
Abstract
A new type of combination ion source has been devised. It unites two complementary ionization methods, i.e., liquid injection field desorption/ionization (LIFDI) and electrospray ionization (ESI). This LIFDI-ESI combination ion source has been constructed for a Fourier transform ion cyclotron resonance (FT-ICR) mass spectrometer. The LIFDI-ESI combination ion source can be switched between the LIFDI and ESI modes of operation within 15 min without breaking the vacuum. The source design and its operation are described. LIFDI-FT-ICR spectra of the ionic liquid trihexyl(tetradecyl)-phosphonium tris(pentafluoroethyl)-trifluorophosphate, polyethylene glycol 600, 2,3,4-tridodecyloxy-benzaldehyde, and [60]fullerene are described.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
In the 1970s, field desorption (FD) became known as the first soft desorption/ionization method in mass spectrometry (MS) [1–4]. As documented in the literature [5] and proven by our own experience [6–13], FD-MS performs equally well for the analysis of nonpolar, polar, and ionic compounds. To effect the soft ionization of atoms and molecules, strong electric fields in the order of 1010 V m–1 (1 V Å–1) are employed [3, 5, 14]. The process of field ionization (FI) is applicable to gaseous (FI mode) as well as condensed phase analytes residing on the emitter surface (FD mode). Analyte molecules are thus ionized very softly, i.e., without noteworthy excess internal energy.
Since the introduction of liquid injection field desorption/ionization (LIFDI), performing the FD experiment has largely been simplified [9, 15, 16] because sample solutions are directly transferred onto the emitter inside the ion source. The basic operation of LIFDI is described elsewhere [9, 10]. LIFDI offers major advantages over conventional FD as it allows for (1) sample transfer to the field emitter under inert conditions, and (2) reduced sample load that results in accelerated measurement cycles. LIFDI has been applied to the analysis of fullerene derivatives [17, 18], transitions metal complexes [10, 12, 13], and other compounds highly reactive towards moisture and air [9, 19].
LIFDI, when combined with the extraordinary resolving power and mass accuracy of Fourier transform ion cyclotron resonance (FT-ICR) instruments, shows impressive capabilities in petroleomics [20–25]. Here, the strength of LIFDI (and FD) is based on the fact that the field ionization process generates ions from nonpolar neutral molecules. The molecules would not be accessible by ESI, for example, an aspect rendering the combination of ESI and LIFDI as an attractive pair of complementary ionization methods.
LIFDI has been available on several types of mass spectrometers including FT-ICR, but to date it has not been adapted to the Bruker Apex series (Bruker Daltonik GmbH, Bremen, Germany) of FT-ICR instruments featuring the manufacturer’s Dual Source MTP. A previous design of this combination source, which was switchable from electrospray ionization (ESI) [26] to matrix-assisted laser desorption/ionization (MALDI) [27], required parts of the ion optical system to be mechanically moved for switching between MALDI and ESI modes and vice versa [28]. The more recently redesigned Bruker ESI-to-MALDI switchable Dual Source MTP for FT-ICR instruments focuses ions from both the ESI and the (MA)LDI, respectively, into an ion funnel [29, 30]. A common housing operated under rough pump vacuum conditions encloses the ultrasonic expansion emerging from the ion desolvation capillary of the ES interface, the MALDI plume, and an ion funnel that receives and focuses ions being delivered from either process. Under these conditions, the residual gas is still capable of thermalizing the ions (p. 88 in [31]), a property particularly valuable to cool the fast ions emerging from the MALDI target. This ion source is even capable of simultaneously admitting ions from its ESI and (MA)LDI units into the ion funnel—an admittedly rare mode of operation that has recently been demonstrated to allow for ion–molecule reactions of laser-desorbed neutrals with electrosprayed ions [32].
After removal of the rear panel bearing the MALDI sample stage from the housing, the remaining assembly is ready to take any other ion delivering device in this position. Thus, we have modified the commercial Dual Source MTP to become a LIFDI-ESI combination ion source. Construction, modes of operation, and performance of our prototype LIFDI-ESI combination ion source are detailed in this manuscript.
