
Abstract
Tris(2,4,6-trimethoxyphenyl) phosphonium acetyl (TMPP-Ac) was previously introduced to improve the mass spectrometric sequence analysis of peptides by fixing a permanent charge at the N-termini. However, peptides containing arginine residues did not fragment efficiently after TMPP-Ac modification. In this work, we combine charge derivatization with photodissociation. The fragmentation of TMPP-derivatized peptides is greatly improved and a series of N-terminal fragments is generated with complete sequence information. Arginine has a special effect on the fragmentation of the TMPP tagged peptides when it is the N-terminal peptide residue. Theoretical and experimental results suggest that this is due to hydrogen transfer from the charged N-terminus to the hydrogen-deficient peptide sequence.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
Peptide sequencing by tandem mass spectrometry has become a well-established methodology [1, 2]. It is normally accomplished by comparing experimental fragmentation spectra with those theoretically predicted for all of the peptides in a protein or genomic database. Although this approach is quite effective, it can fail due to the occurrence of post-translational modifications, unknown mutations, or sequence errors in the database [3, 4]. Peptide de novo sequencing involves directly determining sequences from MS/MS spectra, independent of any database information [2, 5].
Interpretation of peptide MS-MS spectra can be complex. Low-energy activation, including both collision induced dissociation (CID) [6–9] and post-source decay (PSD) [7, 10], leads to mobilization of attached protons that weakens backbone amide bonds and facilitates peptide fragmentation [8]. As a result, gas-phase fragmentation of protonated peptides is typically dominated by cleavages of backbone amide bonds that yield b- and y-type ions. Ideally, a series of b ions or y ions would provide complete sequencing information. However, spectra often contain too few peaks. Because of enhanced cleavage C-terminal to acidic residues and N-terminal to proline [6, 11–13], the fragments corresponding to these cleavages can dominate spectra while others are hard to observe. Furthermore, appearance of other types of fragment ions, such as internal ions, neutral losses, N-terminal an, cn, and dn ions and C-terminal xn, zn, vn, and wn ions can excessively complicate spectra.
For a singly protonated peptide, arginine can sequester an otherwise mobile proton thereby simplifying MS/MS spectra. For example, if arginine is at the N-terminus of a peptide, its spectrum will be dominated by N-terminal fragment ions. However, this kind of charge sequestering is not absolute and a proton can still move around to some extent. To address these problems, several types of peptide derivatization schemes have been developed in the past 20 years [14–21]. Charge tags, based on phosphonium or quaternary ammonium ions, may be helpful in improving sensitivity for detecting peptides that may not readily ionize. They may also simplify the fragmentation of peptide ions, thereby facilitating interpretation of mass spectra [7, 22]. Charge tagged peptides have a fixed positive charge at one end, so in principle, only N-terminal or C-terminal fragments should be formed through charge-remote mechanisms [7, 12, 23].
A question that has arisen during the investigation of charge tagged peptides is whether the charge is really fixed. Trimethylammonium butyric acid N-hydroxysuccinimidyl ester (TMAB-NHS) [24, 25] is one reagent that can introduce a quaternary ammonium group on the N-termini of peptides. It has been previously reported that these modifications improve peptide detection sensitivity, and that mainly N-terminal fragments are observed in MALDI-PSD spectra of charged peptides, leading to simpler fragmentation patterns. However, we have demonstrated that TMAB-tagged peptides can lose a trimethylamine in the CID process generating a mobile proton [26]. In this case, both N- and C-terminal fragment ions appear in spectra, complicating peak identifications.
Another significant problem is that the low-energy collisional fragmentation of derivatized peptides can be inefficient [20]. For example, the reagent tris(trimethoxyphenyl) phosphonium acetic acid N-hydroxysuccinimidyl ester (TMPP-Ac-OSu) can introduce a positive phosphonium ion on the N-termini of peptides [16, 27, 28]. It does not decompose to produce a mobile proton like TMAB. Several studies demonstrated that the phosphonium ion is absolutely fixed at the N-terminus, and N-terminal an ions are produced [16–19, 29]. Recent studies also indicate that the fragmentation efficiency of arginine-containing charge-tagged peptides is low [20, 28, 30]. In fact, barely any sequence information can be obtained. The result that arginine-containing charge-tagged peptides behave differently than other peptides is surprising, since if the charge is sequestered in the tag, all fragments should be generated by a charge-remote mechanism and the basicity of residues should not affect the peptide fragmentation. Nevertheless, because C-terminal arginine-containing peptides are commonly generated by trypsin digestion, different approaches have been investigated to facilitate the fragmentation of TMPP derivatized peptides [28, 30]. Gebler et al. [28] improved their fragmentation by removing the C-terminal basic residue. Tsunasawa et al. [30] recently modified the arginine guanidino group with acetylacetone to reduce its basicity. They observed that when the arginine is at the N-terminus or in the middle of the peptide, this was effective. However, if the arginine is at the C-terminus of the peptide, their TMPP tagged peptides still fragmented poorly. The mechanism for this arginine effect is still not clear. In short, low energy collisional fragmentation of TMPP-tagged peptides still remains problematic.
