase is generally identified with high sequence coverage (>85%) and has a broad range of peptide products from 5 to 23 amino acids long, which could be confidently assigned. Simultaneously, we also labeled standard bovine six protein mix digest to evaluate the efficiency of dual labeling in a multi-protein scenario.
Materials and Methods
Trypsin gold and chymotrypsin of mass spectrometry grade were obtained from Promega (Madison, Wisconsin, USA). α-Cyano-4-hydroxycinnamic acid (HCCA) was obtained from Sigma Aldrich (St. Louis, MO, USA). Six bovine protein tryptic digest equal molar mix (P/N PTD/00001/63) was obtained from Michrom Bioresources (Auburn, CA, USA), acetic anhydride, triethylamine, methanol, and acetonitrile of analytical grade were sourced locally (SpectroChem, India).
Protein Purification
Bacillus subtilis lipase was cloned in pET21b, and purified upon expression in E. coli BL21 (DE3) as described earlier [58]. Purified protein was stored in 2 mM glycine-NaOH buffer (pH 10.0) at –20 °C. Protein purity was checked on SDS-PAGE. Protein quantitation was carried out by the modified Lowry method [59].
Proteolysis
Purified lipase (1 μg ~52 picomoles) was treated with 25 ng of Trypsin Gold and the total reaction volume was adjusted to 30 μL with 50 mM ammonium bicarbonate. The reaction mixture was incubated at 37 °C for 18 h. Samples were then vacuum dried and stored at –20 °C, till further use.
Acetylation
Acetylation was carried out according to the published protocol with slight modification [54]. Acetylation mix was prepared by adding 12 μL of acetic anhydride to 83 μL of methanol, and 5 μL of triethylamine was added to this mix. Two μL of the freshly prepared acetylation mix was added to trypsin digested peptides; the concentration of 17 fmol/μL bovine six protein mix was used. The reaction mixture was allowed to stand at room temperature for 10 min. Samples were then vacuum dried and stored at –20 °C till further use.
Guanidinylation
Guanidinylation and desalting of the peptide samples were carried out according to the published protocol [60]. A stock solution of O-methylisourea was prepared by dissolving 0.05 g in 51 μL of water. The guanidinylation reaction mixture was prepared by mixing a 5-μL aliquot of digested protein with 5.5 μL of 7 N NH4OH and 1.5 μL of O-methylisourea stock solution. After incubating the reaction mixture at 65 °C for 5–10 min, the reaction was terminated by adding 15 μL of 10% TFA (v/v). The acidified reaction mixture was partially dried in a speed-vac to a final volume of 10 μL. Guanidinylated peptides were desalted by using Zip tips (Millipore) packed with the C-18 matrix as per manufacturer’s recommendations.
Dual Labeling
In the case of dual labeling method, peptides were subjected to guanidinylation followed by acetylation (as described above), vacuum dried, and the dual labeled peptides stored at –20 °C, till further use.
Mass Spectrometry
nLC-ESI MS/MS Unlabeled, acetylated, guanidinylated, and dual labeled trypsin digested peptides were resuspended in 10 μL of 5% ACN containing 0.1% formic acid. Peptides were fractionated on nanoflow LC system (Easy nLCII; Proxeon Biosystems, Odense, Denmark) using Bio Basic C-18 Pico Frit nanocapillary column (75 μm × 10 cm; New Objective, Woburn, MA, USA) with a 60 min linear gradient 0%–100% B, 5% ACN with 0.1% formic acid (solvent A), and 95% ACN with 0.1% formic acid (solvent B)] at a flow rate of 200 nL/min and analyzed on LTQ Orbitrap Velos (Thermo Scientific, San Jose, CA, USA); 1.7 kV was applied for ionization. Full scan MS with a mass window 300–2000 Da were acquired after accumulation to target value of 1*E6 in FT mode. FT resolution was set to 60,000, top 20 peptides with two or more charge state were isolated to a target value of 5000, and fragmented in a linear ion trap with a normalized collision energy of 35% in CID mode. Fragment ions were scanned in a low-pressure ion trap at a scan rate of 33,333 am/s, and the minimum threshold of ion selection for MS/MS was set at 500 counts. Ion accumulation time was set at 500 ms for MS and 25 ms for MS/MS. Activation time of 10 ms and q value of 0.25 was used [61].
Data Analysis
Sequest Search
Sequest HT search was performed using Proteome Discoverer 1.4 ver. 1.4.0.288 platform; Thermo Scientific Inc. Raw files of unlabeled and labeled peptides of lipase were analyzed against Bacillus subtilis from NCBInr database, along with a database of common contaminants. Bovine six protein mix was analyzed against Bos tauras from NCBInr database and also against curated bovine database from Uniprot. In the case of unacetylated peptides of lipase, methionine oxidation, deamidation of asparagine/glutamine were taken as variable modification. Trypsin as a protease, with two missed cleavages, the charge of +2 to +3, mass range from 3500 to 5000 Da, peptide length of ≥4 aminoacids were considered for spectral matching. Peptide mass tolerance of 10 ppm and fragment mass tolerance of 0.6 Da were applied. For acetylated peptides of lipase, search included N-terminal acetylation as fixed modification and lysine/histidine/serine/threonine/tyrosine/cysteine acetylation, methionine oxidation as variable modifications. In the case of guanidinylated peptides, guanidinylation of lysine was considered as fixed modification, deamidation of asparagine and glutamine, oxidation of methionine was considered as variable modifications. In the case of dual labeled peptides, guanidinylation of lysine was taken as additional fixed modification; the remaining search parameters are the same as that of acetylation. Target FDR value of 0.01 was achieved by including only high confidence peptide hits, score versus charge state (Xcorr) as filters.