2 Experimental
2.1 FT-ICR Instrument
All experiments were performed on a Bruker Apex-Qe Fourier transform ion cyclotron resonance mass spectrometer (Bruker Daltonik GmbH, Bremen, Germany) equipped with a 9.4 T superconducting magnet. The analyzer of this instrument, actually a quadrupole-ICR hybrid, comprises a rf-only hexapole (h1), a selection RF/DC quadrupole (Q), a second rf-only hexapole (h2), and a high voltage (3 keV) ion transfer flight tube extracting and accelerating ions from h2 through the magnetic field gradient and decelerating them before trapping them at low kinetic energies inside the ICR cell. Either of the two RF-only hexapoles, h1 and h2, can be used for ion accumulation. The quadrupole, Q, can be operated in rf-only mode or in RF/DC mode to select precursor ions for tandem MS experiments. Then h2 is employed for collision-induced dissociation (CID) and subsequent accumulation of the resulting fragment ions. A stream of argon is constantly admitted to h2 at 0.5–2.0 l s–1 for ion cooling, which improves the trapping efficiency of this device. The argon also serves as the collision gas for CID, under the conditions that an appropriate offset voltage is applied to effect activating collisions.
In the original ESI-to-MALDI switchable combination ion source (Dual Source MTP), electrospray ions emerging from the transfer capillary into the first vacuum stage are deflected at a 90° angle into the first ion funnel in front of h1. Deflection of the ion flight path is effected by means of both an attracting potential on the ion funnel with respect to the exit of the transfer capillary and a deflection plate on the opposite side. Ions from LDI or MALDI are generated behind this deflection plate, accelerated towards it and passed through a central hole on-axis with the ion funnel.
2.2 Attaching the LIFDI Source
From the standard Dual Source MTP the rear panel bearing the MALDI sample stage was dismounted and replaced with the custom-built LIFDI source (Supplementary Material, Figures S1, S2). The supply for the offset voltage of the MALDI target was disconnected and electrically insulated. The deflection plate was dismounted from the ion funnel to give a wider access for the LIFDI probe that was positioned on-axis with respect to the ion funnel. After closing the rear panel, the LIFDI probe was slightly inserted between the first electrodes of the ion funnel.
2.3 LIFDI Source
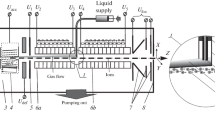
A schematic of the LIFDI setup is shown in Figure 1 and a photograph of the open source housing is shown in Figure 2. The LIFDI probe was inserted via a VAT separation valve (VAT Vakuumventile AG, Haag, Switzerland) through the custom built rear panel bearing an additional turbomolecular pump on its atmospheric side and a guiding for the probe on its vacuum side. A white LED for illumination and a mirror to observe the emitter by a CCD camera with microscopic optics mounted outside was attached to this part as well. During the LIFDI operation a high vacuum of 3–4 × 10–5 mbar as measured by a Balzers IKR 250 ion gauge (GLIFDI) connected to a Balzers TPG 252 controller (Pfeiffer Vacuum GmbH, Asslar, Germany) was maintained by a Balzers TPU 240 turbomolecular pump (TPLIFDI). TPLIFDI was backed by a Balzers Duo 2.5 rotary pump (RPLIFDI). To achieve high vacuum the entrance of the ESI transfer capillary was sealed with a silicon septum. As soon as the pressure had dropped to <10–3 mbar the E2M30 rotary pump (RP1) usually evacuating the ion source housing was separated from it by an additional valve (VLIFDI). With TPLIFDI switched off, VLIFDI opened, and the septum removed the pressure rose to 1.8 mbar as measured by a Balzers Compact Pirani Gauge (G1). The system would then be ready for ESI mode (for photographs of the standard source housing and the entire LIFDI setup see Supplementary Material Figures S1–S3.)

Schematic of the LIFDI-ESI combination source in LIFDI mode of operation. The MALDI unit of the commercial source was replaced by the LIFDI unit, the probe of which is being inserted up to the entrance region of the first ion funnel

Photograph of the open ion source housing with the LIFDI source attached. The ion source housing is spacious (inner dimensions are 35 cm width, 27 cm height, 17 cm length). In this photograph the LIFDI probe has been pushed in even further than it would be in operation so as to show the probe tip with the emitter. It is retracted for closing the panel and then pushed in again so as to position the emitter into the entrance of the ion funnel
The high voltage for FI/FD and the emission-controlled emitter heating current (EHC) were delivered by a Linden TIC-EHC-Programmer (Linden CMS, Leeste, Germany) set to the maximum emission of 4 × 10–8 A, a maximum EHC rate of 30 mA min–1, and a negative potential on the counter electrode of −10.0 to −10.5 kV. By creating the ionizing electric field by virtue of the negative potential on the counter electrode the emitter itself could be operated close to ground potential in order to ensure low ion kinetic energies at the entrance of the ion funnel. The deceleration of the ion beam emerging through the slotted counter electrode was effected by a pair of rectangular electrodes facing each other and aligned in parallel to the ion beam. These focusing/beam steering electrodes were set close to ground potential. Typical settings were an emitter DC offset of +25.0 V and a DC voltage for the ion beam steering electrodes of −5.5 V. The DC voltages were delivered by two Manson MPD-6015 DC regulated power supplies (Manson, Hong Kong, China).
