
Abstract
Peptidoglycan recognition proteins (PGRPs) participate in insect defense against bacterial pathogens by recognizing bacterial cell wall peptidoglycans (PGNs). Here, we identified the PGRP-LB gene in the white-backed planthopper Sogatella furcifera (SfPGRP-LB). SfPGRP-LB is a secreted protein with a typical PGN-binding domain and five conserved amino acid (aa) residues required for amidase activity. Expression analysis showed that the SfPGRP-LB transcript levels were significantly higher in the midgut than in other tissues. Silencing SfPGRP-LB with dsRNA significantly downregulated the expression of Toll pathway genes Toll and Dorsal and Imd pathway genes Imd and Relish after Escherichia coli challenge. However, only Toll and Dorsal expressions were downregulated after Staphylococcus aureus challenge. E. coli and S. aureus challenges rapidly and strongly upregulated SfPGRP-LB expression. Recombinantly expressed SfPGRP-LB (rSfPGRP-LB) had strong affinities for E. coli Dap-type PGN and S. aureus Lys-type PGN and agglutinated the bacteria. However, rSfPGRP-LB inhibited S. aureus but not E. coli growth. Furthermore, rSfPGRP-LB had amidase activity, degraded Lys-type PGN, and destroyed S. aureus cell walls but had no such effects on E. coli Dap-type PGN. Thus, SfPGRP-LB recognizes and binds various bacterial PGNs but only has amidase activity against Lys-type PGN.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
The immune system enables insects to adapt to their ambient environment, contend with biotic stress, and resist various microbial pathogens. Our current understanding of insect immunity is based mainly on analyses of species with complete metamorphosis such as Drosophila melanogaster (Diptera: Drosophilidae) (Dziarski and Gupta 2018; Gottar et al. 2002; Hillyer 2016), Aedes aegypti (Diptera: Culicidae) (Koh et al. 2018; Ramirez et al. 2019; Wang et al. 2018), and Bombyx mori (Lepidoptera: Bombyxidae) (Chen et al. 2018a; Chen and Lu 2018; Li et al. 2019). However, subsequent genome sequencing has clarified immune system function in other insect species as well (Bao et al. 2013; Chen et al. 2018b; Laughton et al. 2011). Studies on completely metamorphic insects revealed that the first step in their immune response is pathogen detection via different pattern recognition receptors. Peptidoglycan recognition proteins (PGRPs) are the major insect pathogen pattern recognition receptors and confer protection against bacterial challenge.
PGRPs occur in most insect immune recognition systems. They detect and are induced by the peptidoglycans (PGNs) that are abundant in certain bacterial cell walls. The proportion of PGNs in Gram-positive bacterial cell walls is ~ 50–90% of the cell dry weight (DW). In contrast, PGNs comprise only ~ 10% of the cell DW in Gram-negative bacterial cell walls. Most PGRPs have a conserved domain that recognizes and binds bacterial cell wall PGNs. According to the size of the transcript and structure of the domains, insect PGRP can be divided into short extracellular PGRP (shortform PGRPs, PGRP-S) and longform PGRP (longform PGRPs, PGRP L) across or within the membrane (Royet et al. 2011). Insect PGRPs distinguish bacteria by the differences in the third amino acid (aa) of their short-peptide PGNs. PGRP-SA in D. melanogaster can recognize Lys-type PGN, which possesses a lysine residue in the third position of its short peptide. PGRP-SB, PGRP-SC, PGRP-LB, PGRP-LC, and PGRP-LE in D. melanogaster preferentially interact with diaminopimelate residues (Dap type). Most Gram-negative and Gram-positive bacteria have these PGNs (Guan et al. 2004; Leulier et al. 2003; Reiser et al. 2004).
PGRP number and size vary among insect species. Seven short and six long PGRPs were detected in D. melanogaster (Neyen et al. 2016; Royet et al. 2011; Werner et al. 2000), three short and four long PGRPs were identified in Anopheles gambiae (Diptera: Culicidae) and A. aegypti (Tanaka et al. 2008), and six short and six long PGRPs were found in B. mori (Tanaka et al. 2008). Hemimetabolic insect have fewer but more structurally uniform PGRPs than holometabolic insect. Only two long form PGRPs were detected in the brown planthopper Nilaparvata lugens (Hemiptera: Delphacidae) and white-backed planthopper Sogatella furcifera (Hemiptera: Delphacidae) (Bao et al. 2013; Wang et al. 2017). PGRP is deleted in Acyrthosiphon pisum (Homoptera: Aphididae) (International Aphid Genomics 2010).