High energy vacuum ultraviolet photodissociation offers a potential solution. Experimental and theoretical work have shown that peptides absorb light at around 160 and 190 nm associated with backbone chromophores [31–33], so F2 and ArF pulsed lasers, operating at 157 and 193 nm, are two common wavelengths used to excite peptide ions and induce fragmentation [34–38]. In previous studies with 157 nm ultraviolet light [37, 39], we demonstrated that when a proton is sequestered by arginine, a plethora of an ions or xn ions is generated, depending on the position of arginine in the sequence. With a photon energy of 7.9 eV, a mobile proton is not needed to facilitate peptide photodissociation. This should be an important advantage when working with charge tagged peptides.
In this work, peptide ions derivatized with [N-Tris(2,4,6-trimethoxyphenyl) phosphonium-acetyl (TMPP-Ac)] were photodissociated. Their fragmentation is quite efficient and a series of N-terminal fragments is generated with complete sequencing information. The influence of basic residues lysine and arginine in the sequence was investigated. Interestingly, arginine has a special effect on the fragmentation of the TMPP tagged peptides when it is the N-terminal peptide residue.
2 Experimental
2.1 Materials
α-Cyano-4-hydroxycinnamic acid, anhydrous methanol, and acetonitrile were obtained from Aldrich Chemicals (Milwaukee, WI, USA). Formic acid (FA) and trifluoroacetic acid (TFA) were purchased from Mallinckrodt Baker (Phillipsburg, NJ, USA). (N-succinimidyloxycarbonylmethyl) tris(2,4,6-trimethoxyphenyl)phosphonium bromide, angiotensin II (DRVYIHPF), and fibrinopeptide A (ADSGEGDFLAEGGGVR) were purchased from Sigma (St. Louis, MO, USA). Synthetic peptides AIEIFR, FSWAEGQR, FSWAEGQK, KFSWAEGQ, RFSWAEGQ, RAAALAAA, and RSAASLNS were purchased from Sigma-Genosys (The Woodlands, TX, USA). Peptide GSFGASIR was synthesized in-house via solid phase reaction [40].
2.2 Labeling Standard Peptides with TMPP-Ac-OSu
One hundred μg of peptide were dissolved in 100 μL labeling buffer (0.2 M NaHCO3 buffer, pH = 9, 20% acetonitrile) in a 1.6 mL microfuge tube, and 1 mg labeling reagent was added at a molar ratio about 10:1. The mixture was quickly vortexed and allowed to react at room temperature for 30 min. The reaction was quenched by adding 200 uL of water causing the reagent to hydrolyze over the next 30 min. Finally, concentrated formic acid was added to pH = 3 before HPLC purification.
2.3 HPLC
The reaction mixture was separated by reversed phase chromatography on a 2695 Bioalliance HPLC (Waters Corp.) using a Phenomenex C18 column (4.6 mm × 250 mm, 5 μm, 300 Å). The mobile phase contained the following: (1) 0.1% TFA in H2O; (2) 0.1% TFA in acetonitrile. The gradient elution was carried out at a flow rate of 1 mL/min with mobile phase B increasing from 5 to 60% in 30 min.
2.4 Mass Spectrometric Analysis
The labeled sample was diluted to 2 pmol/μL with H2O, and then 0.5 μL of this solution was spotted on a MALDI target plate. When the spots were dry, 0.5 μL of matrix solution (10 mg/mL CHCA in 50% ACN/50% H2O/0.1% trifluoroacetic acid) was applied on top of it.
MALDI mass spectra were recorded with an ABI 4700 TOF-TOF mass spectrometer (Foster City, CA, USA). All measurements were performed in positive reflectron mode. For the photodissociation experiments, an F2 laser (Coherent Lambda Physik, Germany) was connected to the ABI 4700 TOF-TOF mass spectrometer as previously described [41]. Its timing is automatically controlled by a programmable delay generator.
2.5 Computational Details
The geometry optimizations of charge-tagged dipeptides, their reaction intermediates, and products were performed using density functional theory (DFT) with the standard B3LYP hybrid exchange-correlation functional and the 6-31++G(d,p) basis set using the Gaussian suite of programs [42]. Low-energy conformations of the peptides were determined and used in the mechanistic studies. Analytical evaluation of force constants and vibrational frequencies was used to verify the nature of the minima and transition states. In particular, all transition states (TS) have one and only one imaginary frequency, and the corresponding normal coordinates were used to confirm the nature of the reactants and products in each step of the mechanism. Natural bond orbital (NBO) calculations were carried out on the optimized configurations for further analysis.