Peaks 6.0 Search
nLC-ESI MS/MS files of acetylated, guanidinylated, dual labeled, and unacetylated trypsin digested lipase were analyzed in the PEAKS6.0 software with recommended data refining parameters, (i.e., precursor ion tolerance of 10 ppm, fragment ion tolerance of 0.6 Da, and precursor correction with a peptide charge window of +1 to +4). The mass window of 200–5000 and retention time window 0.05–59.95 were used for data analysis. Spectral quality was set at 0.65 as recommended by the manufacturers; –10logP score cut-off was set to run specific scores in order to achieve theoretical peptide FDR of zero. Database search for lipase was carried out against Bacillus subtilis from NCBInr database with the same search settings used in Sequest HT. De novo analysis of the raw files was carried out wherein the modification settings were the same as those used in Sequest HT. The de novo sequences obtained were analyzed for differences in the accuracy of sequence assignments between labeled and unlabeled peptides.
Mascot Search
Unlabeled, acetylated, and dual labeled trypsin peptides were subjected to Mascot search using proteome Discoverer ver. 1.4. Spectra were searched against B. subtilis in the Uniprot database along with contaminants; bovine six protein mix was analyzed against curated Bovine database from Uniprot search settings were same as those used for Sequest HT search. The data files (.DAT) generated from this search were used for peptide fragmentation analysis.
Fragmentation Analysis
Fragment ion occurrence, as well as intensity variability statistics, were assessed for unlabeled and labeled peptides using fragmentation analyzer tool [62] ver. 1.5.14. Using high peptide confidence, 95% significance threshold and peptide score ≥30 Mascot identification files (.DAT) were generated from nLC ESI-MS/MS of unlabeled and labeled lipase digests sample triplicates.
For fragmentation analysis, peptides with +2 charge state with at least 10 peptide spectrum matches per sequence across three nLC ESI-MS/MS replicates were selected. All the possible modifications were considered for analysis. Peptide spectrum matches were assessed for changes in normalized median intensity changes in b-/y-ions upon modification. B-/y-ion relative intensity pattern for unique peptides at each amino acid position as a function of the chemical label was generated, and finally, fragment ion occurrence pattern for b-/y-ions was plotted for labeled and unlabeled samples as a function of C-terminal arginine and C-terminal lysine.
Raw Meat ver. 2.1 Analysis
Alterations in the retention time, ionization and precursor ion intensity (MS2) of the peptides upon labeling was assessed by comparing the raw files of unlabeled and dual labeled samples of bovine six protein mix digest and lipase using Raw Meat ver. 2.1 developed by Vast Scientific in conjuction with Thermo Fisher Scientific BRIMS Center, Cambridge, MA, USA.
Results and Discussion
Labeling Efficiency and Protein Identification
Unlabeled and labeled lipase digests were subjected to nLC ESI-MS/MS. Data analysis by Sequest HT and Peaks 6.0 search engine showed complete sequence coverage for unlabeled lipase, whereas for acetylated, guanidinylated, and dual labeled lipase, the sequence coverage was 97%, 100%, and 97%, respectively, suggesting that the labels individually or in combination did not affect the sequence coverage of the protein significantly. At this stage, we checked the labeling efficiency by considering all the modifications as variable and reanalyzed the data (Supplementary Tables S1–S7). Based on the data analysis and spectral information, labeling efficiency was found to be 100% for individual labels as well as in combination (Supplementary Figure S1–S3).
Normalized Fragment Ion Intensity Increased Upon Labeling
The impact of the chemical labels on the peptide fragmentation was assessed by comparing the normalized median fragment ion intensity for b- and y-type ions. Normalized median ion intensity was generated from triplicate runs for each chemical label. From the normalized ion intensity values, it is evident that both chemical labels (i.e., acetylation and guanidinylation) were efficient in increasing overall b- and y-ion intensity. However, increase in the fragment ion intensities is higher upon dual labeling in comparison with individual labels (Table 1), which could be understood as a combinatorial effect (Supplementary Figure S4).
Dual Labeling Improved Peptide Fragmentation
Elevated normalized b- and y-ion relative intensity upon labeling is suggestive of altered fragmentation at the peptide level. Therefore, fragment ion intensity information was derived from all the peptide spectrum matches for unique peptides of lipase (Supplementary Figures S5–S11). Changes in the normalized intensity of b- and y-ions was plotted as a function of each amino acid. The impact of each chemical label on the fragmentation pattern of the peptide was studied.
-
(1)
ALPGTDPNQK
Fragment ion intensity box plot of unlabeled peptide shows dominant y-ion peaks for proline at the third and seventh positions of the peptide Figure 1a. It is very well established that proline promotes fragmentation towards the N-terminal side of the peptide, resulting in strong y-ion at that position [5, 18, 19], and similarly, relative higher intensity y-ion peak for glycine and glutamate as well as higher intensity b-ion peak for aspartate and leucine, characteristic for a typical ion trap CID MS/MS.
