
Abstract
Under considerations in the current study are reactions of the type \( {[{\text{Mn}}{\left( {\text{LOH}} \right)_{{2}}}]^{{{2} + }}} \to {\left[ {{\text{Mn}}\left( {\text{LO}} \right)} \right]^{ + }} + {\text{LO}}{{\text{H}}_{{2}}}^{ + } \), where the ligand LOH represents water or/and methanol. Preferential proton transfer reactions and loss of any ligand fragments are discussed in the light of ligand polarizability, dipole moment, dissociation energy, proton affinity, differences in ligand-ion ionization energy, and ion radii. The results indicate the proton affinity and dissociation energy of the O–H bond are more important for the overall proton transfer reaction than differences in the first ionization energy of the ligand and the second ionization energy of the metal ion.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
Studies of metal atoms and ions and their chemical reactivity in the gas phase are very similar to, and in many ways mimic, the coordination to ligands in human and plant cells, due to the limited amount of surrounding water or other ligand molecules. Since in vivo experiments are difficult to carry out, gas phase experiments of metal ions together with theoretical investigations can reveal a lot of information about the subtle interplay between a metal ion and different types of coordinating species. Manganese, an element with a wide range of oxidation states from –III to + VII, is found to a large extent in biological systems [1] in which it has been suggested it can act as a Lewis acid in enzymes as well as an oxidation catalyst. [2] Hence, computational studies in this area are extremely useful in order to reveal physical and chemical information about the role of different metal atoms in human or plant cells. Several research groups have shown a large interest in the stability of metal ion-ligand complexes. Stability of complexes can be evaluated and interpreted via physical properties of ligands, metal ions, and by the minimum number of ligands that are required to stabilize the complex. In one experimental study made by Stace and co-workers [3] based on mass-spectrometry, the aim was to investigate the different physical properties of oxygen- and nitrogen-coordinating ligands with the Mg2+-ion. In this study, Stace performed studies for the gas phase stability of [Mg(L)N]2+ complexes using the pick-up technique. Similar studies were performed by Shvartsburg and Siu, [4] in which the number of water molecules required to stabilize various metal dications was explored. The goal was to see if there is a minimum size n min below which metal ion complex dissociation occurs spontaneously through charge transfer or through proton transfer. In another experimental study by Stace et al., [5] different fragmentation pathways for [Mg(CH3OH)N]+ and [Mg(CH3OH)N]2+ complexes were investigated. These fragmentation pathways included numerous reactions of charge-transfer, proton transfer, and neutral ligand loss. One important conclusion from these studies was that the stable complexes can be divided into three different categories. In the first group belong ligands with high first ionization energy (IE) > 12 eV, which makes any charge transfer reaction unfavorable. The critical n min in this group is 1 or 2. Water, carbon dioxide, and acetonitrile belong to this group. In the second group come ligands with a quite small IE, but with sufficiently large polarizability. The minimum value for stabilization of a complex is 2. Members of this group are pyridine, diethyl ether, and butylamine. Finally, the last group contains ligands that display a combination of the different physical properties which, taken together, can stabilize the complex, and the minimum size in this group is 3. It is a well known fact that the difference between the ionization energy of a metal ion and a ligand is a crucial factor in the stabilization of a complex against charge transfer, and Marcus [6] has given a detailed analysis of charge transfer processes in solution. Possible charge transfer processes that can occur in the gas phase by a single molecule and a doubly charged metal ion have been suggested in later work, [7]. The difference between the second ionization energy of the metal ion, M2+, and the first ionization energy of the ligand, has been given as a reason for proton transfer reactions to occur. The magnitude of this difference determines if a charge transfer reaction will be energetically favorable or not. If the difference is large, it will occur spontaneously at contact between a doubly charged metal ion and a neutral ligand.
Using the technique of electrospray ionization, Kebarle and co-workers, [8] investigated the ions of Mg, Ca, Sr, Ba, and many ions of the first row transition metals. Only hydroxide species were detected for Be and Cu, whereas for Mg, Ca, and Sr hydroxide species as well as [M(H2O)N]2+ indicating stable complexes were found. No hydroxide species could be found for Ba.