3 Results and Discussion
3.1 Arginine-Containing Peptides
Both PSD and photodissociation spectra of unlabeled and TMPP-tagged model peptides (arginine- and lysine-containing peptides) were recorded with the same apparatus. Figure 1a displays the MALDI-TOF/TOF PSD spectrum of peptide AIEIFR. y-Type ions dominate the spectrum as expected, since arginine is at the C-terminus of the peptide, but b ions also appear. The b3 ion intensity is particularly high due to enhancement by the acidic Asp [6, 11, 12]. In the photodissociation spectrum (Figure 1b), most of the fragments are x, v, w ions; small a2, b2, and immonium ions are also present. The strong intensities of a2, b2 ions are probably due to their special oxazolone structure [43–45]. In these two spectra, both N- and C-terminal fragment ions appear, but neither appears in a complete series. Figure 1c displays the PSD spectrum of TMPP-labeled AIEIFR. As seen in the Figure 1c, the positive charge group at the N-terminus of the peptides not only eliminates the C-terminal fragment ions as expected, but also eliminates most of other sequence ions. Those containing the TMPP tag are labeled with an asterisk. The only two bn ions observed are formed due to help from the hydrogen on a nearby carboxylic acid: the Glu side chain or the C-terminus. This is common in MS/MS spectra of TMPP derivatives [16]. The most intense peak, at m/z 573.19, is associated with the TMPP tag in its cleaved form [TMPP – CH = C = O]+). Loss of this charged TMPP tag leaves the peptide uncharged; further peptide fragmentation is unobservable. In conclusion, the PSD spectrum has been simplified by charge tag derivatization, but almost no information remains.

MALDI-TOF-TOF PSD spectra of AIEIFR (a) before, (c) after TMPP derivatization and photodissociation spectra of AIEIFR, (b) before, and (d) after TMPP derivatization
The photodissociation spectrum of TMPP-AIEIFR is presented in Figure 1d. A complete series of N-terminal a* ion fragments is observed. d *2 –d *4 Ions generated by the loss of side chain also appear. d-Type fragments can distinguish the isobaric amino acids leucine and isoleucine [4, 38, 39, 46]. The intensities of fragment ions are greatly improved compared with the PSD experiment.
The effect of arginine position in the sequence was next investigated. As noted above, Tsunasawa and coworkers improved fragmentation efficiency by reducing the basicity of arginine by a chemical modification [30]. However, they observed no improvement in fragment ion signal with arginine on the C-terminus, so arginine position in the sequence is evidently crucial. PSD and photodissociation spectra of Angiotensin II(DRVYIHPF) TMPP derivative were next recorded. In the PSD spectrum (Figure 1A in Supplementary Material), there is only one sequence ion, b *1 , corresponding to the enhanced cleavage C-terminal to Asp. The spectrum provides very little information about the peptide. In contrast, photodissociation (Supplementary Figure 1B) increases the intensity of all fragment ions. In particular, a complete an ion series appears in the spectrum.
The last example involves a TMPP-labeled peptide with arginine at its N-terminus, RAAALAAA. Its PSD spectrum (Figure 2a), displays only one peak (m/z = 573.19) because of the loss of the charge tag. In this extreme case, no sequence ions are observed in the spectrum. With photodissociation (Figure 2b), many fragments are detected: a *1 –a *7 ions, d *1 , and d *5 ions. However, we observed a significant decrease in the sequence ion signal. Since the sequence ion peak intensity is relatively low, the vertical scale in much of the spectrum has been expanded five times in order to display these fragment peaks. This significant drop in sequence ion intensity is associated with arginine being at the N-terminus of the peptide.

MALDI-TOF-TOF spectra of TMPP-RAAALAAA (a) PSD and (b) photodissociation
Several other peptides (ADSGEGDFLAEGGGVR, GSFGASIR, FSWAEGQR, RFSWAEGQ, and RSAASLNS) have been derivatized and results similar to those just reported were obtained. Photodissociation does improve the fragmentation efficiency of TMPP tagged peptides independent of where the arginine residue is located in the sequence. However, when arginine is at the N-terminus of the peptide, the intensities of backbone fragment ions are significantly smaller than when it is elsewhere. Fortunately, trypsin is the most commonly used proteolytic enzyme, and arginine rarely appears at peptide N-termini. Therefore, photodissociation of TMPP-tagged peptides should be of general utility.
3.2 Lysine-Containing Peptides
Lysine is a basic amino acid with properties similar to those of arginine [47–49]. Lysine-containing peptides were next investigated. The PSD spectrum of TMPP-FSWGAEGQK (Figure 3a) contains more sequence ions than the previously studied peptides (Figures 1c, 2a, and Supplementary Figures 1A and 2A); a *1 –a *3 and a *6 were observed together with some b-type ions, but they still do not yield a full peptide sequence. With photodissociation (Figure 3b), the intensities of the sequence ions are larger, and an almost complete series of an ions was recorded. The intensities of a *1 and a *3 ions are high since the aromatic residues at these locations (F and W) can absorb the photon energy more efficiently; a4 and a7 are not obvious in the spectrum; both are generated by cleavage C-terminal to glycine. Previous studies [50, 51] have shown that a radical C-terminal to glycine often undergoes secondary fragmentation to form other ions. Some d-type ions (d *2 , d *6 , and d *8 ) also appear in the spectrum because of the side chain loss.