The innate immune response of insects is mainly mediated by two different nuclear factor-κB (NF-κB) signaling pathways, Toll and Imd (Lemaitre et al. 1995). In Drosophila, the main genes involved in the Toll pathway (Tolls, Spatzle, Dorsal and Dif) and Imd pathway (Imd, Relish) can be activated by Gram-positive bacteria, Gram-negative bacteria and fungi through the identification and combination of pathogens and different PGRPs and GNBPs. The expressions of Toll and Imd pathway gene are activated, and the expressions of related antibacterial peptide protein are finally induced to achieve the purpose of antibacterial (Belvin and Anderson 1996; Myllymaki et al. 2014; Ramet 2012; Valanne et al. 2011). For example, PGRP-LE activates the phenoloxidase pro-PPO system and Imd pathway (Takehana et al. 2002). PGRP-SA, PGRP-SC1, and PGRP-SD may induce the Toll pathway (Bischoff et al. 2004; Garver et al. 2006; Michel et al. 2001). PGRP-LA, PGRP-LC, PGRP-LB, and PGRP-LF promote the Imd pathway in response to Gram-negative bacterial challenge (Gendrin et al. 2013; Iatsenko et al. 2016). D. melanogaster harbors one secreted and two cytosolic PGRP-LB isoforms; the former hydrolyzes PGN, whereas the latter regulates regional activation (Charroux et al. 2018). In B. mori, PGRP-LC1, PGRP-S1, and PGRP-S3 expressions are upregulated in response to Gram-positive and Gram-negative bacteria and fungi while PGRP-S2 expression is upregulated by bacteria only (Yang et al. 2015). In B. mori, PGRP-LB and PGRP-L5 expressions are upregulated by fungi; whereas, PGRP-LB and PGRP-L6 participate in the Imd signaling pathway (Tanaka and Sagisaka 2016; Zhan et al. 2018). PGRP-S4 initiates the B. mori immune response by activating the pro-PPO system. PGRP-S5 activates the phenoloxidase pro-PPO system and the Imd signaling pathway (Chen et al. 2016; Yang et al. 2017). In A. aegypti, PGRP-S1, PGRP-SC2, and PGRP-LB expressions are upregulated by both Gram-negative and Gram-positive bacteria. In contrast, PGRP-LA, PGRP-LC, and PGRP-LD are not induced by either bacteria type (Wang and Beerntsen 2015). In Tribolium castaneum (Coleoptera: Paracaridae), PGRP-LA expression is upregulated by Gram-positive and Gram-negative bacteria and PGRP-LC and PGRP-LE are induced mainly by Gram-negative bacteria via the Imd signaling pathway. PGRP-S (short type) did not participate in the T. castaneum immune response (Koyama et al. 2015). In the incompletely metamorphic insect N. lugens, PGRP-LB is induced by Gram-positive and Gram-negative bacteria; whereas, PGRP-LC is only activated by Gram-positive bacteria (Bao et al. 2013).
The white-backed planthopper S. furcifera is an important insect pest on rice (Oryza sativa L.) and is incompletely metamorphic. However, little is known about its immune recognition, pattern-recognition receptor (PRR) signaling pathways, or innate immune response. Here, we used PGRP-LB sequencing data obtained by transcriptomics to investigate the structural and immune function characteristics of SfPGRP-LB. The results of this study will help us better understand the innate immune system of S. furcifera.
Materials and methods
Insects
S. furcifera individuals were acquired from Zhejiang University (Hangzhou, China). They were bred on rice seedlings (var. Taichung Native 1 [TN1]) for > 30 generations in 80-mesh wooden cages (50 cm × 50 cm × 50 cm) in an artificial climate chamber (27 ± 1 °C; 75–80% RH; 16/8 h light:dark photoperiod).
Sequence cloning, phylogenetic tree construction, and structure prediction
The ORF encoding SfPGRP-LB was amplified by PCR with PGRP-LB-full-F and PGRP-LB-full-R (Table 1). The PCR conditions were 95 °C for 5 min followed by 30 cycles at 95 °C for 30 s, 57 °C for 30 s, and 72 °C for 2 min, and a final extension at 72 °C for 10 min. The amplified product was purified using a gel extraction kit (Omega Bio-Tek, Norcross, GA, USA). Purified DNA was ligated into a pGM-T vector (TIANGEN Biotech, Beijing, China) and sequenced completely from both directions (Sangon Biotech, Shanghai, China).
Potential protein transmembrane helices were predicted with TMHMM Server v. 2.0 (http://www.cbs.dtu.dk/services/TMHMM/). Molecular weights and isoelectric points were determined with the ExPaSy ProtParam tool (https://web.expasy.org/protparam/). Open reading frames (ORF) were identified with the EditSeq program in DNAStar. Multiple aa sequence alignments were conducted in ClustalX v. 2.1. Signal peptides were predicted with the SignalP-5.0 Server (http://www.cbs.dtu.dk/services/SignalP/).
The phylogenetic tree was plotted by the neighbor-joining (NJ) method in MEGA v. 7.0. The following PGRPs were used in the phylogenetic analysis: PGRP-LA (D. melanogaster; NP_001261623), PGRP-LB (D. melanogaster; NP_001247054), PGRP-LC (D. melanogaster; NP_001163397), PGRP-LD (D. melanogaster; NP_001027113), PGRP-LE (D. melanogaster; NP_573078), PGRP-LF (D. melanogaster; NP_648299), PGRP-LA (A. aegypti; XP_001655982), PGRP-LB (A. aegypti; XP_021709443), PGRP-LC (A. aegypti; XP_021698612), PGRP-LE (A. aegypti; XP_021704907), PGRP-LA (Bactrocera dorsalis (Diptera: Tephritidae); XP_011210802), PGRP-LB (B. dorsalis; XP_011211382), PGRP-LC (B. dorsalis; XP_011211382), PGRP-LA (T. castaneum; XP_008192537), PGRP-LB (T. castaneum; XP_969556), PGRP-LE (T. castaneum; XP_008192547), PGRP-LB (B. mori; XP_012548100), PGRP-LE (B. mori; XP_004929966), PGRP-LB (N. lugens; AGK40911), PGRP-LC (N. lugens; AGK40912), and PGRP-LB (S. furcifera; MW323547.1).