Two specific reaction pathways were observed; proton transfer, Reaction 1, and neutral ligand loss, Reaction 2.
This group also observed a decreasing maximum number of coordinated water molecules along with a decreasing second ionization energy of the metal. It was suggested that the two important factors for the proton transfer to occur were the second ionization energy of the metal and the bond dissociation energy, ED, between the metal ion and the hydroxyl group, ED(M+-OH). This analysis of Kebarle et al. was later revised in a computational study by Beyer et al. [9] In this study, proton transfer reactions of [M(H2O)2]2+-system, where M = Be, Mg, Ca, Sr, and Ba, were investigated. Beyer discussed the concept and the implications of an ionic bond between the metal ion and the hydroxyl group, rather than a covalent bond considered in earlier studies. The bond dissociation energy between the metal-ion and the hydroxyl group ED(M2+-OH– ) was expressed as a result of three different energetic contributions:
where IE(M+) is the second ionization energy of the metal ion, ED(M+-OH) defined as previously and EA(OH) is the electron affinity of the -OH group. Accordingly, the total energy for the proton transfer reaction ETot for a metal ion-complex can be expressed as:
where Eave[M(H2O)N]2+ and Eave[M(OH(H2O)N-2]+ represents the average binding energy of N water molecules to the metal ion before and after the proton reaction respectively, and EPT consists of two terms, one of them that includes the energy for performing the reaction:
and one additional term ECoulomb, which is the energy released by the Coulomb explosion.
It was found that the proton transfer reaction proceeded via a second shell intermediate, with one water molecule hydrogen bonded in the second shell in order to facilitate the proton transfer. Two transition states were located on the potential energy surface for all ions except Be. The second transition state was found to lower the energy barrier for the proton transfer reaction and thus acted as a loophole for the release of H3O+. In similar studies, [10] the doubly charged ions of Sn, Pb, and Hg were considered for metal ion complexes of type [M(H2O)2]2+ and [M(H2O)4]2+. A second transition state connected by a second shell intermediate proceeding the proton transfer, was also found for the [M(H2O)2]2+-complexes of the work of Stace [10] and in one study by Metz and co-workers [11]. Second shell structure preceding a proton transfer reaction for [Mg(R OH)N]2+-complexes was also discussed in another paper by Stace [12].
1.1 Computational Details
Based on the work of Beyer et al., only [Mn(H2O)2]2+, [Mn(CH3OH)2]2+, and [Mn(H2O)(CH3OH)]2+-complexes are considered. All calculations were done with the Gaussian 03 [13] package, employing the B3LYP [14]-functional with the 6–311 basis set equipped with (3df, 3pd)-polarization functions on all atoms, in order to model the hydrogen bonded system adequately. Frequency calculations were performed on all structures to ensure minima of the potential energy surface. Initial starting geometries for the transition state search with the quasi-Newton synchronous transit-guided method (QST3) in Gaussian 03, were found via a relaxed potential energy scan for the complexes. The scan was performed decreasing the O-Mn-O angle in 20 steps. Intrinsic reaction coordinate calculations (IRC) and an additional structure relaxation confirmed the connection between a minimum and an obtained transition state on the potential energy surface. All the calculations were performed on the Swedish computer clusters Akka and Sarek.
2 Results and Discussion
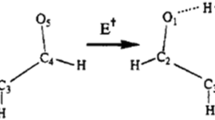
As already pointed out, a second transition state could be found for the [Mn(H2O)2]2+-complex, but only one transition state that connects the reactants and the products could be located for the [Mn(CH3OH)2]2+ and the [Mn(H2O)(CH3OH)]2+-complexes. Despite numerous attempts, no complete proton transfer reaction could be obtained for the H3O+-loss from the [Mn(H2O)(CH3OH)]2+-complex. Figures 1, 2, and 3 show the structures in the reaction sequence for proton transfer reactions and, similarly, Tables 1, 2, and 3 show important bond lengths for these structures.