MALDI-TOF-TOF spectra of TMPP-FSWGAEGQK (a) PSD and (b) photodissociation; MALDI-TOF-TOF spectra of TMPP-KFSWGAEGQ (c) PSD, and (d) photodissociation
In order to investigate whether the position of lysine in the sequence affects the fragment ion intensity, TMPP-KFSWGAEGQ was studied. The TMPP ion (m/z 573.19) dominates the PSD spectrum (Figure 3c) with some sequence ions just above noise level. Photodissociation (Figure 3d) produces more intense sequence ions and an almost complete series of an ions. Thus, photodissociation improves the fragmentation efficiency of TMPP-labeled lysine-containing peptides compared to PSD. In contrast with arginine, there is no significant intensity drop for sequence ions when lysine is at the N-terminus of the peptide.
3.3 Arginine Position Effect
To confirm the effect that arginine position has, we chose peptides (FSWGAEGQR and RFSWGAEGQ) with almost the same sequence as in the previous examples except that lysine was replaced by arginine.
Figure 4a and b display the photodissociation spectra of these two derivatized peptides. The two spectra look totally different. With TMPP-FSWGAEGQR, a complete series of an ions is observed; d *2 , d *6 , and d *8 ions are also present because the loss of amino acid side chains. Only one b ion (b *8 + H2O ions) is formed via a rearrangement at the C-terminus [16, 52]. In contrast, with TMPP-RFSWGAEGQ (Figure 4b), the spectrum is almost blank above m/z 600. Compared with the lysine effects seen in Figure 3b and d, arginine is obviously a much larger perturbation.

MALDI-TOF-TOF photodissociation spectra of (a) TMPP-FSWGAEGQR and (b) TMPP-RFSWGAEGQ
The intensity of the TMPP ion (m/z 573.19) in Figure 4b is worth noting. It becomes almost 10 times stronger than other sequence ions, while it is comparable to other sequence ions when arginine is at the C-terminus of the peptide (Figure 4a). Although formation of the TMPP+ ion fragment is associated with breaking of the amide bond, the backbone amide bond should be stronger than the C–C bond and only weakened when there is a mobile proton nearby. However, no evidence for the production of a mobile proton to break this amide bond has been reported. All the other sequence ions (a *1 –a *3 , a *5 , a *8 , d *2 , d *6 , d *8 , b *8 + H2O) in the MS2 spectra of TMPP-FSWGAEGQR and TMPP-RFSWGAEGQ are generated by C–C bond dissociation by a charge remote mechanism.
One possible explanation for this strong tag loss would involve a salt bridge between the arginine side chain and C-terminal carboxylic acid group. If the N-terminal arginine interacts with the C-terminal end and forms a ring structure, the TMPP group that is outside the ring might be easily cleaved off by photoexcitation. To test this hypothesis, the C-terminal carboxylic acid group was blocked by esterification of TMPP-RAAALAAA. Supplementary Figure 2 compares the photodissociation results before and after esterification. Clearly, there is no significant difference. The tag loss is still dominant in both spectra and all the sequence ions are weak, indicating that the salt bridge between the arginine side chain and C-terminal carboxylic acid group is not responsible for the arginine effect.
3.4 Computational Results and Analysis
In order to understand the experimental observations and elucidate the underlying reaction mechanisms, we have carried out quantum chemical calculations of the peptide structures using DFT methods. In almost all experiments just described, the stable charge tag product ion at m/z 573.19 was formed. This results from the peptide bond decomposition adjacent to the charge tag at the N-terminus. Production of the 573.19 Da mass loss requires that a hydrogen atom must transfer from the charged N-terminus (i.e., from TMPP+ – CH2–CO–), and almost certainly from the acetyl group.
Intramolecular hydrogen transfer is a well-known general unimolecular reaction in organic radical cations, the McLafferty rearrangement being a well-known example. Several studies [17, 29] have been devoted to elucidating the mechanisms of peptide and cation radical unimolecular dissociations. In particular, in the presence of a neighboring charged group, hydrogen atom migration is often feasible, and such a mechanism may be involved in the present case.
We focus our studies on intramolecular hydrogen atom transfer in some prototypical peptide models. Since the TMPP group is relatively large (C27H33O9P, 70 atoms) it was simulated by the parent system PH3, which is presumed to have electronic effects similar to those of TMPP [53]. (The ECD fragmentation of doubly-charged TMPP-tagged peptide ions has previously been studied by Chamot-Rooke and collaborators [53, 54]). The interactions between the charged phosphonium ion and peptides were modeled in conjugates that had PH +3 – CH2 –CO– linked to dipeptides AA, AR, and RA.
Structures of PH +3 – CH2–CO–AA and the charge tag fragment ion PH +3 – CH–CO were calculated as shown in Figure 5. PH +3 – CH2–CO–AA has elongated P–C and C–C bonds (1.83 and 1.53 Å, respectively) compared with the corresponding values in the PH +3 –CH–CO charge tag fragment (1.75 and 1.34 Å, respectively). It is clear that the product is best described as the resonance form PH +3 – CH = C = O. In the absence of any hydrogen migration, the strength of the C–N peptide bond in PH +3 – CH2–CO–AA is evaluated to be 66 kcal/mol. This is the minimum energy that must be input to create the radical products (PH +3 – CH2–CO and a peptide radical) via direct peptide bond cleavage. In the presence of hydrogen migration, however, more stable closed-shell products PH +3 – CH = C = O and a peptide can be formed, requiring significantly less energy.