2.4 Switching Between ESI and LIFDI
Switching back and forth between ESI and LIFDI could be accomplished in 10–15 min. Details of the procedure are provided as Supplementary Material.
2.5 Acquisition of LIFDI Mass Spectra
Sample solutions of liquid trihexyl(tetradecyl)-phosphonium tris(pentafluoroethyl)-trifluorophosphate (1 mg mL–1 in MeOH), polyethylene glycol 600 (1 mg ml–1 in MeOH), 2,3,4-tridodecyloxy-benzaldehyde (0.5 mg ml–1 in tetrahydrofurane), and [60]fullerene (almost saturated in toluene) were prepared. The solutions were prepared onto the emitter following a procedure commonly used in LIFDI [9, 10, 13].
Activated tungsten wire (13 μm) LIFDI emitters (Linden CMS, Leeste, Germany) were employed. During the experiments, the EHC was ramped from 0 to 60 mA at rates up to 30 mA min–1. The emitter heating was run under emission-control at 2–4 × 10–8 mA emission. Baking at an EHC of 80–85 mA was used to clean the emitter after each measurement. The emitter high voltage and EHC were controlled using a TIC-EHC-Programmer unit (Linden CMS, Leeste, Germany).
Ions were accumulated in the collision hexapole for 0.5–5.0 s and then transferred into the ICR cell. Ions over the m/z 87–1600 range were excited and detected using standard settings as established in ESI mode. Data acquisition was performed in broadband mode with 1 MB data points. Typically, 16–64 transients were accumulated for one magnitude mode spectrum; the end of data acquisition was determined by the sudden drop in the TIC after most of the sample had been desorbed. External mass calibration was performed on sodium adduct ions of polyethylene glycol (PEG 600) delivered by LIFDI. The instrument was controlled by the Bruker ApexControl software (ver. 3.0.0) and data analysis was performed using the Bruker DataAnalysis software (ver.4.0).
3 Results and Discussion
The liquid injection field desorption/ionization-electrospray ionization (LIFDI-ESI) combination ion source as described here is the result of several different approaches to the subject, which shall not be discussed in depth. Nonetheless, it seems worth noting that any of the LIFDI-ESI combination ion source variants we have tried was basically able to deliver ions from the field emitter to the ICR cell, of course at highly varying efficiencies. All preliminary designs made use of a LIFDI probe that was inserted on-axis with respect to the ion funnel in order to generate ions roughly at the same position where MALDI ions are being produced in the standard Dual Source MTP. Apart from this common feature, the main alterations between those designs were (1) the insertion depth of the emitter from positions slightly in front of to several millimeters inside the ion funnel, (2) the actual pressure in the voluminous ion source housing ranging from 2 × 10–2 to 3 × 10–5 mbar, (3) the design of the LIFDI probe tip with different shapes and alignments of the electrodes for beam focusing and steering, and (4) the construction of the mechanic components serving as an additional vacuum system, the vacuum lock, the guiding for the probe, and the emitter observation optics.
The limited low-mass capabilities of the Apex-Qe instrument caused by the transmission characteristics of the quadrupole section essentially prohibit its application to analytes below m/z 180. Therefore, initial experiments could not be performed as one would usually do in FD-MS, i.e., by tuning on long-lasting, almost time-invariant molecular ion signals as generated from acetone or toluene when admitted from a reference inlet system. Instead, one had to rely on a compound that would deliver ions with an FI-like continuity while desorbing rather constantly from the emitter at a low to moderate emitter heating current. From previous work, the persistence of the cation signal of trihexyl(tetradecyl)-phosphonium tris(pentafluoroethyl)-trifluorophosphate, an ionic liquid (IL) seemed appropriate [11]. In fact, the [C32H68P]+ phosphonium ion signal, m/z 483.50532 (theoretical value), could immediately be observed as soon as the potential difference between the emitter and the counter electrode exceeded 4 kV. Then, at the full potential difference of 10.5 kV, the signal persisted for several minutes, thereby allowing tuning of the instrument.