Figure 1 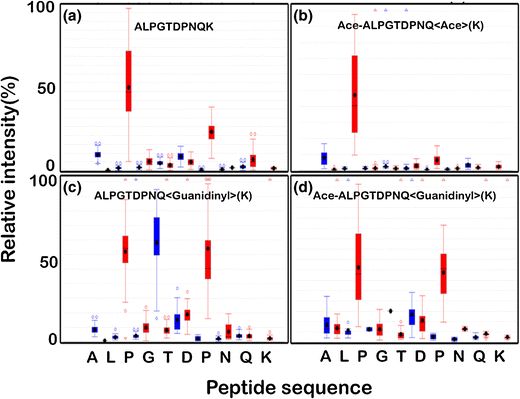
A box plot of the fragment ion and corresponding relative intensity for (a) unlabeled (519 PSM’s), (b) acetylated (92 PSM’s), (c) guanidinylated (110 PSM’s), and (d) dual labeled (127 PSM’s) peptide “ALPGTDPNQK”; b- and y-fragment ions are represented in blue and red color, respectively
There was a reversal in the b- and y-ion intensities for aspartate and glutamate residues upon acetylation Figure 1b. Guanidinylation resulted in stronger y-ion intensity for asparagine and aspartate residues, and higher b-ion intensity was observed at threonine residue. Overall, higher intensity peaks could be seen for threonine, glycine, and aspartate upon guanidinylation Figure 1c. Although proline effect is still prominent for all the labeled peptides, each label did influence the peptide fragmentation pattern, whereas dual labeling showed a cumulative effect of acetylation and guanidinylation, Figure 1d.
-
(2)
KVDIVAHSMGGANTLYYIK
This peptide presents an interesting case where basic residues are present at the termini and the middle of the peptide in the form of lysine and histidine, respectively. This allows us to monitor the influence of these residues on peptide fragmentation with and without labeling. All the amino acids were represented in the MS/MS spectra for the unlabeled peptide. Higher fragment ion intensities were observed for residues close to the histidine residue Figure 2(a). This could be attributed to the higher basicity of histidine residue in comparison to lysine [23].
Acetylated peptide had increased b-ion relative intensity, especially towards the C-terminal. Lowered peptide basicity upon di-acetylation of lysine at peptide N-terminal seemingly suppressed the histidine effect while improving fragmentation at glycine and tyrosine residues, Figure 2b. Guanidinylation resulted in b- and y-ion representation prominently at the amino terminus of the peptide. Increase in peptide basicity due to guanidinylation of lysine to homoarginine resulted in a dominant y-ion representation, Figure 2c. Dual labeling resulted in acetyl, guanidyl lysine at peptide amino termini and guanidinylation of carboxyl terminus. Dual labeling resulted in the equitable fragment ion distribution across the entire peptide length Figure 2d. Thus a balance of peptide basicity upon dual labeling allowed optimized fragment ion occurrence in this peptide.
Figure 2 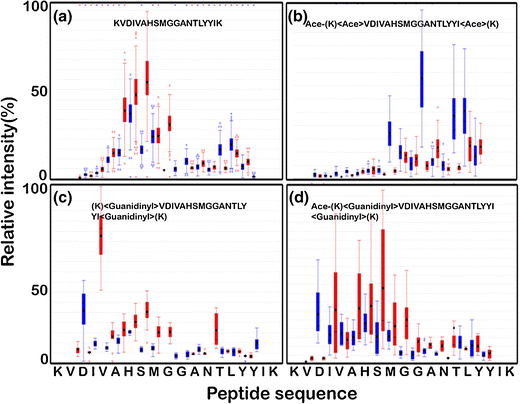
A box plot of the fragment ion and corresponding relative intensity for (a) unlabeled (192 PSM’s), (b) acetylated (33 PSM’s), (c) guanidinylated (11 PSM’s), and (d) dual labeled (30 PSM’s) peptide “KVDIVAHSMGGANTLYYIK”; b- and y-fragment ions are represented in blue and red color, respectively


Dual Labeling Increased the Accuracy of De Novo Sequence Annotation
Improvement in accuracy of de novo amino acid sequence annotation upon labeling was compared for unlabeled and labeled peptides of lipase using PEAKS 6.0 algorithm. The confidence of amino acid assignment of a peptide is judged with the help of two statistical parameters. They are, TLC (total local confidence), which measures the probability of correct amino acid assignment in a peptide and ALC (average local confidence), which indicates the sum total of percentage (or probability) of correct amino acid assignment in the sequence (TLC/peptide length*100).
Peptide assignments generally were found to be very close to the actual sequence for both unlabeled and dual labeled peptides. Although there are errors in identification of a correct amino acid at a given position, number of mass fits (N = G + G, Q = G + A, S + Y = C + F, K = Q,W = E + G, (M + 16) = F, etc.) for amino acids were lower in dual labeled peptides (13) compared with unlabeled peptides (28) because of increased fragment ion occurrence. Dual labeling, thus, improved the accuracy of the de novo fragment ion annotation compared with unlabeled peptides (Table 2).
Due to the complexity involved in de novo sequencing, the studies of this kind will help in the development of de novo sequencing algorithms. Even in large scale protein identification studies, it is prudent to manually evaluate the spectra to confirm the results (on crucial peptides or PTMs). Phenylalanine and oxidized methionine have identical mass but differ in fragmentation [63] and that helps in distinguishing these residues, Acetylation is useful in differentiating lysine and glutamine.
Fragment Ion Occurrence was Balanced upon Dual Labeling
Higher accuracy in de novo sequencing suggests that dual labeling improved the fragmentation of peptides. The effect of dual labeling on fragment ion occurrence was evaluated for peptides with C-terminal arginine and C-terminal lysine. It is hypothesized that acetylation improves b-ion representation in peptides with arginine at the carboxyl terminus, whereas for peptides with lysine at the carboxyl terminus, a dual impact is expected; (1) guanidinylation should improve the ionization efficiency, (2) acetylation is expected to increase b-ion representation. In total, spectra with balanced representation of b- and y-ions are expected.