Optimized structures of PH +3 – CH2 – CO – AA and the charge tag fragment product
Our analysis thus suggests that in the absence of a mobile proton, a hydrogen migration itself is able to weaken the peptide bond adjacent to the charge tag. To gain more insight into the nature of the hydrogen transport, we analyzed the reaction paths involving PH +3 – CH2–CO–AA, PH +3 – CH2–CO–AR, and PH +3 – CH2–CO–RA peptides to establish the transition state energy barriers for hydrogen migration. We investigated several possible reaction mechanisms for hydrogen migration. First we explored a one-step pathway (not shown) involving a direct hydrogen transfer from the methylene group to the amide nitrogen. Such a reaction proceeds via a four-membered cyclic TS with an associated high energy barrier (e.g., 60 kcal/mol for PH +3 – CH2–CO–AA), and is unlikely. Then we explored a two-step pathway (not shown) involving a hydrogen transfer from the methylene group onto the adjacent amide carboxyl followed by a hydrogen transfer to the amide nitrogen. However, both reaction steps again involve four-membered cyclic transition states. The resulting energy barriers, though lower than the direct one-step migration pathway, are still quite high (e.g., 48 and 44 kcal/mol for the two reaction steps for PH +3 – CH2–CO–AA). In addition, the rate-determining first step did not show any selectivity between PH +3 – CH2–CO–AA, PH +3 – CH2–CO–AR, and PH +3 – CH2–CO–RA, yielding essentially the same barrier in all three cases. This is not consistent with the experimental observations.
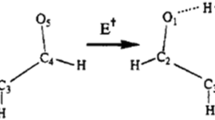
Finally, we considered a reaction pathway that does not involve such unstable four-membered cyclic structures. The key structures (minima and transfer states) involved in this reaction path for PH +3 – CH2–CO–AA are illustrated in Figure 6. As seen in Figure 6, this two-step reaction pathway involves a hydrogen transfer from the methylene group onto the alanine carboxyl, proceeding through a seven-membered cyclic transition state (TS), followed by a five-membered cyclic TS for the hydrogen transfer to the amide nitrogen. The energy barriers for the two reaction steps are substantially lower due to the stability of the larger rings (e.g., 24 and 16 kcal/mol for the two reaction steps for PH +3 – CH2–CO–AA). Hydrogen atom migration to the amide nitrogen site results in substantial bond length changes in the peptide bonds. For example, the C–C bond in the charge tag shrinks from 1.53 to 1.34 Å as it gains double bond character. More significantly, the peptide C–N bond increases from a value of 1.34 Å in PH +3 – CH2–CO–AA to 1.56 Å after hydrogen atom migration to the nitrogen. This is clear evidence of the weakening of the peptide bond after hydrogen atom migration from the adjacent carbon. An important consequence is that hydrogen migration facilitates the resulting amide bond cleavage.

Hydrogen migration and associated reaction path for PH +3 – CH2 – CO – AA
Hydrogen atom transport pathways have also been calculated for the dipeptide systems PH +3 – CH2–CO–AR and PH +3 – CH2–CO–RA. Relative energies of the different peptide dissociations via hydrogen migration are reported in Table 1. The barriers for hydrogen atom migration are qualitatively similar to what was described earlier for PH +3 – CH2–CO–AA. A more careful analysis of these reactions, however, provides a clue to understand the experimentally observed sequence dependence of the fragmentation efficiency. The PH +3 – CH2–CO–RA reaction path has a comparatively lower barrier by 2–4 kcal/mol (Table 1) than the PH +3 – CH2–CO–AA and PH +3 – CH2–CO–AR migration paths, presumably due to the high basicity side chain neighboring the charge tag. The possible effect on the transition states for hydrogen atom migration was studied using the natural population charge analysis (NPA) of oxygen and nitrogen atoms of the charge-tagged peptides. NPA atomic charge at the recipient amide group oxygen and nitrogen atoms were calculated for the reactants, transition states, intermediates, and products at the B3LYP/6-31++G(d,p) level. For the rate-determining Reactant → TS1 hydrogen atom migration, the charge on the recipient oxygen atom changes from −0.66 to −0.68 for PH +3 CH2 – CO – AA, −0.67 to −0.68 for PH +3 – CH2–CO–AR, and −0.65 to −0.69 for PH +3 – CH2–CO–RA. A larger difference in the charge of oxygen atoms proximate to arginine indicates a polarizing effect leading to a more favorable hydrogen migration. Thus, it appears that the hydrogen migration pathway can be particularly efficient in peptides containing arginine adjacent to the charge tag. These observations are sufficient to explain the arginine effect observed in the experimental spectra for various peptide sequences.