RNA isolation and cDNA synthesis
Total RNA was extracted from S. furcifera with an RNA extraction kit (Axygen Scientific Inc., Union City, CA, USA). First-strand cDNA was synthesized with the PrimeScript™ IV first-strand cDNA synthesis mix (Takara Biomedical Technology Co. Ltd., Dalian, China). One microgram first-strand cDNA served as a template for PCR and qRT-PCR.
qRT-PCR analysis
The qRT-PCR amplifications were performed with TB Green® Premix Ex Taq™ II (Takara Biomedical Technology Co. Ltd., Dalian, China). A β-actin (GenBank ID: ALO78726.1) cDNA fragment was amplified as an internal control using β-actin-RTF/β-actin-RTR primers (Table 1). Each qRT-RCR reaction system was cycled in 20 μL volume consisting of 2 μL of cDNA sample, 1 μL of each 5 μM primer, 10 μL of 2 × TB Green Premix Ex Taq II, and 6 μL of ddH2O. All qRT-PCR reactions were performed in triplicate in a CFX96 real-time PCR detection system (Bio-Rad Laboratories, Hercules, CA, USA). The cycling protocol was 95 °C for 30 s followed by 40 cycles of 95 °C for 5 s and 60 °C for 30 s. Relative target gene expression was determined by the comparative 2−ΔΔCT method [ΔΔCT = ΔCT (target) − ΔCT (calibrator)](Livak and Schmittgen 2001).
Tissue distribution analysis
To investigate the tissue distribution of SfPGRP-LB mRNA, total RNA was extracted from the head, midgut, thorax, epidermis, fat body, spermary, and ovary using fifth instars and adults. The organs were rinsed several times in 1 × phosphate-buffered saline (PBS, 1.47 mM KH2PO4, 8.1 mM NaH2PO4, 137 mM NaCl, and 2.68 mM KCl; pH 7.4) and mixed with those from 30 to 80 adults. Organs were used in high-quality RNA extraction and cDNA synthesis. The qRT-PCR-specific primers PGRP-LBRTF/PGRP-LBRTR (Table 1) with 103-bp amplicons were designed according to full-length SfPGRP-LB cDNA.
Bacterial challenge
Gram-negative E. coli strain (K12) and Gram-positive S. aureus strain (ATCC6538) were used to infect S. furcifera. E. coli and S. aureus were cultured in LB medium at 37 °C with shaking at 200 rpm until OD600 = 0.6. The bacteria were sedimented at 37 °C by centrifugation at 5000×g for 10 min, washed, resuspended in 1 × PBS to a density of 5 × 108 CFU mL−1, and heat-killed by boiling for 30 min.
For the immune challenge experiment, fourth-instar S. furcifera larvae (day 1) were randomly assigned to the E. coli, S. aureus, and PBS treatment groups. Each larva was anesthetized by 10 s exposure to 5 mPa CO2 and positioned supine in the grooves of an agarose gel placement plate. Then, 0.5 μL of devitalized bacterial suspension or PBS solution was injected with a FemtoJet microinjection system (Eppendorf, Hamburg, Germany) into the abdominal segment junction between the second and third appendages (Chen et al. 2018b). The treated larvae were raised in an artificial incubation chamber and fed fresh rice seedlings. Samples were collected at 0, 6, 12, and 24 and 48 h post-injection and there were three replicates per time point. RNA extraction from midgut and fat body, cDNA synthesis, and qRT-PCR were conducted and specific qRT-PCR primers (Table 1) were prepared as previously described.
RNA interference
SfPGRP-LB and GFP (GenBank ID: KU306402.1) cDNA fragments were amplified by PCR using primers containing the T7 promoter (5′–TAATACGACTCACTATAGGG–3; Table 1). The products were purified and used as templates to synthesize dsRNA in a T7 high-yield RNA transcription kit (Vazyme Biotech Co. Ltd., Nanjing, China). The reactions were conducted as follows: 95 °C for 5 min followed by 30 cycles at 95 °C for 30 s, 57 °C for 30 s, and 72 °C for 30 s, and a final extension at 72 °C for 10 min. The dsRNA products were dissolved in ultrapure water and their concentration and purity were determined with a NanoDrop 2000 spectrophotometer (Thermo Fisher Scientific, Waltham, MA, USA) and by 1.5% agarose gel electrophoresis, respectively. Fourth-instar nymphs were either microinjected with dsSfPGRP-LB (treatment) or dsGFP (control). Then, 200 ng of dsRNA from SfPGRP-LB or GFP was injected into the segment between the second and third appendages with a FemtoJet microinjection system (Eppendorf, Hamburg, Germany) (Chen et al. 2018b). After microinjection, each nymph group was fed TN1 rice under the previously described conditions.
Effects of SFPGRP-LB on Toll and Imd pathways in vivo
To determine whether SfPGRP-LB modulates the expression of genes involved in the Toll and Imd pathways after bacterial challenge, the Toll, Imd, Dorsal, and Relish transcription levels were measured and compared among the dsSfPGRP-LB group injected with E. coli (dsSfPGRP-LB + E. coli), dsSfPGRP-LB group injected with S. aureus (dsSfPGRP-LB + S. aureus), dsGFP group injected with E. coli (dsGFP + E. coli), and dsGFP group injected with S. aureus (dsGFP + S. aureus). The specific qRT-PCR primers are shown in Table 1.