Normalized fragment ion occurrence pattern of unlabeled and labeled peptides was compared to examine the impact of individual labels on the peptide fragmentation. In the case of unlabeled peptides, normalized y-ion occurrence percentage is higher compared with b-ions through the peptide length as seen in a typical ion trap CID, Figure 3a. Acetylation of the peptides resulted in substantial increase in b-ion occurrence, with a concomitant decrease in y-ion occurrence, especially after the first 7–8 ions. Thus tandem mass spectra of an acetylated peptide are expected to be dominated by b-ions with high-intensity y-ions, Figure 3b.

Comparison plot of variation in b- (blue) and y- (red) ion occurrence for trypsin digested peptides of lipase. Unlabeled peptides (20 unique peptides, 2686 PSM’s) (a), acetylated peptides (23 unique peptides, 1802 PSM’s) (b), guanidinylated peptides (25 unique peptides, 1993 PSM’s) (c), and dual labeled peptides (30 unique peptides, 2709 PSM’s) (d). Y-axis is the occurrence of ions (%) and the x-axis is fragment number. Dashed lines are neutral loss ions y-NH3 (pink), yellow (y-H2O), purple (b-H2O), and turquoise (b-NH3)
Guanidinylation increases the peptide basicity due to the conversion of lysine to homoarginine. While this does not affect the peptides with C-terminal arginine (except missed cleavage products), peptides with C-terminal lysine are expected to show fragmentation behavior similar to C-terminal arginine peptides. Guanidinylated peptides had higher y-ion occurrence percentage compared with b-ion up to 15 ions. Guanidinylation showed marginal improvement in b- and y-ion intensities. When coupled with higher y-ion occurrence percentage, strong y-ion appearance with few b-ions of high intensity can be expected in typical guanidinylated peptide spectra, Figure 3c. Dual labeling had a combinatorial effect where there is higher y-ion occurrence at the peptide N-terminal. However, b-ion occurrence percentage increased with the fragment length suggesting that acetylation tilted the balance of fragment ion appearance towards b-ion; b- and y-type ion representation was much more balanced for dual labeled peptides for the first 15 fragment ions, Figure 3d.
Fragment ion occurrence pattern for unlabeled and labeled peptides was further examined by comparing the ion distribution for peptides with lysine and arginine at the C-terminal. C-terminal arginine peptides showed the highest difference in b- and y-ion occurrence percentage, Figure 4a. A similar pattern is seen in guanidinylated peptides, Figure 4c, since guanidinylation does not affect the peptides with arginine at C-terminal. Acetylation-induced increase in b-ion occurrence is clearly evident, Figure 4b and d, which balances the b- and y-ion representation in the MS /MS spectra up to 12 fragment ions.

Comparison plot of variation in b- (blue) and y- (red) ion occurrence for peptides with C-terminal arginine (peptides with internal lysine and terminal peptide included). Unlabeled (eight unique peptides, 823 PSM’s) (a), acetylated (11 unique peptides, 1257 PSM’s) (b), guanidinylated (15 unique peptides, 1005 PSM’s) (c), and dual labeled (17 unique peptides, 1774 PSM’s) (d). Y-axis is the occurrence of ions (%) and the x-axis is fragment number. Dashed lines are neutral loss ions Y-NH3 (pink), yellow (y-H2O), purple (b-H2O), and turquoise (b-NH3)
Similarly, for peptides with lysine at C-termini, y-ion occurrence was slightly higher for unlabeled peptides, Figure 5a; guanidinylation increased this difference, Figure 5c. On the other hand, acetylation skewed the balance of fragment ion occurrence towards b-ions, Figure 5b. Dual labeling resulted in equitable b- and y-ion occurrence without compromising the ion intensity, Figure 5d.

Comparison plot of variation in b (blue) and y (red) ion occurrence for peptides with C-terminal lysine (peptide with internal arginine are included). Unlabeled (12 unique peptides, 1862 PSM’s) (a), acetylated (12 unique peptides, 545 PSM’s) (b), guanidinylated (10 unique peptides, 988 PSM’s) (c), and dual labeled (13 unique peptides, 935 PSM’s) (d). Y-axis is the occurrence of ions (%) and the x-axis is fragment number. Dashed lines are neutral loss ions Y-NH3 (pink), yellow (y-H2O), purple (b-H2O), and turquoise (b-NH3)
Overall, y-type ions were over-represented in the unlabeled peptides; the balance shifted to increased b-ion occurrence upon acetylation. Although guanidinylation had improved the balance of b- and y-ion occurrence, its impact on overall fragment ion intensity was less. It is the dual labeling that clearly improved the balance of b- and y-ion representation and also had a substantial increase in overall fragment ion intensities. This is also reflected in the increased accuracy of de novo amino acid annotation.