Calculations have also been carried out in the absence of the charge on the tag group attached to the peptides. The calculated barrier heights for intramolecular hydrogen transfer for comparable reactions are significantly higher (by 15–20 kcal/mol). Hence, in the absence of charge, intramolecular hydrogen transfer leading to peptide bond breaking is unlikely. In conclusion, the charge tag facilitates a hydrogen atom shift from the methylene carbon atom to the carbonyl oxygen and subsequently to the amide nitrogen, and this enables amide bond dissociation.
3.5 An Alternative Charge Tag Linkage
In order to further probe the mechanism involving the carboxylated intermediate and possibly improve the charge tag, the carbonyl group was replaced with CH2 by preparing ethyl tris(2,4,6-trimethoxyphenyl) phosphonium (ethyl-TMPP) derivatives. Unfortunately, a significant loss of the ethyl-TMPP moiety was still observed through a reverse Michael-type reaction [55], and no improvement was achieved.
4 Conclusions
Photodissociation (157 nm) provides a way to solve the problem of the low fragmentation efficiency of charge tagged peptides that is caused by the lack of a mobile proton. In general, a complete series of N-terminal fragment ions is formed, providing more sequence information. When arginine is at the N-terminus of the peptide, sequence ion intensity is significantly decreased and the loss of the tag becomes dominate in the spectrum. We believe that efficient loss of the tag is induced by peptide bond dissociation involving a hydrogen atom migration. Quantum chemical calculations have been carried out for a stepwise hydrogen atom migration reaction pathway involving a carboxylated intermediate. Energy barriers are found to be lower when arginine is present at the N-termini of peptides. In this case, instead of peptide backbone bonds being broken, the precursor ion quickly decomposes and loses the TMPP tag.
References
Aebersold, R., Mann, M.: Mass spectrometry-based proteomics. Nature 422(6928), 198–207 (2002)
Aebersold, R., Goodlett, D.R.: Mass spectrometry in proteomics. Chem. Rev. 101(2), 269–295 (2001)
Cox, J., Hubner, N.C., Mann, M.: How much peptide sequence information is contained in ion trap tandem mass spectra? J. Am. Soc. Mass Spectrom. 19(12), 1813–1820 (2008)
Zhang, L.Y., Reilly, J.P.: Peptide de novo sequencing using 157 nm photodissociation in a tandem time-of-flight mass spectrometer. Anal. Chem. 82(3), 898–908 (2010)
Taylor, J.A., Johnson, R.S.: Implementation and uses of automated de novo peptide sequencing by tandem mass spectrometry. Anal. Chem. 73(11), 2594–2604 (2001)
Burlet, O., Orkiszewski, R.S., Ballard, K.D., Gaskell, S.J.: Charge promotion of low-energy fragmentations of peptide ions. Rapid Commun. Mass Spectrom. 6(11), 658–662 (1992)
Johnson, R.S., Martin, S.A., Biemann, K.: Collision-induced fragmentation of (M + H)+ ions of peptides—side chain-specific sequence ions. Int. J. Mass Spectrom. Ion Processes 86, 137–154 (1988)
Wysocki, V.H., Tsaprailis, G., Smith, L.L., Breci, L.A.: Special feature: Commentary—mobile and localized protons: a framework for understanding peptide dissociation. J. Mass Spectrom. 35(12), 1399–1406 (2000)
Waugh, R.J., Bowie, J.H., Gross, M.L.: Collision-induced dissociations of deprotonated peptides. Dipeptides containing methionine or cysteine. Rapid Commun. Mass Spectrom. 7(7), 623–625 (1993)
Kaufmann, R., Kirsch, D., Spengler, B.: Sequencing of peptides in a time-of-flight mass spectrometer: evaluation of postsource decay following matrix-assisted laser desorption ionisation (MALDI). Int. J. Mass Spectrom. Ion Processes 131, 355–385 (1994)
Qin, J., Chait, B.T.: Preferential fragmentation of protonated gas-phase peptide ions adjacent to acidic amino-acid residues. J. Am. Chem. Soc. 117(19), 5411–5412 (1995)
Yu, W., Vath, J.E., Huberty, M.C., Martin, S.A.: Identification of the facile gas-phase cleavage of the Asp Pro and Asp Xxx peptide bonds in matrix-assisted laser-desorption time-of-flight mass-spectrometry. Anal. Chem. 65(21), 3015–3023 (1993)
Tsaprailis, G., Nair, H., Somogyi, Á., Wysocki, V.H., Zhong, W., Futrell, J.H., Summerfield, S.G., Gaskell, S.J.: Influence of secondary structure on the fragmentation of protonated peptides. J. Am. Chem. Soc. 121(22), 5142–5154 (1999)