Recombinant vector construction
The ORF encoding SfPGRP-LB was amplified by PCR with PGRP-LB-protein-F/PGRP-LB-protein-R primers (Table 1). The PCR conditions were 95 °C for 5 min followed by 30 cycles at 95 °C for 30 s, 57 °C for 30 s, and 72 °C for 30 s, and a final extension at 72 °C for 10 min. The PCR product was digested with the restriction enzymes BamHI and XhoI (Takara Biomedical Technology Co. Ltd., Dalian, China) and ligated to the BamHI/XhoI site of the pET28a (+) vector (Novagen, Darmstadt, Germany). The recombinant vectors transformed BL21-competent cells for protein expression.
Recombinant protein purification
BL21 colonies were incubated in fresh LB medium at 37 °C with shaking at 200 rpm until OD600 = 0.6. Then 0.5 mM isopropyl-β-dithiogalactoside was added to induce PGRP-LB expression with poly-His tags at the C-terminus. After incubation at 200 rpm for 4 h, the cells were collected and resuspended in Buffer B (8 M urea, 50 mM Tris–HCl, and 300 mM NaCl; pH 8.0). After sonication on ice for 10 min (5 s on, 10 s off), the supernatant was collected by centrifugation at 12,000×g and 4 °C for 10 min, mixed with nickel nitrilotriacetate (Ni–NTA; Sangon Biotech, Shanghai, China) and gently shaken at 4 °C overnight. The bound protein was eluted in PBS-T buffer with 20, 50, and 100 mM imidazole.
Western blotting assay
Purified recombinant rSfPGRP-LB was separated by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to a polyvinylidene fluoride (PVDF) membrane. Western blotting was conducted using mouse primary and goat anti-rabbit immunoglobulin G (IgG)-conjugated horseradish peroxidase secondary antibodies (Sangon Biotech, Shanghai, China). Western blot signals were developed in an enhanced chemiluminescence (ECL) detection kit (Bio-Rad Laboratories, Hercules, CA, USA) and photographed with a Molecular Imager® ChemiDoc™ XRS + system (Bio-Rad Laboratories, Hercules, CA, USA). β-actin polyclonal rabbit serum was used to ensure equal protein loading.
Amidase activity assay
Amidase activity was measured as described in a previous report (Yang et al. 2019) with some modifications. Briefly, Dap-type PGN from E.coli and Lys-type PGN from S. aureus were purchased from Sigma-Aldrich (Merck KgAA, Darmstadt, Germany). Twenty micrograms of bovine serum albumin (BSA; Sangon Biotech, Shanghai, China), 20 µg of rSfPGRP-LB, or 20 μg of lysozyme (Sangon Biotech, Shanghai, China) was resuspended in 100 μL of PBS, respectively, and mixed with 100 μL of reaction buffer (2 mM ZnSO4, 40 mM MgCl2, and 100 mM Tris HCl; pH 7.9). Then, the suspensions were incubated at 37 °C for 60, 120, or 180 min, respectively. Afterward, 40 μL of this mixture was added to 100 μL of 1 M NaOH and the suspension was incubated at 37 °C for 30 min. Then, 10 μL of 0.5 M H2SO4 and 1 mL of 98% (v/v) H2SO4 were added in succession and the mixtures were boiled for 5 min. After cooling, 10 μL of 4% (w/v) CuSO4·5H2O and 20 μL of 95% (v/v) ethanol containing 1.5% (v/v) p-hydroxydiphenyl were added and the mixture was incubated at 30 °C for 30 min. Then, 200 μL of this mixture was taken and its OD560 was measured in a CMax Plus multiskan (Molecular Devices LLC, San Jose, CA, USA).
Agglutination assay
Bacterial agglutination was performed as previously described (Dawadi et al. 2018) with minor modifications. Briefly, E. coli and S. aureus were incubated overnight, inoculated into LB medium at 1:100 ratio, and incubated at 37 °C with shaking at 200 rpm until OD600 = 0.4–0.6. The bacteria were then centrifuged at 5,000 × g for 5 min, washed, collected in 500 µL of 1 × PBS, and diluted to 10–5 ×. A 10-μL aliquot was mixed with 50 μL of rSfPGRP-LB (0.45 μg/μL) and 10 μL of Tris buffer (2 mM ZnSO4, 40 mM MgCl2, and 100 mM Tris–HCl; pH 7.9) and placed in oscillating incubator at 37 °C with shaking at 150 rpm for 3 h. Bacterial distribution was observed under an inverted microscope system (Olympus Corp., Tokyo, Japan). For the control, 50 μL of Tris buffer replaced the rSfPGRP-LB.