Proton transfer reaction for a [M(H2O)2]2+-complex. These are the different structures during the proton transfer reaction \( {[{\text{Mn}}{({{\text{H}}_{{2}}}{\text{O}})_{{2}}}]^{{{2} + }}} \to {\left[ {\text{MnOH}} \right]^{ + }} + {{\text{H}}_{{3}}}{{\text{O}}^{ + }} \)

Proton transfer for a [Mn(CH3OH)2]2+ -complex

Proton transfer reaction for a [Mn(CH3OH)(H2O)]2+-complex. These are the different structures during the proton transfer reaction \( {[{\text{Mn}}({\text{C}}{{\text{H}}_{{3}}}{\text{OH}})({{\text{H}}_{{2}}}{\text{O}})]^{{{2} + }}} \to {\left[ {\text{MnOH}} \right]^{ + }} + {\text{C}}{{\text{H}}_{{3}}}{\text{O}}{{\text{H}}_{{2}}}^{ + } \)
Energy reaction profiles for proton transfer reactions have been calculated for the [Mn(H2O)2]2+, [Mn(CH3OH)2]2+, and the [Mn(H2O)(CH3OH)]2+-complexes, respectively. These profiles are shown in Figures 4, 5, and 6. Also shown are the energy barriers for electron transfer reactions and for neutral ligand loss. In all reactions, proton transfer reactions are always lower in energy than neutral ligand loss and electron transfer and, hence, more likely to be observed in experiment. Table 4 show different physical properties for water and methanol molecules in the gas phase, which will be discussed in context to the shape of the energy profiles. Figure 4 shows the energy profile for proton transfer of a [Mn(H2O)2]2+-complex, which agrees very well with the corresponding energy profile for a [Mg(H2O)2]2+-complex from the study of Beyer et al. [9].

Energy profile for the reaction \( {[{\text{Mn}}{({{\text{H}}_{{2}}}{\text{O}})_{{2}}}]^{{{2} + }}} \to {\left[ {\text{MnOH}} \right]^{ + }} + {{\text{H}}_{{3}}}{{\text{O}}^{ + }} \)

Energy profile for the reaction \( {[{\text{Mn}}{({\text{C}}{{\text{H}}_{{3}}}{\text{OH}})_{{2}}}]^{{{2} + }}} \to {[{\text{Mn}}({\text{C}}{{\text{H}}_{{3}}}{\text{O}})]^{ + }} + {\text{C}}{{\text{H}}_{{3}}}{\text{O}}{{\text{H}}_{{2}}}^{ + } \)

Energy profile for the reactions \( {[{\text{Mn}}({\text{C}}{{\text{H}}_{{3}}}{\text{OH}})({{\text{H}}_{{2}}}{\text{O}})]^{{{2} + }}} \to {[{\text{Mn}}({\text{C}}{{\text{H}}_{{3}}}{\text{O}})]^{ + }} + {{\text{H}}_{{3}}}{{\text{O}}^{ + }} \) and \( {[{\text{Mn}}({\text{C}}{{\text{H}}_{{3}}}{\text{OH}})({{\text{H}}_{{2}}}{\text{O}})]^{{{2} + }}} \to {\left[ {{\text{Mn}}\left( {\text{OH}} \right)} \right]^{ + }} + {\text{C}}{{\text{H}}_{{3}}}{\text{O}}{{\text{H}}_{{2}}}^{ + } \)
Taking the initial [M(H2O)2]2+-complex as a zero point, the first transition point, TS1, lies higher for [Mg(H2O)2]2+ at 152.9 kJmol–1 than that for the [Mn(H2O)2]2+-complex at 115.7 kJmol–1. The second shell intermediate structure lie at 131.9 kJmol–1 for [Mg(H2O)2]2+-complex, whereas the corresponding point for the [Mn(H2O)2]2+-complex only reaches 83.6 kJmol–1. Finally, the last transition structure, TS2, displays a reversed behavior compared with TS1, which stays at 165.0 kJmol–1 for [Mg(H2O)2]2+ and 102.6 kJmol–1 for [Mn(H2O)2]2+. This is surprising since the second ionization energy for Mg is slightly lower than that of Mn (15.0 eV for Mg and 15.6 eV for Mn), i.e., the energy difference for Mg and H2O is larger than that of Mn and H2O. Previous studies [3] suggested that a large energy difference between the second ionization energy of the metal ion and the first ionization energy of the ligand would result in a spontaneous charge transfer reaction. Hence, a larger difference should result in lower energy barriers which is not in accordance with the date presented here, where the transition states for the [Mg(H2O)2]2+-complex lies higher above the zero-point than the [Mn(H2O)2]2+-complex. One of the conclusions from the study of Beyer et al. [9] was that the transition states are correlated to the ionic radius, rather than to differences in ionization energies between the metal ion and the ligand. This correlation can also be found in this study, since the data can be better understood when the ionic radii for Mg2+ and Mn2+ are taken into account. These radii are shown in Table 5. Also shown in Figure 4 is the energy barriers for neutral ligand loss at 300 kJmol–1 and the electron transfer reaction at approximately 200 kJmol–1. Similar barrier heights for neutral ligand loss and electron transfer were also seen in the study of Beyer et al., which strongly indicates a low accessibility for these two reactions. Electron transfer was not detected or observed in the studies of Kebarle et al. [8].