Keough, T., Youngquist, R.S., Lacey, M.P.: A method for high-sensitivity peptide sequencing using postsource decay matrix-assisted laser desorption ionization mass spectrometry. Proc. Natl. Acad. Sci. U.S.A. 96(13), 7131–7136 (1999)
Keough, T., Youngquist, R.S., Lacey, M.P.: Sulfonic acid derivatives for peptide sequencing. Anal. Chem. 75(7), 156a–165a (2003)
Huang, Z.-H., Wu, J., Roth, K.D.W., Yang, Y., Gage, D.A., Watson, J.T.: A picomole-scale method for charge derivatization of peptides for sequence analysis by mass spectrometry. Anal. Chem. 69(2), 137–144 (1997)
Liao, P.C., Huang, Z.H., Allison, J.: Charge remote fragmentation of peptides following attachment of a fixed positive charge: A matrix-assisted laser desorption/ionization postsource decay study. J. Am. Soc. Mass Spectrom. 8(5), 501–509 (1997)
Huang, Z.-H., Shen, T., Wu, J., Gage, D.A., Watson, J.T.: Protein sequencing by matrix-sssisted laser desorption ionization-postsource decay-mass spectrometry analysis of the N-Tris(2,4,6-trimethoxyphenyl)phosphine-acetylated tryptic digests. Anal. Biochem. 268(2), 305–317 (1999)
Shen, T.L., Allison, J.: Interpretation of matrix-assisted laser desorption/ionization postsource decay spectra of charge-derivatized peptides: some examples of tris[(2,4,6-trimethoxyphenyl) phosphonium]-tagged proteolytic digestion products of phosphoenolpyruvate carboxykinase. J. Am. Soc. Mass Spectrom. 11(2), 145–152 (2000)
Pashkova, A., Chen, H.-S., Rejtar, T., Zang, X., Giese, R., Andreev, V., Moskovets, E., Karger, B.L.: Coumarin tags for analysis of peptides by MALDI-TOF MS and MS/MS. 2. Alexa Fluor 350 tag for increased peptide and protein identification by LC-MALDI-TOF/TOF MS. Anal. Chem. 77(7), 2085–2096 (2005)
Peters, E.C., Horn, D.M., Tully, D.C., Brock, A.: A novel multifunctional labeling reagent for enhanced protein characterization with mass spectrometry. Rapid Commun. Mass Spectrom. 15(24), 2387–2392 (2001)
Roth, K.D.W., Huang, Z.H., Sadagopan, N., Watson, J.T.: Charge derivatization of peptides for analysis by mass spectrometry. Mass Spectrom. Rev. 17(4), 255–274 (1998)
Gross, M.L.: Charge-remote fragmentations—method, mechanism, and applications. Int. J. Mass Spectrom. Ion Processes 118, 137–165 (1992)
Che, F.Y., Fricker, L.D.: Quantitative peptidomics of mouse pituitary: comparison of different stable isotopic tags. J. Mass Spectrom. 40(2), 238–249 (2005)
Mirzaei, H., Regnier, F.: Enhancing electrospray ionization efficiency of peptides by derivatization. Anal. Chem. 78(12), 4175–4183 (2006)
He, Y., Reilly, J.P.: Does a charge tag really provide a fixed charge? Angew. Chem. Int. Ed. 47(13), 2463–2465 (2008)
Shen, T.L., Huang, Z.H., Laivenieks, M., Zeikus, J.G., Gage, D.A., Allison, J.: Evaluation of charge derivatization of a proteolytic protein digest for improved mass spectrometric analysis: de novo sequencing by matrix-assisted laser desorption/ionization post-source decay mass spectrometry. J. Mass Spectrom. 34(11), 1154–1165 (1999)
Chen, W., Lee, P.J., Shion, H., Ellor, N., Gebler, J.C.: Improving de novo sequencing of peptides using a charged tag and C-terminal digestion. Anal. Chem. 79(4), 1583–1590 (2007)
Sadagopan, N., Watson, J.T.: Mass spectrometric evidence for mechanisms of fragmentation of charge-derivatized peptides. J. Am. Soc. Mass Spectrom. 12(4), 399–409 (2001)
Kuyama, H., Sonomura, K., Shima, K., Nishimura, O., Tsunasawa, S.: An improved method for de novo sequencing of arginine-containing, N-tris(2,4,6-trimethoxyphenyl)phosphonium-acetylated peptides. Rapid Commun. Mass Spectrom. 22(13), 2063–2072 (2008)
Clark, L.B.: Polarization assignments in the vacuum UV spectra of the primary amide, carboxyl, and peptide groups. J. Am. Chem. Soc. 117(30), 7974–7986 (1995)
Peterson, D.L., Simpson, W.T.: Polarized electronic absorption spectrum of amides with assignments of transitions. J. Am. Chem. Soc. 79(10), 2375–2382 (1957)
Woody, R.W., Koslowski, A.: Recent developments in the electronic spectroscopy of amides and alpha-helical polypeptides. Biophys. Chem. 101, 535–551 (2002)
Bowers, W.D., Delbert, S.S., Hunter, R.L., Mclver, R.T.: Fragmentation of oligopeptide ions using ultraviolet-laser radiation and Fourier-transform mass-spectrometry. J. Am. Chem. Soc. 106(23), 7288–7289 (1984)
Morgan, J.W., Russell, D.H.: Comparative studies of 193-nm photodissociation and TOF-TOFMS analysis of bradykinin analogues: The effects of charge site(s) and fragmentation timescales. J. Am. Soc. Mass Spectrom. 17(5), 721–729 (2006)