Scanning electron microscopy
Escherichia coli and S. aureus were incubated at 37 °C with shaking at 200 rpm until OD600 = 0.4–0.6. For each treatment group, 2 mL of bacterial suspension was centrifuged at 5000×g for 5 min and the cells were collected and washed with 1 × PBS. The E. coli or S. aureus was resuspended in 500 μL of 1 × PBS and mixed with 150 μL of 1 × PBS, 150 μL of rSfPGRP-LB (0.45 μg/μL), or 150 μL of ampicillin (0.45 μg/μL). All treatment groups were incubated at 25 °C for 12 h and washed with 1 × PBS. Then, 200 μL of prechilled 2.5% (v/v) glutaraldehyde was added to the cells and the suspensions were incubated at 4 °C overnight. The bacteria were rinsed with 1 × PBS and centrifuged at 4 °C for 20 min and the supernatant was discarded. The bacteria were dehydrated twice with an ethanol gradient (30% (v/v), 50% (v/v), 70% (v/v), 80% (v/v), 95% (v/v), and 100% (v/v)) and 100% isoamyl acetate. The cells were resuspended, incubated at 4 °C for 20 min, and centrifuged at 5000×g at 4 °C for 20 min to collect the pellet. The dehydrated samples were immersed in isoamyl acetate and subjected to zero-point drying. The dried samples were coated and examined and photographed under a scanning electron microscope (SEM) (Hitachi, Tokyo, Japan).
Inhibition zone assay
SfPGRP-LB antibacterial activity was determined using an inhibition zone assay. E. coli and S. aureus were incubated in LB broth at 37 °C until OD600 = 0.6. Each 100-μL bacterial suspension was spread onto LB agar plates. Filter disks 5 mm in diameter were soaked in 0.45 μg/μL rSfPGRP-LB and set on the media. The plates were incubated at 37 °C until bacterial growth was visible. Ampicillin (0.45 μg/μL) and BSA (0.45 μg/μL) were the positive and negative controls, respectively.
Antimicrobial cell counting kit-8 (CCK-8) assay
The rSfPGRP-LB antibacterial activity was determined using a CCK-8 (Glpbio, Montclair, CA, USA) assay (Yang et al. 2018). In brief, 1 mL of E. coli and S. aureus was incubated overnight in LB medium and centrifuged at 5000×g at 37 °C for 10 min. The bacterial cells were collected, washed thrice, and resuspended in 1 mL of 1 × PBS. Then, 0.5 μL of bacterial suspension was added to 80 μL of a liquid LB medium followed by 15 μL of rSfPGRP-LB (0.45 μg/μL), 15 μL of BSA (0.45 μg/μL) or 15 μL of ampicillin (0.45 μg/μL) and 5 μL of CCK-8 reagent (the main ingredient is the tetrazolium salt WST-8). The blank control consisted of 5 μL of CCK-8 reagent plus 95 μL of LB medium. All reaction systems were incubated in a 96-well microplate (Thomas Scientific, Swedesboro, NJ, USA) at 37 °C. OD450 was measured in a CMax Plus multiskan (Molecular Devices LLC, San Jose, CA, USA) at 4, 6, and 8 h.
Statistical analysis
The mRNA expression levels were determined by one-way ANOVA and Student’s t test in SPSS v. 19.0 (IBM Corp., Armonk, NY, USA). Differences between treatment means were considered statistically significant at p < 0.05.
Results
SfPGRP-LB sequence and evolutionary analyses
We identified and cloned full length cDNA of SfPGRP-LB by searching the S. furcifera transcriptome database (BioSample accession: SAMN12612920) with BLASTX at e-value = 10–5 and using PCR, respectively. The nucleotide sequence of SfPGRP-LB obtained by transcriptome sequencing was exactly the same as the result of PCR full-length amplification (S1.seq). SfPGRP-LB is long and resembles D. melanogaster PGRP-LB. SfPGRP-LB cDNA has a 606-bp ORF encoding a putative 201-aa protein (GenBank ID: MW323547.1). The predicted MW of SfPGRP-LB is ~ 22.60 kDa and its theoretical pI = 6.39. SfPGRP-LB has five Zn2+-dependent amidase active sites (H–Y–H–T–C) (Fig. 1A). This protein might have amidase activity in the presence of Zn2+. SfPGRP-LB had no transmembrane region but did have a signal peptide at the protein N-terminus (aa: 1–20) (Fig. 1A).

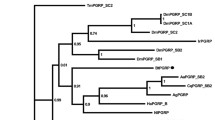
SfPGRP-LB gene sequence analysis. A Alignment of PGRP amino acidsequences. SfPGRP-LB: S. furcifera PGRP-LB; DmPGRP-LB: D. melanogaster PGRP-LB; DmPGRP-LC: D. melanogaster PGRP-LC; T7 lysozyme: bacteriophage T7 lysozyme. The signal peptide region is marked by blueline; H–Y–H–T–C residues are denoted by asterisks; the GW and R residues determining peptidoglycan-binding specificity are marked by three red dots above the alignment. B Phylogenetic tree was constructed by NJ (bootstrap test; 1000 replicates) from 21 insect PGRP proteins. Numbers of branch lengths are shown beside branches
To clarify the evolutionary position of SfPGRP-LB, we constructed a phylogenetic tree using the sequence of SfPGRP-LB and other insect PGRP proteins available. Twenty-one PGRP aa sequences from seven insect species were used in the sequence alignment and evolutionary analysis. A phylogenetic tree was constructed by the NJ method and showed that all PGRP proteins identified here were in groups PGRP-LA to PGRP-LF and SfPGRP-LB was in PGRP-LB. Moreover, SfPGRP-LB has the highest homology with N. lugens PGRP-LB (sequence similarity, 80%), which belongs to the same planthopper family (Fig. 1B).