A LIFDI spectrum of the IL as obtained with one of the preliminary versions of the LIFDI-ESI combination source at 1.2 × 10–2 mbar is shown in Figure S4. Even this spectrum features a resolution of R = 56,000 for the analyte peaks and a mass accuracy of 1.1 ppm for the cation, [C32H68P]+, m/z 483.50476. This demonstrates that ion desorption from the field emitter can even occur at a moderate vacuum of just 1.2 × 10–2 mbar, which is considered an extremely high pressure for FD-MS. Furthermore, it indicates that mass accuracy as well as resolving power of the particular FT-ICR instrument are basically independent from the mode of analyte ion generation.
Nonetheless, further experiments indicated that a better vacuum was needed to generate and transfer molecular ions into the ICR cell. Our investigations revealed that odd-electron ions, such as molecular ions, could not be observed under poor vacuum conditions. From this perspective, the circumstances that the even-electron IL ion rather than toluene molecular ion initially had to be used turned out really fortunate. Obviously, ions formed by FI of toluene would not have been detected under these conditions.
From here on forward, we will focus our discussion on the source design and the parameters as described in the Experimental section, summarized in Table 1, and illustrated in Figures 1 and 2. As the project progressed, it became obvious that in particular the conditions for the operation of the ion funnel and the trapping voltages of the collision and accumulation hexapole (h2) required major adjustments compared to those applicable under standard ESI conditions.
The number of lenses and rf multipoles along the ion optical axis through the ion funnel and the hQh section is relatively large, and the relevant values of these settings for ESI and LIFDI modes are summarized and compared in Table 1. The parameters are named and grouped according to the Apex Control 3.0.0 software.
After having established a set of parameters a broadband LIFDI-FT-ICR mass spectrum of polyethylene glycol 600 could be obtained. Until that moment, mass calibration had simply been copied from the underlying ESI method file. After that, based on the LIFDI spectrum of PEG 600, a more accurate calibration could be established for subsequent work. Using this external calibration, the accurate m/z values of a subsequent spectrum delivered formulas for the [M + Na]+ ions of PEG 600 (Figure 3). On the list, including 8 to 16 mer, the largest deviation between the measured and the calculated m/z values was only 0.4 ppm. The resolution at m/z 613 was R = 45,000, which was equivalent to the one obtained in ESI mode when the same range and transient size was selected.

Broadband LIFDI-FT-ICR mass spectrum of polyethylene glycol 600 after external calibration (based on the same compound). The list of formulas derived from the accurate m/z values is inserted for [M + Na]+ ions of 8 to the 16 mer; it shows only 0.4 ppm as the largest deviation between measured and calculated m/z. The resolution at m/z 613 is R = 45,000
Interestingly, using an earlier version of the LIFDI source, even at 1.2 × 10–2 mbar a broadband LIFDI-FT-ICR mass spectrum of PEG 600 was obtained (Figure S7). This spectrum already showed good mass accuracy of about 1 ppm as exemplified by the list of formulas derived from the accurate m/z values of the [M + Na]+ and the [M + K]+ ions of 9 to 16 mer. The resolution at m/z 613 was R = 45,000 as well. However, the spectrum obtained at 1.2 × 10–2 mbar differed from that in Figure 3 in that the 5-fold amount of sample applied onto the emitter only resulted in a maximum intensity by one order of magnitude lower than at high vacuum. As the actual ion current could be monitored on the TIC-EHC-Programmer, it was obvious that these losses in signal intensity at 1.2 × 10–2 mbar were not due to suppression of ion desorption itself. Instead, there was a problem of ion transmission within this setup, arguably caused by ion losses in the vicinity of the emitter as a result of an increased number of collisions. In addition, fragmentation of desorbed ions could result in low-mass fragment ions not accessible by the mass spectrometer and, hence, were lost for analysis.
Next, we analyzed 2,3,4-tridodecyloxy-benzaldehyde as a representative nonpolar compound as this would yield a molecular ion rather than an adduct ion in FD-MS. The broadband LIFDI-FT-ICR mass spectrum of 2,3,4-tridodecyloxy-benzaldehyde exhibited a reasonably strong molecular ion peak at m/z 658.58964 (calc. for C43H78O4: m/z 658.58946, –0.3 ppm; Figure 4). The insert of Figure 4 shows an expansion to reveal the isotopic pattern of the \( {{\text{M}}^{{ + \bullet }}} \) ion signal. Whereas the reference FD spectrum obtained on our magnetic sector instrument was virtually free of fragment ion peaks, the spectrum on the FT-ICR instrument exhibited two intensive fragment ion peaks. The fragment ions arose by loss of dodecene, C12H24, from \( {{\text{M}}^{{ + \bullet }}} \) to form the fragment ion at m/z 490.40155 (calc. for C31H54O4: m/z 490.40166, 0.2 ppm). The process can readily be explained by a variant of the McLafferty rearrangement that is common for phenylalkanes (p. 296 in [33]). The resulting fragment ion, which is still an odd-electron positive ion, then loses another dodecene molecule to yield the second generation fragment ion at m/z 322.21331 (calc. for C19H30O4: m/z 322.21386, 1.7 ppm). The comparatively low mass accuracy of the fragment ion at m/z 322.21331 is due to the fact that this was outside the range of reference peaks used to build to mass calibration file.