Choi, K.M., Yoon, S.H., Sun, M.L., Oh, J.Y., Moon, J.H., Kim, M.S.: Characteristics of photodissociation at 193 nm of singly protonated peptides generated by matrix-assisted laser desorption ionization (MALDI). J. Am. Soc. Mass Spectrom. 17(12), 1643–1653 (2006)
Thompson, M.S., Cui, W.D., Reilly, J.P.: Fragmentation of singly charged peptide ions by photodissociation at lambda = 157 nm. Angew. Chem. Int. Ed. 43(36), 4791–4794 (2004)
Reilly, J.P.: Ultraviolet photofragmentation of biomolecular ions. Mass Spectrom. Rev. 28(3), 425–447 (2009)
Cui, W.D., Thompson, M.S., Reilly, J.P.: Pathways of peptide ion fragmentation induced by vacuum ultraviolet light. J. Am. Soc. Mass Spectrom. 16(8), 1384–1398 (2005)
Wellings, D.A., Atherton, E.: Standard Fmoc protocols. Solid Phase Pept. Synth. 289, 44–67 (1997)
Zhang, L.Y., Reilly, J.P.: Peptide photodissociation with 157 nm light in a commercial tandem time-of-flight mass spectrometer. Anal. Chem. 81(18), 7829–7838 (2009)
Frisch, M.J.: Gaussian 09, Revision A.02. Gaussian, Inc, Wallingford CT (2009)
Cordero, M.M., Houser, J.J., Wesdemiotis, C.: The neutral products formed during backbone fragmentations of protonated peptides in tandem mass-spectrometry. Anal. Chem. 65(11), 1594–1601 (1993)
Paizs, B., Lendvay, G., Vekey, K., Suhai, S.: Formation of b(2)(+) ions from protonated peptides: an ab initio study. Rapid Commun. Mass Spectrom. 13(6), 525–533 (1999)
Harrison, A.G.: To b or not to b: The ongoing saga of peptide b ions. Mass Spectrom. Rev. 28(4), 640–654 (2009)
Johnson, R.S., Martin, S.A., Biemann, K., Stults, J.T., Watson, J.T.: Novel fragmentation process of peptides by collision-induced decomposition in a tandem mass-spectrometer—differentiation of leucine and isoleucine. Anal. Chem. 59(21), 2621–2625 (1987)
Harrison, A.G.: The gas-phase basicities and proton affinities of amino acids and peptides. Mass Spectrom. Rev. 16(4), 201–217 (1997)
Bliznyuk, A.A., Schaefer, H.F., Amster, I.J.: Proton affinities of lysine and histidine: a theoretical consideration of the discrepancy between experimental results from the kinetic and bracketing methods. J. Am. Chem. Soc. 115(12), 5149–5154 (1993)
Carr, S.R., Cassady, C.J.: Gas-phase basicities of histidine and lysine and their selected di- and tripeptides. J. Am. Soc. Mass Spectrom. 7(12), 1203–1210 (1996)
Zhang, L.Y., Cui, W.D., Thompson, M.S., Reilly, J.P.: Structures of a-type ions formed in the 157 nm photodissociation of singly-charged peptide ions. J. Am. Soc. Mass Spectrom. 17(9), 1315–1321 (2006)
Zhang, L.Y., Reilly, J.P.: Radical-driven dissociation of odd-electron peptide radical ions produced in 157 nm photodissociation. J. Am. Soc. Mass Spectrom. 20(7), 1378–1390 (2009)
Ballard, K.D., Gaskell, S.J.: Intramolecular oxygen-18 isotopic exchange in the gas phase observed during the tandem mass spectrometric analysis of peptides. J. Am. Chem. Soc. 114(1), 64–71 (1992)
Chamot-Rooke, J., Malosse, C., Frison, G., Turecek, F.: Electron capture in charge-tagged peptides. Evidence for the role of excited electronic states. J. Am. Soc. Mass Spectrom. 18(12), 2146–2161 (2007)
Chamot-Rooke, J., van der Rest, G., Dalleu, A., Bay, S., Lemoine, J.: The combination of electron capture dissociation and fixed charge derivatization increases sequence coverage for O-glycosylated and O-phosphorylated peptides. J. Am. Soc. Mass Spectrom. 18(8), 1405–1413 (2007)
Bunk, D.M., Macfarlane, R.D.: Derivatization to enhance sequence-specific fragmentation of peptides and proteins. Int. J. Mass Spectrom. Ion Processes 126, 123–136 (1993)
Acknowledgment
The authors acknowledge support for this work by the National Science Foundation (grants CHE-1012855 and CHE-0911454). They are thankful to one of the reviewers for suggesting the mechanism involving a methylene hydrogen transfer to the remote amide carboxyl group.
Author information
Authors and Affiliations
Corresponding author
Electronic Supplementary Materials
Below is the link to the electronic supplementary material.
ESM 1
(DOCX 181 kb)
Rights and permissions
About this article
Cite this article
He, Y., Parthasarathi, R., Raghavachari, K. et al. Photodissociation of Charge Tagged Peptides. J. Am. Soc. Mass Spectrom. 23, 1182–1190 (2012). https://doi.org/10.1007/s13361-012-0379-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13361-012-0379-x