SfPGRP-LB tissue expression analyses
SfPGRP-LB mRNA expression was detected in the head, thorax, epidermis, fat body, midgut, spermary, and ovary. It was highly expressed in the male and female midgut and to a lesser degree in the female fat body but its expression levels were low in the head, thorax, spermary, and ovary (Fig. 2).

SfPGRP-LB expression patterns. SfPGRP-LB expression in midgut, thorax, epidermis, head, fat body, ovary, and spermary. β-actin was internal control. Black bars represent standard deviation (SD)
SfPGRP-LB expression analysis in response to bacterial challenge
Bacteria-induced SfPGRP-LB expression profiles were analyzed to determine whether SfPGRP-LB has immune-related function in Gram-positive and Gram-negative bacteria. Whether in the midgut or in the fat body, the mRNA expression of SfPGRP-LB increased under the challenge of different pathogens. Especially in the midgut, after 6 h of E. coli and S. aureus induction, the expression level of SfPGRP-LB increased significantly, and continued to increase at 12–48 h. In the fat body, the expression of SfPGRP-LB did not increase until 12 h after the bacterial challenge, and the rate of increase was lower than that in the midgut (Fig. 3).

Analysis on the expression level of SfPGRP-LB in fat body a and midgut b after E. coli or S. aureus challenge, respectively. PBS was negative control. Asterisks above bars indicate statistically significant differences between E. coli and S. aureus injection groups and control group (p < 0.05). Black bars represent standard deviation (SD)
Relationship between SfPGRP-LB and NF-κB pathway
To clarify NF-κB pathway regulation by SfPGRP-LB, we measured the Toll, Imd, Dorsal, and Relish transcript levels in the dsSfPGRP-LB + E. coli and dsSfPGRP-LB + S. aureus treatment groups. The transcript level of SfPGRP-LB substantially declined by 87–98% at 12–72 h after dsSfPGRP-LB injection compared with that after dsGFP injection group (Fig. 4A). Additionally, the Toll, Imd, Dorsal, and Relish mRNA levels were significantly downregulated in the dsSfPGRP-LB + E. coli injection group at 24 and 72 h compared with those in the dsGFP + E. coli injection group (p < 0.05) (Fig. 4B). However, only the Toll and Dorsal mRNA levels were downregulated in the dsSfPGRP-LB + S. aureus injection group at 24 and 72 h compared with those in the dsGFP + S. aureus injection group (p < 0.05) (Fig. 4C).

qRT-PCR analysis of Toll, Imd, Dorsal, and Relish after dsSfPGRP-LB injection and bacterial challenge. A SfPGRP-LB silencing efficiency. B Relative Toll, Imd, Dorsal, and Relish transcription after E. coli challenge in dsSfPGRP-LB and dsGFP groups. C Relative Toll, Imd, Dorsal, and Relish transcription after S. aureus challenge in dsSfPGRP-LB and dsGFP groups. Asterisks above bars indicate statistically significant differences between E. coli or S. aureus injection groups and control group (p < 0.05). Black bars represent standard deviation (SD)
Protein purification
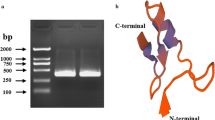
To identify the immune-related functions of SfPGRP-LB, recombinant protein was expressed in E. coli. Purified recombinant protein was detected by Coomassie brilliant blue (CBB) staining and western blotting. The rSfPGRP-LB target was detected with anti-HisTag antibody and appeared as a single ~ 23 kDa band (Fig. S2).
SfPGRP amidase activity
Sequencing demonstrated that SfPGRP-LB has five sites vital to amidase activity. Hence, E. coli Dap-type and S. aureus Lys-type PGN served as substrates to analyze rSfPGRP-LB amidase activity. After 60–180 min, rSfPGRP-LB had PGN-lytic type amidase activity on Lys-type PGN. However, rSfPGRP-LB could not degrade Dap-type PGN (Fig. 5).

Amidase activity assay. Dap-type and Lys-type PGNs treated with 20 μg of BSA, 20 μg of rSfPGRP-LB, or 20 μg of lysozyme. PGN degradation detected by changes in OD560. Asterisks above bars indicate statistically significant differences between lysozyme or rSfPGRP-LB groups and BSA group (p < 0.05). Black bars represent standard deviation (SD)
rSfPGRP-LB agglutination analysis
The rSfPGRP-LB agglutination test results are shown in Fig. 6. After 3 h of incubation, the E. coli and S. aureus in the control groups (Fig. 6A and B) were diffuse and did not agglutinate. In contrast, E. coli (Fig. 6C) and S. aureus (Fig. 6D) treated with rSfPGRP-LB were bound to Lys- and Dap-type PGN. Thus, SfPGRP-LB agglutinates various bacteria.

Recombinant SfPGRP-LB agglutination. A E. coli treated with Tris (ZnCl2). B S. aureus treated with Tris (ZnCl2). C E. coli treated with rSfPGRP-LB (ZnCl2). D S. aureus treated with rSfPGRP-LB (ZnCl2)
Effects of rSfPGRP-LB on bacterial morphology
After 12 h of incubation with rSfPGRP-LB, S. aureus cells were destroyed and their cell content had leaked. Nevertheless, there was no obvious damage to E. coli cells subjected to rSfPGRP-LB. No pore formation or leakage was detected in BSA-treated E. coli or S. aureus. Ampicillin-treated E. coli and S. aureus presented with cell damage (Fig. 7). Hence, SfPGRP-LB severely damaged S. aureus membrane integrity.