Broadband LIFDI-FT-ICR mass spectrum of 2,3,4-tridodecyloxy-benzaldehyde. The molecular ion, m/z 658.58964 (calc. for C43H78O4: m/z 658.58946, –0.3 ppm), undergoes loss of dodecene to form the fragment ion at m/z 490.40155 (calc. for C31H54O4: m/z 490.40166, 0.2 ppm), that loses another dodecene to yield the second generation fragment ion at m/z 322.21331 (calc. for C19H30O4: m/z 322.21386, 1.7 ppm). The insert shows an expansion to reveal the isotopic pattern of the \( {{\text{M}}^{{ + \bullet }}} \) peak
In addition to mass accuracy and mass resolution, LIFDI mass spectra obtained using a FT-ICR instrument should preferably show the low level of fragmentation or its complete absence as known from LIFDI spectra on sector and time-of-flight instruments. The distinguished influence of the type of ions created, i.e., relatively stable even-electron positive ions on one hand versus readily dissociating odd-electron positive ions on the other, points towards an unintentional source of ion activation. The apparent lack of softness may, to some extent, be attributed to the ionization process carried out under non-optimum vacuum conditions. The ions are at keV kinetic energies in the immediate vicinity of the emitter until they become decelerated by the action of the opposed electric field between the counter electrode and the focusing electrodes on the probe tip. Thus, they may suffer from collision-induced fragmentation during the few first millimeters of their flight paths. The majority of dissociations, however, appear related to the ion life-time requirements of the FT-ICR instrument. Here, the most rapid transfer of ions from the emitter into the accumulation hexapole still takes milliseconds rather than microseconds on a sector instrument. The accumulation in h2 prior to m/z analysis even lasts in the order of seconds, resulting in a time scale expansion of roughly five orders of magnitude compared with “classic” FD-MS instrumentation.
Collision-induced dissociations in h2 appeared to be the bottleneck to achieve reasonable softness of the overall procedure. The degree of CID should mainly be influenced by (1) the potential differences used for ion trapping and ejection from h2 and (2) the amount of argon gas used for thermalization to improve the trapping efficiency of the storage device. Consequently, tuning conditions along the ion optical path from the source up to the ICR cell have been explored. Broadband LIFDI-FT-ICR mass spectra of 2,3,4-tridodecyloxy-benzaldehyde were acquired at constant gas flow of 0.7 l s–1 into h2 while reducing the set of trapping voltages (entrance lens trap, entrance lens extract, and exit lens trap; Table 1) from 12 to 9 V (Figure S8). Fragmentation of \( {{\text{M}}^{{ + \bullet }}} \), m/z 658.5899, was clearly reduced when shallow trapping wells were used. Unfortunately, the overall intensity also dropped significantly. For example, while the relative abundance of the \( {{\text{M}}^{{ + \bullet }}} \) ion could be improved from 26% at 12 V trapping to 78% at 9 V, the intensity of the signals dropped by a factor of more than 5.
To check the influence of the argon gas flow into h2 another set of spectra was acquired at constant trapping voltage of 10 V while decreasing the gas flow from 1.1 to 0.3 l s–1. Although fragmentation of \( {{\text{M}}^{{ + \bullet }}} \) was effectively reduced at low gas flow, the overall decrease in intensity was tremendous. For example, \( {{\text{M}}^{{ + \bullet }}} \) exhibited a relative intensity of just 29% at 1.1 l s–1 but it became the base peak of the spectrum acquired at 0.3 l s–1, however, the absolute signal intensity dropped from 3.5 × 106 to 3 × 105 counts (Figure S9).
Obviously, the advantages of suppressed fragmentation are more than counterbalanced by a severe loss in overall intensity of the spectra. To this end it deemed to be reasonable to establish a set of parameters that would deliver effective trapping and thus ion intensity at an acceptable level of fragmentation.