Dual Labeling of Six Protein Mix Improves the Accuracy of Protein Identification
It was clear from the de novo sequence analysis as well as fragmentation analysis that dual labeling of lipase improved the efficiency of peptide fragmentation. Nonetheless, it is important to ascertain universality of this procedure across different types of proteins. Bovine six protein mix is routinely used in mass spectrometry as a protein standard for evaluating the quality of the nLC-ESI MS/MS procedures. Unlabeled and dual labeled six protein mix were subjected to nLC-ESI MS/MS followed by Sequest HT search. Under ideal conditions, a bovine six protein mix is expected to give six protein hits. However, six hitchhikers are routinely identified upon database search (ABRF 2013) [64]. We have analyzed raw files of unlabeled and labeled six protein mix against Uniprot and NCBInr databases to evaluate accuracy and database dependency on protein identification (Supplementary Tables S8–S11). Sequence coverage obtained was slightly lower for dual labeled sample compared with unlabeled proteins. This could be attributable to the peptide loss associated with multiple desalting and resuspension steps and also due to changes in the peptide solubility upon modification. However, more importantly, the number of unique peptides is critical for protein identification and, in fact, total unique peptides in the digest increased from 48 to 53 upon dual labeling.
The number of protein dentifications were dependent on the database on which search was executed. Uniprot database had a higher number of protein identifications compared with NCBInr database. Sequest HT search of unlabeled and dual labeled peptides of bovine six protein against bovine species in Uniprot database gave 15 and 9 protein identifications, respectively, whereas 13 and 8 proteins were identified when the search was done using NCBInr Bos taurus database (Table 3).
While all the component proteins of bovine six protein mix were identified for both unlabeled and dual labeled samples, the accuracy of protein identification for bovine six protein mix improved upon dual labeling as the number of proteins identified (9 and 8) is close to the expected value (6), indicating a reduced number of false positives. Based on analysis of MS2 precursor ion intensities by Raw Neat ver. 2.1, a decrease in ion count and intensity was observed. Fragment ion intensities were higher for labeled peptides despite a marginal decrease in MS2 precursor ion intensities, as shown in Supplementary Figure S12. While MS2 precursor ion intensity certainly increases the chances of fragmentation, labeling is clearly increasing the relative ion intensity and occurrence, which are central to peptide/protein identification. The trade-off between MS2 precursor ion intensity versus increase in fragment ion relative intensity/occurrence is evident. By dual labeling of peptides, we were able to eliminate at least three to four common intrusive protein identifications. Optimization of peptide fragmentation by dual labeling has augmented increase in accuracy of protein identification.
Fragment Ion Occurrence was Balanced upon Dual Labeling for Six Protein Mix
The reason for the improvement in accuracy of protein inference accuracy was investigated further by checking the fragment ion occurrence differences for dual labeled and unlabeled samples. Similar to lipase, for bovine six protein mixture, also the fragment ion profile is largely dominated by b- and y-ions; y-ion occurrence percentage was higher compared with b-ions typical of an ion trap CID Figure 6a.

Comparison plot of variation in b- (blue) and y- (red) ion occurrence for peptides of bovine digest six protein mix unlabeled (a), dual labeled (b). Y-axis is the occurrence of ions (%) and the x-axis is fragment number. Dashed lines are neutral loss ions Y-NH3 (pink), yellow (y-H2O), purple (b-H2O), and turquoise (b-NH3)
However, the equivalent occurrence of both b- and y-ions was much clearer in the dual labeled bovine six protein mix compared with lipase Figure 6b. Thus, dual labeling of bovine six protein mixture improved the fragment ion occurrence with an improved protein inference accuracy as the final outcome.
Earlier studies employing chemical labels for improving fragment ion occurrence/intensity largely focused on enhancing or suppressing the representation of a particular fragment ion; however, the study presented here focused on equivalent representation of both types of fragment ions in the MS/MS spectra. It would be interesting to investigate the effect of this dual modification method in other fragmentation modes like HCD, ETD etc.
Conclusions
The efficiency of acetylation, guanidinylation, and their combination as routine chemical tools in peptide fragmentation was investigated. Labeled and unlabeled peptide digests were subjected to nLC ESI-MS/MS database search, de novo sequencing, and fragmentation analysis. From this analysis, it was clear that dual labeling was most efficient in improving the relative intensity and occurrence of the fragment ions in the spectra. This resulted in improved de novo sequence annotation accuracy and protein inference without compromising the sequence coverage. High efficiency, safety, and ease of these protocols make the dual labeling (acetylation and guanidinylation) of peptides the most attractive validation tool for de novo annotation as well database-dependent proteomic workflows.