Morphological changes in bacteria. E. coli and S. aureus treated with BSA, rSfPGRP-LB, or ampicillin and examined under SEM
SfPGRP-LB antibacterial activity
SfPGRP-LB has key residues that enable it to cleave PGNs and inhibit bacterial growth. We performed inhibition zone and CCK-8 assays to evaluate bacterial growth in the presence of rSfPGRP-LB. After rSfPGRP-LB application, inhibition zones expanded on each bacterial plate. rSfPGRP-LB had antibacterial efficacy against S. aureus (Fig. 8A) but not E. coli. Ampicillin had strong antibacterial efficacy against E. coli and S. aureus.

Antibacterial activity analysis. A rSfPGRP-LB activity against E. coli and S. aureus determined by inhibition zones. E-1 and S-1: 0.45 mg/mL BSA were negative controls. E-2 and S-2: 0.45 mg/mL rSfPGRP-LB, and E-3 and S-3: 0.45 mg/mL ampicillin were positive controls. B rSfPGRP-LB antibacterial efficacy measured by CCK-8 assay in liquid culture. CK: bacteria-free liquid culture. 0.45 mg/mL BSA: negative control. 0.45 mg/mL ampicillin: positive control. Asterisks above bars indicate statistically significant differences relative to ampicillin (p < 0.05). Black bars represent standard deviation (SD)
We monitored bacterial growth under various applications using 4, 6, and 8 h CCK-8 assays. In the 0.45 mg/mL BSA protein treatment group (negative control), OD450 = 1.2–1.5 for E. coli and S. aureus in 8 h. In the 0.45 mg/mL ampicillin treatment groups (positive controls), OD450 = 0.1–0.2 for E. coli and S. aureus (Fig. 8B). In the cultures treated with 0.45 mg/mL rSfPGRP-LB, OD450 = 1.2 for E. coli and OD450 = 0.3 for S. aureus after 8 h (Fig. 8B). Hence, rSfPGRP-LB could significantly inhibit the growth of S. aureus.
Discussion
PGRP has been detected in mollusks, insects, echinoderms, zebrafish, frogs, mice, and humans.
In the innate immune response, insect PGRP is a PRR bindingbacterial peptidoglycan and activating the Toll and Imd signaling pathways (Gottar et al. 2002; Ramet et al. 2002). It also hydrolyzes amide bonds in bacterial peptidoglycan, has bactericidal activity (Cheng et al. 1994), acts as an opsonin, and promotes phagocytosis (Garver et al. 2006; Royet et al. 2011). The PGRP gene family is relatively conserved among insects. Certain PGRPs have a ~ 160-aa domain structurally homologous to bacteriophage T7 lysozyme/Zn-dependent N–acetylmuramoyl–L–alanine amidase (Liu et al. 2001; Mellroth et al. 2003; Werner et al. 2000). The sequence analysis (Fig. 1) revealed that SfPGRP-LB has all the T7 lysozyme amidase activity sites required for Zn2+ binding. As SfPGRP-LB protein was thought to have amidase activity against bacterial peptidoglycan, we analyzed tissue-specific and bacteria-induced SfPGRP-LB expression profiles. SfPGRP-LB expression was highly upregulated in the midgut (Fig. 2). Thus, it operates mainly in the intestinal tract which may be a challenge route in S. furcifera. The gut is one of the most important interfaces between insects and the internal and external environment. However, owing to its special function, the gut is often infected by external pathogenic bacteria (Huang et al. 2015; Zheng et al. 2017). In Rhynchophorus ferrugineus, PGRP-LB is of great significance for the development of new management strategies to destroy intestinal pathogenic bacteria (Dawadi et al. 2018). In our study, the mRNA expression of SfPGRP-LB was highly upregulated in the midgut (Fig. 2). In addition, E. coli and S. aureus injections significantly upregulated SfPGRP-LB expression and the rate of increase in fat body was lower than that in the midgut (Fig. 3); thus, we supposed SfPGRP-LB may prevent bacteria from penetrating midgut cells. A previous study showed that PGRP induction confers resistance to microbial pathogen invasion. After E. coli or S. aureus injection, PGRP-B and PGRP-C mRNA levels are upregulated in Helicoverpa armigera (Lepidoptera: Noctuidae) (Yang et al. 2013). In addition, PGRP-S5 transcription significantly increases when B. mori is infected with Pseudomonas aeruginosa and S. aureus (Chen et al. 2014). E. coli and Bacillus subtilis challenges significantly upregulate PGRP-LB expression in N. lugens (Bao et al. 2013). These results suggest that SfPGRP-LB plays vital roles in natural immune responses to pathogen invasion.
PGRPs recognize and bind pathogens and initiate downstream immune responses such as Toll and Imd pathway activation and antimicrobial peptide biosynthesis (Garver et al. 2006; Michel et al. 2001). The Toll pathway is activated mainly by Lys-type PGN, whereas, Dap-type PGN activates the Imd pathway (Leulier et al. 2003). Drosophila PGRP-SCla recognizes invading E. coli and Micrococcus luteus and activates the Toll pathway (Garver et al. 2006). Drosophila PGRP-LC and PGRP-LE recognize Dap-type PGN on Gram-negative bacterial cell wall and activate the Imd pathway (Choe et al. 2002; Gottar et al. 2002; Iatsenko et al. 2016; Kurata 2010). When dsSfGFRP-LB was injected into S. furcifera and induced by E. coli, Toll and Dorsal (Toll pathway) and Imd and Relish (Imd pathway) expressions were significantly upregulated compared with that in the control. When dsSfGFRP-LB was injected into S. furcifera and induced by S. aureus, Toll and Dorsal (Toll pathway) expressions were declined relative to the control, whereas Imd and Relish (Imd pathway) expression did not significantly change (Fig. 4). Therefore, SfGFRP-LB may participate in E. coli resistance via the Toll and Imd pathways. In contrast, SfGFRP-LB resistance to S. aureus is mediated by the Toll pathway.