The data presented in Table 4 show that the proton affinity is higher for the methanol molecule than for the water molecule, which can explain the higher energy barrier for the proton transfer reaction of the “methanol only” complex, shown in Figure 5. On the other hand, the first ionization energy, IE, is considerably lower for methanol than for water. This implies a larger difference between the ionization energy of the ligand and the second ionization energy of the metal ion, and this energy difference gives a higher possibility for electron transfer within the complex. Interestingly, the barrier for electron transfer reaction is only about 90 kJmol–1, a result from this larger energy difference.
It is also reasonable to suspect that the hydrogen in the O–H group in the methanol molecule is more acidic than the hydrogen atoms in the [Mn(H2O)2]2+-complex. If electron density is withdrawn from the hydrogen atom on the hydroxyl group in the [Mn(CH3OH)2]2+ complex, the proton transfer reaction can proceed more readily and therefore the energy barrier is lowered to some extent. The net effect is shown in Figure 5, where a small increase of the energy barrier for proton reaction can be seen compared with the “water-only” complex. An elevated level for the proton transfer reaction in [Co(CH3OH)N]2+ complexes was also confirmed in the work of Metz and coworkers [17].
The energy barrier for neutral ligand loss is raised to an even higher level, reflecting a higher binding energy for methanol than for water [18]. Finally, the energy barrier for the mixed ligand complex in Figure 6 follows the same pattern as the other observations in this study. Two transition states were located for loss of H3O+. However, no successful IRC calculation could connect the products with any of the two transition states found and, hence, this reaction pathway was ruled out.
The lowest transition state for loss of H3O+ from the [Mn(H2O)(CH3OH)]2+ complex lies approximately 19 kJmol–1 above the transition state for loss of CH3OH +2 , which is indicated by the dotted line in Figure 6. Furthermore, neutral ligand loss and electron transfer reactions for methanol or water from the [Mn(H2O)(CH3OH)]2+ complex are shown in Figure 6. Only electron transfer from the H2O molecule seems to be accessible at 66 kJmol–1 above the zero-point. Electron transfer reaction and loss of CH3OH+ and neutral ligand loss of either methanol or water all shows reactions barrier above 200 kJmol–1, which makes these reactions unfavorable. Three transition states could be found for CH3OH +2 , but only one of them proved to be a true transition state for the reaction considered. This is a manifestation of the higher proton affinity in combination with a higher bond dissociation energy ED(RO-H) of the methanol molecule, which makes loss of CH3OH +2 more favorable than loss of H3O+. The reaction pathways discussed above might not be the only possible ones, and other reaction/fragmentation pathways could be more accessible than the pathways considered in this study. For example, when investigating fragmentation pathways arising from [Mn(CH3OH)N]2+ complexes, Stace et al. [5] found one of the prominent signals in the MIKES spectra coming from [Mg(OH)(CH3OH)]+, which implies loss of CH +3 . Hence, one possible explanation for the absence of H3O+ loss from the mixed ligand complex in this study could be an indication of the presence for other more accessible pathways not dealt with in the current study.