References
Domon, B., Aebersold, R.: Options and considerations when selecting a quantitative proteomics strategy. Nat. Biotechnol. 28, 710–721 (2010)
Frese, C.K., Altelaar, A.F.M., Hennrich, M.L., Nolting, D., Zeller, M., Griep-Raming, J., Heck, A.J.R., Mohammed, S.: Improved peptide identification by targeted fragmentation using CID, HCD, and ETD on an LTQ-Orbitrap Velos. J. Proteome Res. 10, 2377–2388 (2011)
Huang, Y., Tseng, G.C., Yuan, S., Pasa-Tolic, L., Lipton, M.S., Smith, R.D., Wysocki, V.H.: A data-mining scheme for identifying peptide structural motifs responsible for different MS/MS fragmentation intensity patterns. J. Proteome Res. 7, 70–79 (2008)
Nair, H., Wysocki, V.H.: Are peptides without basic residues protonated primarily at the amino terminus? Int. J. Mass Spectrom. 174, 95–100 (1998)
Breci, L.A., Tabb, D.L., Yates III, J.R., Wysocki, V.H.: Cleavage N-terminal to proline: analysis of a database of peptide tandem mass spectra. Anal. Chem. 75, 1963–1971 (2003)
Huang, Y., Triscari, J.M., Pasa-Tolic, L., Anderson, G.A., Lipton, M.S., Smith, R.D., Wysocki, V.H.: Dissociation behavior of doubly-charged tryptic peptides: correlation of gas-phase cleavage abundance with ramachandran plots. J. Am. Chem. Soc. 126, 3034–3035 (2004)
Tabb, D.L., Huang, Y., Wysocki, V.H., Yates III, J.R.: Influence of basic residue content on fragment ion peak intensities in low-energy collision-induced dissociation spectra of peptides. Anal. Chem. 76, 1243–1248 (2004)
Dongre, A.R., Jones, J.L., Somogyi, A., Wysocki, V.H.: Influence of peptide composition, gas-phase basicity, and chemical modification on fragmentation efficiency: evidence for the mobile proton model. J. Am. Chem. Soc. 118, 8365–8374 (1996)
Gu, C., Tsaprailis, G., Breci, L., Wysocki, V.H.: Selective gas-phase cleavage at the peptide bond C-terminal to aspartic acid in fixed-charge derivatives of Asp-containing peptides. Anal. Chem. 72, 5804–5813 (2000)
Tabb, D.L., Smith, L.L., Breci, L.A., Wysocki, V.H., Lin, D., Yates III, J.R.: Statistical characterization of ion trap tandem mass spectra from doubly charged tryptic peptides. Anal. Chem. 75, 1155–1163 (2003)
Huang, Y., Triscari, J.M., Tseng, G.C., Pasa-Tolic, L., Lipton, M.S., Smith, R.D., Wysocki, V.H.: Statistical characterization of the charge state and residue dependence of low-energy CID peptide dissociation patterns. Anal. Chem. 77, 5800–5813 (2005)
Morrison, L., Somogyi, A., Wysocki, V.H.: The influence glutamic acid in protonated b –> b formation from VGEIG and related analogs. Int. J. Mass Spectrom. 325/327, 139–149 (2012)
Wysocki, V.H., Tsaprailis, G., Smith, L.L., Breci, L.A.: Mobile and localized protons: a framework for understanding peptide dissociation. J. Mass Spectrom. 35, 1399–1406 (2000)
Ballard, K.D., Gaskell, S.J.: Dehydration of peptide [M+H](+) ions in the gas phase. J. Am. Soc. Mass Spectrom. 4, 477–481 (1993)
Burlet, O., Yang, C.Y., Gaskell, S.J.: Influence of cysteine to cysteic acid oxidation on the collision-activated decomposition of protonated peptides: evidence for intraionic interactions. J. Am. Soc. Mass Spectrom. 3, 337–344 (1992)
Burlet, O., Orkiszewski, R.S., Ballard, K.D., Gaskell, S.J.: Charge promotion of low-energy fragmentations of peptide ions. Rapid Commun. Mass Spectrom. 6, 658–662 (1992)
Cox, K.A., Gaskell, S.J., Morris, M., Whiting, A.: Role of the site of protonation in the low-energy decompositions of gas-phase peptide ions. J. Am. Soc. Mass Spectrom. 7, 522–531 (1996)
Bleiholder, C., Suhai, S., Harrison, A.G., Paizs, B.: Towards understanding the tandem mass spectra of protonated oligopeptides. 2: the proline effect in collision-induced dissociation of protonated Ala-Ala-Xxx-Pro-Ala (Xxx = Ala, Ser, Leu, Val, Phe, and Trp). J. Am. Soc. Mass Spectrom. 22, 1032–1039 (2011)
Harrison, A.G., Young, A.B.: Fragmentation reactions of deprotonated peptides containing proline. The proline effect. J. Mass Spectrom. 40, 1173–1186 (2005)
Medzihradszky, K.F., Trinidad, J.C.: Unusual fragmentation of Pro-Ser/Thr-containing peptides detected in collision-induced dissociation spectra. J. Am. Soc. Mass Spectrom. 23, 602–607 (2012)
Herrmann, K.A., Wysocki, V.H., Vorpagel, E.R.: Computational investigation and hydrogen/deuterium exchange of the fixed charge derivative tris(2,4,6-trimethoxyphenyl) phosphonium: implications for the aspartic acid cleavage mechanism. J. Am. Soc. Mass Spectrom. 16, 1067–1080 (2005)
Mak, M., Mezo, G., Skribanek, Z., Hudecz, F.: Stability of Asp–Pro bond under high and low energy collision induced dissociation conditions in the immunodominant epitope region of herpes simplex virion glycoprotein D. Rapid Commun. Mass Spectrom. 12, 837–842 (1998)
Tsaprailis, G., Nair, H., Zhong, W., Kuppannan, K., Futrell, J.H., Wysocki, V.H.: A mechanistic investigation of the enhanced cleavage at histidine in the gas-phase dissociation of protonated peptides. Anal. Chem. 76, 2083–2094 (2004)