Based on such settings the broadband LIFDI-FT-ICR mass spectrum of [60]fullerene has been obtained (Figure S10). The emitter was loaded with four repetitive dips lasting 3 s each. Assuming a volume of 50–60 nL per dip [9] and a concentration of about 0.5 mg ml–1 of C60 in toluene, this corresponds to 0.1–0.15 μg of sample consumed per measurement. Desorption of \( {{\text{C}}_{{{6}0}}}^{{ + \bullet }} \), m/z 719.9997, occurred from 45 < EHC < 65 mA. Argon was admitted to h2 at 1.25 l s–1, and trapping voltages of 17 V were employed. The spectrum exhibited the monoisotopic molecular ion, \( {{\text{C}}_{{{6}0}}}^{{ + \bullet }} \), and three carbon isotopic peaks with very good mass accuracy. One obtained for monoisotopic molecular ion, \( ^{{{12}}}{{\text{C}}_{{{6}0}}}^{{ + \bullet }} \), m/z 719.99977, (calc. for \( ^{{{12}}}{{\text{C}}_{{{6}0}}}^{{ + \bullet }} \): m/z 719.99945, –0.4 ppm), and for the carbon isotopic peaks, i.e., \( ^{{{12}}}{{\text{C}}_{{{59}}}}^{{{13}}}{{\text{C}}^{{ + \bullet }}} \), m/z 721.00300 (calc. for \( ^{{{12}}}{{\text{C}}_{{{59}}}}^{{{13}}}{{\text{C}}^{{ + \bullet }}} \): m/z 721.00281, –0.3 ppm), \( ^{{{12}}}{{\text{C}}_{{{58}}}}^{{{13}}}{{\text{C}}_{{2}}}^{{ + \bullet }} \), m/z 722.00627 (calc. for \( ^{{{12}}}{{\text{C}}_{{{58}}}}^{{{13}}}{{\text{C}}_{{2}}}^{{ + \bullet }} \): m/z 722.00616, –0.2 ppm), and \( ^{{{12}}}{{\text{C}}_{{{57}}}}^{{{13}}}{{\text{C}}_{{3}}}^{{ + \bullet }} \), m/z 723.01018 (calc. for \( ^{{{12}}}{{\text{C}}_{{{57}}}}^{{{13}}}{{\text{C}}_{{3}}}^{{ + \bullet }} \): m/z 723.00952, –0.9 ppm). As can be seen from the comparison between experimental and theoretical isotopic pattern, all of these peaks corresponded very well to their calculated m/z values and expected intensities. The doubly charged ion, \( {{\text{C}}_{{{6}0}}}^{{{2} + }} \), m/z 360, was neither observed in this spectrum nor in a LIFDI-FT-ICR spectrum published by others [20].
4 Conclusions
A LIFDI-ESI combination ion source for a FT-ICR mass spectrometer has been devised and its capabilities in the LIFDI mode have been demonstrated. The instrument’s operation in ESI mode has not been altered to a notable extent. Ionic and highly polar compounds yield abundant even-electron positive ions in the LIFDI mode of this new combination ion source. Nonpolar compounds forming odd-electron positive ions are still difficult to analyze. This can be attributed to the increased ion life-time requirements of the FT-ICR instrument allowing more time for fragmentation compared with established configurations for LIFDI on magnetic sector or time-of-flight instruments. Even rapid transfer of ions from the emitter into the accumulation hexapole takes milliseconds rather than microseconds. The accumulation prior to m/z analysis even lasts in the order of seconds, resulting in a time scale expansion of roughly five orders of magnitude compared with “classic” FD-MS instrumentation. Currently, tuning conditions along the ion optical path from the source up to the ICR cell and other parameters related to the operation of the accumulation hexapole are being explored. The first set of experiments to achieve greater softness of this series of ion-guiding and ion-storing steps along the hQh section are promising. We expect noteworthy improvements for the analysis of nonpolar compounds with this specific LIFDI-FT-ICR instrument configuration in near future.
References
Beckey, H.D.: Field Desorption Mass Spectrometry: A Technique for the Study of Thermally Unstable Substances of Low Volatility. Int. J. Mass Spectrom. Ion Phys. 2, 500–503 (1969)
Beckey, H.D., Schulten, H.-R.: Field Desorption Mass Spectrometry. Angew. Chem 87, 425–438 (1975)
Beckey, H.D.: Principles of Field Desorption and Field Ionization Mass Spectrometry. Pergamon Press, Oxford (1977)
Schulten, H.-R.: Recent Advances in Field Desorption Mass Spectrometry. Adv. Mass Spectrom. 7A, 83–97 (1978)
Prókai, L.: Field Desorption Mass Spectrometry. Marcel Dekker, New York (1990)
Siegler, F.; Wolff, J. J.; Gross, J. H. Analysis of Ferrocenyl Compounds by LR and HR Field Desorption Mass Spectrometry. Adv. Mass Spectrom. 1998, 14, B083140–1-B083140/21.