2.1 Natural Population Charges
In order to analyze the importance of charge transfer during the proton transfer reaction, natural charges on all atoms in the complexes for the reactions, have been investigated and are shown in Tables 6, 7, and 8, respectively. Beyer et al. made a similar investigation and found that the charge on the metal ion only changed by 0.1 e for Ca and the other metal ions. The conclusion was that charge transfer was not the driving force for the proton transfer reaction. The charges on the atoms in Tables 6, 7, and 8 reveal that the biggest change in charges occurs for the two oxygen atoms and the manganese atom. Although a larger change can be observed in the current study in comparison to the values reported by Beyer et al. [9], the charge transfer is relatively small and, hence, the difference in second and first ionization energies of the metal ion and the ligand play a minor role for the proton transfer reaction. However, the ionization energies might have a crucial impact on charge transfer reactions.
As a consequence of the minor change in natural charge on the atoms, the two most important factors that determine if a proton transfer reaction will occur are the proton affinity PA, and the bond dissociation energy ED of the O–H bond. Furthermore, no conclusion or prediction can be made here if the factors change their importance or influence with a changing number of ligands in the complex.
3 Conclusions
Proton transfer reactions have been investigated for the smallest possible of ligands size, and the results have been evaluated in the light of ionization energies of ligand and metal ion, proton affinity, and ion size. The results show that proton affinity and dissociation energy of the O–H bond are believed to be more important for the overall proton transfer reaction than differences in the first ionization energy of the ligand and the second ionization energy of the metal ion. For a “water-only” complex, proton transfer is facilitated by two reaction barriers via a second shell structure, similar to what has been found for hydrated [Mg(H2O)2]2+ complex by Beyer [9]. For a “methanol-only” complex only one reaction barrier could be located with a slightly higher reaction barrier than water containing complexes. For a mixed complex, only loss of CH3OH +2 could be found, no complete reaction pathway was found for loss of H3O+. For all complexes, proton transfer reaction is found to be lower in energy compared with neutral ligand loss or electron transfer reaction. With these results, more complementary information has been shown in conjunction with the previous studies of proton transfer and mixed ligand complexes made by Stace and Beyer [9, 18].
References
Frieden, E. New perspectives on the essential trace elements. J. Chem. Ed., 62, 11, 917–923 (1985)
da Silva, J. R. R. F.; Williams, R. J. P. The biological chemistry of the elements: the inorganic chemistry of life. Clarendon Press; Oxford University Press: Oxford, UK/New York, NY. pp. 370–386 (1991)
Walker, N., Dobson, M., Wright, R., Barran, P., Murrell, J., Stace, A.: A gas-phase study of the coordination of Mg2+ with oxygen- and nitrogen-containing ligands. J. Am. Chem. Soc. 122, 11138–11145 (2000)
Shvartsburg, A., Siu, K.: Is there a minimum size for aqueous doubly charged metal cations? J. Am. Chem. Soc. 123, 10071–10075 (2001)
Woodward, C.; M. D. A.J.; Stace, A. Chemistry of Mg+ and Mg2+ in association with methanol clusters. J. Phys. Chem. A 101, 2279–2287 (1997)