van Dongen, W.D., Ruijters, H.F., Luinge, H.J., Heerma, W., Haverkamp, J.: Statistical analysis of mass spectral data obtained from singly protonated peptides under high-energy collision-induced dissociation conditions. J. Mass Spectrom. 31, 1156–1162 (1996)
Somasundaram, P., Koudelka, T., Linke, D., Tholey, A.: C-terminal charge-reversal derivatization and parallel use of multiple proteases facilitates identification of protein C-termini by C-terminomics. J. Proteome Res. 15, 1369–1378 (2016)
Leitner, A., Lindner, W.: Chemistry meets proteomics: the use of chemical tagging reactions for MS-based proteomics. Proteomics 6, 5418–5434 (2006)
Kimmel, J.R.: Guanidination of proteins. Methods Enzymol. 11, 584–589 (1967)
Barlow, C.K., O'Hair, R.A.: Gas-phase peptide fragmentation: how understanding the fundamentals provides a springboard to developing new chemistry and novel proteomic tools. J. Mass Spectrom. 43, 1301–1319 (2008)
Roth, K.D., Huang, Z., Sadagopan, N., Watson, J.T.: Charge derivatization of peptides for analysis by mass spectrometry. Mass Spectrom. Rev. 17, 255–274 (1998)
Garcia-Murria, M.J., Valero, M.L., Sanchez Del Pino, M.M.: Simple chemical tools to expand the range of proteomics applications. J. Proteome 74, 137–150 (2011)
Hughes, C., Ma, B., Lajoie, G.A.: De novo sequencing methods in proteomics. Methods Mol. Biol. 604, 105–121 (2010)
Ma, B., Johnson, R.: De novo sequencing and homology searching. Mol. Cell. Proteomics 11, O111 (2012)
Ma, B.: Novor: real-time peptide de Novo sequencing software. J. Am. Soc. Mass Spectrom. 26, 1885–1894 (2015)
Devabhaktuni, A., Elias, J.E.: Application of de novo sequencing to large-scale complex proteomics data sets. J. Proteome Res. 15, 732–742 (2016)
Carabetta, V.J., Li, T., Shakya, A., Greco, T.M., Cristea, I.M.: Integrating Lys-N proteolysis and N-terminal guanidination for improved fragmentation and relative quantification of singly-charged ions. J. Am. Soc. Mass Spectrom. 21, 1050–1060 (2010)
Cockrill, S.L., Foster, K.L., Wildsmith, J., Goodrich, A.R., Dapron, J.G., Hassell, T.C., Kappel, W.K., Scott, G.B.: Efficient micro-recovery and guanidination of peptides directly from MALDI target spots. Biotechniques 38, 301–304 (2005)
Hennrich, M.L., Boersema, P.J., van den Toorn, H., Mischerikow, N., Heck, A.J., Mohammed, S.: Effect of chemical modifications on peptide fragmentation behavior upon electron transfer induced dissociation. Anal. Chem. 81, 7814–7822 (2009)
Alley Jr., W.R., Mechref, Y., Klouckova, I., Novotny, M.V.: Improved collision-induced dissociation analysis of peptides by matrix-assisted laser desorption/ionization tandem time-of-flight mass spectrometry through 3-sulfobenzoic acid succinimidyl ester labeling. J. Proteome Res. 6, 124–132 (2007)
Lee, Y.H., Kim, M.S., Choie, W.S., Min, H.K., Lee, S.W.: Highly informative proteome analysis by combining improved N-terminal sulfonation for de novo peptide sequencing and online capillary reverse-phase liquid chromatography/tandem mass spectrometry. Proteomics 4, 1684–1694 (2004)
Samyn, B., Debyser, G., Sergeant, K., Devreese, B., Van, B.J.: A case study of de novo sequence analysis of N-sulfonated peptides by MALDI TOF/TOF mass spectrometry. J. Am. Soc. Mass Spectrom. 15, 1838–1852 (2004)
Samgina, T.Y., Kovalev, S.V., Gorshkov, V.A., Artemenko, K.A., Poljakov, N.B., Lebedev, A.T.: N-terminal tagging strategy for de novo sequencing of short peptides by ESI-MS/MS and MALDI-MS/MS. J. Am. Soc. Mass Spectrom. 21, 104–111 (2010)
Miyashita, M., Hanai, Y., Awane, H., Yoshikawa, T., Miyagawa, H.: Improving peptide fragmentation by N-terminal derivatization with high proton affinity. Rapid Commun. Mass Spectrom. 25, 1130–1140 (2011)
Waldera-Lupa, D.M., Stefanski, A., Meyer, H.E., Stuhler, K.: The fate of b-ions in the two worlds of collision-induced dissociation. Biochim. Biophys. Acta 1834, 2843–2848 (2013)
Harrison, A.G.: Cyclization of peptide b9 ions. J. Am. Soc. Mass Spectrom. 20, 2248–2253 (2009)
Lau, K.W., Hart, S.R., Lynch, J.A., Wong, S.C., Hubbard, S.J., Gaskell, S.J.: Observations on the detection of b- and y-type ions in the collisionally activated decomposition spectra of protonated peptides. Rapid Commun. Mass Spectrom. 23, 1508–1514 (2009)
Atik, A.E., Yalcin, T.: A systematic study of acidic peptides for b-type sequence scrambling. J. Am. Soc. Mass Spectrom. 22, 38–48 (2011)
Chen, X., Yu, L., Steill, J.D., Oomens, J., Polfer, N.C.: Effect of peptide fragment size on the propensity of cyclization in collision-induced dissociation: oligoglycine b(2)–b(8). J. Am. Chem. Soc. 131, 18272–18282 (2009)
Erlekam, U., Bythell, B.J., Scuderi, D., Van Stipdonk, M., Paizs, B., Maitre, P.: Infrared spectroscopy of fragments of protonated peptides: direct evidence for macrocyclic structures of b5 ions. J. Am. Chem. Soc. 131, 11503–11508 (2009)