Gross, J.H., Weidner, S.M.: Influence of Electric Field Strength and Emitter Temperature on Dehydrogenation and C–C Cleavage in Field Desorption Mass Spectrometry of Polyethylene Oligomers. Eur. J. Mass Spectrom. 6, 11–17 (2000)
Gross, J.H., Vékey, K., Dallos, A.: Field Desorption Mass Spectrometry of Large Multiply Branched Saturated Hydrocarbons. J. Mass Spectrom. 36, 522–528 (2001)
Linden, H.B.: Liquid Injection Field Desorption Ionization: a New Tool for Soft Ionization of Samples Including Air-Sensitive Catalysts and Non-Polar Hydrocarbons. Eur. J. Mass Spectrom. 10, 459–468 (2004)
Gross, J.H., Nieth, N., Linden, H.B., Blumbach, U., Richter, F.J., Tauchert, M.E., Tompers, R., Hofmann, P.: Liquid Injection Field Desorption/Ionization of Reactive Transition Metal Complexes. Anal. Bioanal. Chem. 386, 52–58 (2006)
Gross, J.H.: Liquid Injection Field Desorption/Ionization Mass Spectrometry of Ionic Liquids. J. Am. Chem. Soc. Mass Spectrom. 18, 2254–2262 (2007)
Monillas, W.H., Yap, G.P.H., Theopold, K.H.: A Tale of Two Isomers: A Stable Phenyl Hydride and a High-Spin (S = 3) Benzene Complex of Chromium. Angew. Chem. Int. Ed. 46, 6692–6694 (2007)
Langlotz, B.K., Fillol, J.L., Gross, J.H., Wadepohl, H., Gade, L.H.: Living Radical Polymerization of Acrylates Mediated by 1,3-Bis(2-Pyridylimino)Isoindolatocobalt(II) Complexes: Monitoring the Chain Growth at the Metal. Chem. Eur. J. 14, 10267–10279 (2008)
Inghram, M.G., Gomer, R.: Mass-Spectrometric Investigation of the Field Emission of Positive Ions. Z. Naturforsch. 10A, 863–872 (1955)
Linden, H. B. Sensitivity Improvement by 2–3 Orders of Magnitude and Significantly Raised Sample Throughput, Even Under Inert Conditions, by In-Source Liquid Injection FD; ASMS: Chicago, 2001; pp. MPA 024.
Linden, H. B. Quick Soft Analysis of Sensitive Samples Under Inert Conditions by In-Source Liquid Injection FD; ASMS: Orlando, 2002; pp. MPL 373.
Talyzin, A.V., Tsybin, Y.O., Schaub, T.M., Mauron, P., Shulga, Y.M., Zuettel, A., Sundqvist, B., Marshall, A.G.: Composition of Hydrofullerene Mixtures Produced by C60 Reaction with Hydrogen Gas Revealed by High-Resolution Mass Spectrometry. J. Phys. Chem. B 109, 12742–12747 (2005)
Talyzin, A.V., Tsybin, Y.O., Peera, A.A., Schaub, T.M., Marshall, A.G., Sundqvist, B., Mauron, P., Zuettel, A., Billups, W.E.: Synthesis of C59Hx and C58Hx Fullerenes Stabilized by Hydrogen. J. Phys. Chem. B 109, 5403–5405 (2005)
Dransfield, T.A., Nazir, R., Perutz, R.N., Whitwood, A.C.: Liquid Injection Field Desorption/Ionization of Transition Metal Fluoride Complexes. J. Fluorine Chem. 131, 1213–1217 (2010)
Schaub, T.M., Hendrickson, C.L., Qian, K., Quinn, J.P., Marshall, A.G.: High-Resolution Field Desorption/Ionization Fourier Transform Ion Cyclotron Resonance Mass Analysis of Nonpolar Molecules. Anal. Chem. 75, 2172–2176 (2003)
Schaub, T.M., Linden, H.B., Hendrickson, C.L., Marshall, A.G.: Continuous-Flow Sample Introduction for Field Desorption/Ionization Mass Spectrometry. Rapid Commun. Mass Spectrom. 18, 1641–1644 (2004)
Schaub, T.M., Rodgers, R.P., Marshall, A.G., Qian, K., Green, L.A., Olmstead, W.N.: Speciation of Aromatic Compounds in Petroleum Refinery Streams by Continuous Flow Field Desorption Ionization FT-ICR Mass Spectrometry. Energy Fuels 19, 1566–1573 (2005)