M. R. A. Chemical and electrochemical electron-transfer theory. Ann. Rev. Phys. Chem. 15, 155–196 (1964)
Corongiu, G., Clementi, E.: Study of Structure of Molecular Complexes. 16. Doubly Charged Cations Interacting with Water. J. Chem.Phys. 69, 4885–4887 (1978)
Jayaweera, P., Blades, A., Ikonomou, M., Kebarle, P.: Production and Study in the Gas-Phase of Multiply Charged Solvated or Coordinated Metal-Ions. J. Am. Chem. Soc. 112, 2452–2454 (1990)
Beyer, M., Williams, E., Bondybey, V.: Unimolecular reactions of dihydrated alkaline earth metal dications [M(H2O)2]2+, M = Be, Mg, Ca, Sr, and Ba: Salt-bridge mechanism in the proton-transfer reaction [M(H2O)2]2+ → MOH+ + H3O+. J. Am. Chem. 121, 1565–1573 (1999)
Cox, H., Stace, A.: Molecular view of the anomalous acidities of Sn2+, Pb2+, and Hg2+. J. Am. Chem. Soc. 126, 3939–3947 (2004)
Metz, R.B.: Optical spectroscopy and photodissociation dynamics of multiply charged ions Int. J. Mass Spectrom. 235, 131–143 (2004)
Wu, B., Duncombe, B., Stace, A.: The solvation of Mg2+ with gas-phase clusters composed of alcohol molecules. J. Phys. Chem. A 112, 2182–2191 (2008)
Frisch, M.J., Trucks, G.W., Schlegel, H.B., Scuseria, G.E., Robb, M.A., Cheeseman, J.R., Montgomery, J.A., Vreven Jr., T., Kudin, K.N., Burant, J.C., Millam, J.M., Iyengar, S.S., Tomasi, J., Barone, V., Mennucci, B., Cossi, M., Scalmani, G., Rega, N., Petersson, G.A., Nakatsuji, H., Hada, M., Ehara, M., Toyota, K., Fukuda, R., Hasegawa, J., Ishida, M., Nakajima, T., Honda, Y., Kitao, O., Nakai, M.I.H., Klene, M., Li, X., Knox, J.E., Hratchian, H.P., Cross, J.B., Bakken, V., Adamo, C., Jaramillo, J., Gomperts, R., Stratmann, R.E., Yazyev, O., Austin, A.J., Cammi, R., Pomelli, C., Ochterski, J.W., Ayala, P.Y., Morokuma, K., Voth, G.A., Salvador, P., Dannenberg, J.J., Zakrzewski, V.G., Dapprich, S., Daniels, A.D., Strain, M.C., Farkas, O., Malick, D.K., Rabuck, A.D., Raghavachari, K., Foresman, J.B., Ortiz, J.V., Cui, Q., Baboul, A.G., Cliord, S., Cioslowski, J., Stefanov, B.B., Liu, G., Liashenko, A., Piskorz, P., Komaromi, I., Martin, R.L., Fox, D.J., Keith, T., Al-Laham, M.A., Peng, C.Y., Nanayakkara, A., Challacombe, M., Gill, P.M.W., Johnson, B., Chen, W., Wong, M.W., Gonzalez, C., Pople, J.A.: Gaussian 03, Revision C.02. Gaussian, Inc, Wallingford, CT (2004)
Becke, A.: D.; Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 98, 5648–5652 (1993)
Lide, D. CRC Handbook of Chemistry and Physics, Internet Version 2005, http://www.hbcpnetbase.com. CRC Press: Boca Raton, FL, 2005.
Shannon, R.: Revised Effective Ionic Radii and Systematic Studies of Interatomic Distances in Halides and Chalcogenides. Acta Crystallogr. A32, 751–767 (1976)
Thompson, C.J., Faherty, K.P., Stringer, K.L., Metz, R.B.: Electronic spectroscopy and photodissociation dynamics of Co2+-methanol clusters: [Co(CH3OH)]2+ (n = 4–7). Phys. Chem., Chem. Phys. 7, 814–818 (2005)
Duncombe, B., Rydén, J., Puskar, L., Cox, H., Stace, A.: A Gas-Phase Study of the Preferential Solvation of Mn2+ in Mixed Water/Methanol Clusters. J. Am. Soc. Mass Spectrom. 19, 520–530 (2008)
Acknowledgment
The authors thank SNIC for allocated computational time on the computer clusters Akka and Sarek.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(BIB 10 kb)
Rights and permissions
About this article
Cite this article
Rydén, J., Öberg, S. Proton Transfer Reactions for Methanol and Water Containing Manganese Ion Complexes. J. Am. Soc. Mass Spectrom. 22, 2276–2281 (2011). https://doi.org/10.1007/s13361-011-0260-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13361-011-0260-3