Harrison, A.G.: Peptide sequence scrambling through cyclization of b(5) ions. J. Am. Soc. Mass Spectrom. 19, 1776–1780 (2008)
Harrison, A.G.: To b or not to b: the ongoing saga of peptide b ions. Mass Spectrom. Rev. 28, 640–654 (2009)
Harrison, A.G.: Fragmentation reactions of b(5) and a (5) ions containing proline—the structures of a(5) ions. J. Am. Soc. Mass Spectrom. 23, 594–601 (2012)
Harrison, A.G., Young, A.B., Bleiholder, C., Suhai, S., Paizs, B.: Scrambling of sequence information in collision-induced dissociation of peptides. J. Am. Chem. Soc. 128, 10364–10365 (2006)
Yague, J., Paradela, A., Ramos, M., Ogueta, S., Marina, A., Barahona, F., Lopez de Castro, J.A., Vazquez, J.: Peptide rearrangement during quadrupole ion trap fragmentation: added complexity to MS/MS spectra. Anal. Chem. 75, 1524–1535 (2003)
Kulkarni, H.M., Ramesh, V., Srinivas, R., Jagannadham, M.V.: Acetylating tryptic peptides enhances b ion intensity in MALDI-TOF/TOF: implications in peptide sequencing and identification of proteins in an Antarctic bacterium Pseudomonas syringae. Proteom. Insights 3, 1–16 (2010)
Cupo, P., El-Deiry, W., Whitney, P.L., Awad, W.M.: Stabilization of proteins by guanidination. J. Biol. Chem. 255, 10828–10833 (1980)
Brancia, F.L., Oliver, S.G., Gaskell, S.J.: Improved matrix-assisted laser desorption/ionization mass spectrometric analysis of tryptic hydrolysates of proteins following guanidination of lysine-containing peptides. Rapid Commun. Mass Spectrom. 14, 2070–2073 (2000)
Liu, X., Zhang, J., Zheng, Z., Yang, X., Jia, W., Li, L., Gong, Y., Cai, Y., Zhu, Y., He, F.: A systematic N-terminal peptide quantitative labeling strategy for differential proteomic analysis. Proteom. Clin. Appl. 4, 633–643 (2010)
Acharya, P., Rao, N.M.: Stability studies on a lipase from Bacillus subtilis in guanidinium chloride. J. Protein Chem. 22, 51–60 (2003)
Markwell, M.A., Hass, S.M., Tolbert, N.E., Bieber, L.L.: Protein determination in membrane and lipoprotein samples: manual and automated procedures. Methods Enzymol. 72, 296–303 (1981)
Beardsley, R.L., Reilly, J.P.: Optimization of guanidination procedures for MALDI mass mapping. Anal. Chem. 74, 1884–1890 (2002)
Bhanuramanand, K., Ahmad, S., Rao, N.M.: Engineering deamidation-susceptible asparagines leads to improved stability to thermal cycling in a lipase. Protein Sci. 23, 1479–1490 (2014)
Barsnes, H., Eidhammer, I., Martens, L.: FragmentationAnalyzer: an open-source tool to analyze MS/MS fragmentation data. Proteomics 10, 1087–1090 (2010)
Jagannadham, M.V.: Identifying the sequence and distinguishing the oxidized methionine from phenylalanine peptides by MALDI TOF/TOF mass spectrometry in an Antarctic bacterium Pseudomonas syringae. Proteom. Insights 2, 27–31 (2009)
Andacht, T.M., Bennett, K.L., Bystrom, C., Dangott, L., Leszyk, J., Molina, H., Moritz, R.L., Phinney, B.S., Thompson, J.W., Williams, J., Elortza, F., Chambers, M., Tabb, D., Bunger, M.: The 2012/2013 PRG Study: assessing longitudinal variability in routine peptide LC-MS/MS analysis. J. Biomol. Tech. 24, S69 (2013)
Acknowledgements
The authors acknowledge support for this work by a Network grant on BioDiscovery (BSC0120). K.B.R. and S.R.K. acknowledge the fellowship from UGC and CSIR. The authors thank Ms. Kameswari for her input in the preparation of the manuscript.
Author information
Authors and Affiliations
Corresponding author
Additional information
K. Bhanuramanand and Sankara Rao Kola contributed equally to this work.
Electronic supplementary material
Tables: Sequest HT Search output files for lipase with labels as fixed and variable modifications, unlabeled and dual labeled bovine six protein mix digest searched against NCBInr and Uniprot databases in Excel files.
Figures: S1–S3 : Tandem mass spectra of labeled and unlabeled peptides of lipase; S4: median intensity of b- and y-ions derived from nLC ESI-MS/MS spectra; S5–S11: A box plot of the fragment ion and corresponding relative intensities for peptides of lipase; S12: Comparison of MS2 precursor ion intensity of unlabeled and labeled lipase and bovine six protien mix.
ESM 1
(DOCX 4078 kb)
Table S1
(XLSX 40 kb)
Table S2
(XLSX 74 kb)
Table S3
(XLSX 57 kb)
Table S4
(XLSX 33 kb)
Table S5
(XLSX 49 kb)
Table S6
(XLSX 33 kb)
Table S7
(XLSX 45 kb)
Table S8
(XLSX 32 kb)
Table S9
(XLSX 19 kb)
Table S10
(XLSX 33 kb)
Table S11
(XLSX 23 kb)
Table S12
(XLSX 32 kb)
Rights and permissions
About this article

Cite this article
Kuchibhotla, B., Kola, S.R., Medicherla, J. et al. Combinatorial Labeling Method for Improving Peptide Fragmentation in Mass Spectrometry. J. Am. Soc. Mass Spectrom. 28, 1216–1226 (2017). https://doi.org/10.1007/s13361-017-1606-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13361-017-1606-2