Schaub, T.M., Hendrickson, C.L., Quinn, J.P., Rodgers, R.P., Marshall, A.G.: Instrumentation and Method for Ultrahigh Resolution Field Desorption Ionization Fourier Transform Ion Cyclotron Resonance Mass Spectrometry of Nonpolar Species. Anal. Chem. 77, 1317–1324 (2005)
Fu, J., Klein, G.C., Smith, D.F., Kim, S., Rodgers, R.P., Hendrickson, C.L., Marshall, A.G.: Comprehensive Compositional Analysis of Hydrotreated and Untreated Nitrogen-Concentrated Fractions from Syncrude Oil by Electron Ionization, Field Desorption Ionization, and Electrospray Ionization Ultrahigh-Resolution FT-ICR Mass Spectrometry. Energy Fuels 20, 1235–1241 (2006)
Smith, D.F., Schaub, T.M., Rodgers, R.P., Hendrickson, C.L., Marshall, A.G.: Automated Liquid Injection Field Desorption/Ionization for Fourier Transform Ion Cyclotron Resonance Mass Spectrometry. Anal. Chem. 80, 7379–7382 (2008)
Applied Electrospray Mass Spectrometry; Pramanik, B. N.; Ganguly, A. K.; Gross, M. L., Eds.; Marcel Dekker: New York, 2002.
MALDI-MS. A Practical Guide to Instrumentation, Methods and Applications; Hillenkamp, F.; Peter-Katalinic, J., Eds.; Wiley-VCH: Weinheim, 2007.
Baykut, G., Fuchser, J., Witt, M., Weiss, G., Gosteli, C.: A Combined Ion Source for Fast Switching Between Electrospray and Matrix-Assisted Laser Desorption/Ionization in Fourier Transform Ion Cyclotron Resonance Mass Spectrometry. Rapid Commun. Mass Spectrom. 16, 1631–1641 (2002)
Shaffer, S.A., Tang, K., Anderson, G.A., Prior, D.C., Udseth, H.R., Smith, R.D.: A Novel Ion Funnel for Focusing Ions at Elevated Pressure Using Electrospray Ionization Mass Spectrometry. Rapid Commun. Mass Spectrom. 11, 1813–1817 (1997)
Shaffer, S.A., Prior, D.C., Anderson, G.A., Udseth, H.R., Smith, R.D.: An Ion Funnel Interface for Improved Ion Focusing and Sensitivity Using Electrospray Ionization Mass Spectrometry. Anal. Chem. 70, 4111–4119 (1998)
Schalley, C.A., Springer, A.: Mass Spectrometry and Gas-Phase Chemistry of Non-Covalent Complexes. John Wiley and Sons, Inc., Hoboken (2009)
Ganza, V., Gross, J.H.: Gas-Phase Reactions of Laser-Desorbed Molecules with Ions From Electrospray. Eur. J. Mass Spectrom. 16, 479–487 (2010)
Gross, J.H.: Mass Spectrometry—A Textbook, 2nd edn. Springer-Verlag, Heidelberg (2011)
Acknowledgments
A gift of ionic liquids from Dr. W.-R. Pitner (Merck KGaA, Darmstadt, Germany), a sample of [60]fullerene from Professor W. Krätschmer (MPI für Kernphysik, Heidelberg), and one of 2,3,4-tridodecyloxy-benzaldehyde from Professor U. Bunz (Institute of Organic Chemistry, Heidelberg University) are gratefully acknowledged. The authors thank the Collaborative Research Centre (SFB 623 at the Faculty of Chemistry and Earth Sciences, University of Heidelberg) for using the FT-ICR instrument, and Bruker Daltonik GmbH (Bremen) for discussion and technical support.
Author information
Authors and Affiliations
Corresponding author
Additional information
Dedicated to Professor J. J. Veith on the occasion of his 70th birthday.
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(PDF 5793 kb)
Rights and permissions
About this article
Cite this article
Linden, H.B., Gross, J.H. A Liquid Injection Field Desorption/Ionization-Electrospray Ionization Combination Source for a Fourier Transform Ion Cyclotron Resonance Mass Spectrometer. J. Am. Soc. Mass Spectrom. 22, 2137–2144 (2011). https://doi.org/10.1007/s13361-011-0259-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13361-011-0259-9