A previous study demonstrated that PGRP proteins cleaving bacterial peptidoglycan might have antibacterial activity. PGRP-LC of Bombus lantschouensis (Hymenoptera: Apoidea) directly binds Dap-type PGN and responds to E. coli challenge (Liu et al. 2020). The same is true for PGRP-L protein in humans, mice, and Amphioxus (Li et al. 2012; Wang et al. 2003; Yang et al. 2010). PGRP proteins are bactericidal factors in immunization. However, not all proteins with amidase activity are bactericidal. Drosophila PGRP-SC has a conserved lactamase protein structure with no direct antibacterial activity (Mellroth and Steiner 2006). To verify whether SfPGRP-LB has amidase activity, we used Dap-type and Lys-type PGN as research objects. The rSfPGRP-LB protein hydrolyzed only Lys-type PGN (Fig. 5). In Drosophila, PGRP-LB, PGRP -SC1(a/b), and PGRP-SC2 are known or predicted to have amidase activity, and they also modulate activation of the Imd pathway (Bischoff et al. 2006; Mellroth et al. 2003; Paredes et al. 2011; Royet et al. 2011; Zaidman-Remy et al. 2006, 2011). In our study, PGRP-LB had amidase activity and resisted pathogen challenge through the NF-κB pathway. In Drosophila, there are 13 members of the PGRP family, and the division of labor is more detailed. In S. furcifera, there are only two family members of PGRP (PGRP-LB and PGRP-LC); thus, the function may be more comprehensive.
Certain PGRPs recognize and bind PGNs in bacterial cell walls and promote aggregation. B. mori PGRP-S5 binds PGNs and promotes E. coli and P. aeruginosa aggregation (Chen et al. 2014). In the presence of Zn2+, H. armigera PGRP-B and PGRP-C induce E. coli and S. aureus aggregation (Yang et al. 2013). In the present study, rSfPGRP-LB bound E. coli and S. aureus in vitro and promoted E. coli and S. aureus agglutination (Fig. 6). The E. coli and S. aureus activity tests showed that rSfPGRP-LB significantly inhibited the growth of S. aureus but not that of E. coli (Fig. 8). Moreover, rPGRP-LB damaged the cell walls of S. aureus but not those of E. coli (Fig. 7). For these reasons, we speculated that the antibacterial/bactericidal mechanisms of rSfPGRP-LB differ between E. coli and S. aureus.
In conclusion, our results suggest that SfPGRP-LB regulates the balance between the invasion of pathogenic bacteria and reproduction of pathogenic bacteria in the gut. In this process, SfPGRP-LB may have multiple functions. First, it prevents the overproduction of Gram-positive bacteria Lys-type PGN by degrading it and realizes the innate immune response of S. furcifera to intestinal Gram-positive bacteria. Second, it acts as a PRR to activate the NF-κB signaling pathway to resist the growth of pathogenic bacteria in the gut.
References
Bao YY, Qu LY, Zhao D, Chen LB, Jin HY, Xu LM, Cheng JA, Zhang CX (2013) The genome- and transcriptome-wide analysis of innate immunity in the brown planthopper Nilaparvata Lugens. BMC Genom 14:160. https://doi.org/10.1186/1471-2164-14-160
Belvin MP, Anderson KV (1996) A conserved signaling pathway: the Drosophila toll-dorsal pathway. Annu Rev Cell Dev Biol 12:393–416. https://doi.org/10.1146/annurev.cellbio.12.1.393
Bischoff V, Vignal C, Boneca IG, Michel T, Hoffmann JA, Royet J (2004) Function of the Drosophila pattern-recognition receptor PGRP-SD in the detection of Gram-positive bacteria. Nat Immunol 5:1175–1180. https://doi.org/10.1038/ni1123
Bischoff V, Vignal C, Duvic B, Boneca IG, Hoffmann JA, Royet J (2006) Downregulation of the Drosophila immune response by peptidoglycan-recognition proteins SC1 and SC2. PLoS Pathog 2:e14. https://doi.org/10.1371/journal.ppat.0020014
Charroux B, Capo F, Kurz CL, Peslier S, Chaduli D, Viallat-Lieutaud A, Royet J (2018) Cytosolic and secreted peptidoglycan-degrading enzymes in Drosophila respectively control local and systemic immune responses to microbiota. Cell Host Microbe 23(215–228):e214. https://doi.org/10.1016/j.chom.2017.12.007
Chen K, Lu Z (2018) Immune responses to bacterial and fungal infections in the silkworm, Bombyx mori. Dev Comp Immunol 83:3–11. https://doi.org/10.1016/j.dci.2017.12.024
Chen K, Liu C, He Y, Jiang H, Lu Z (2014) A short-type peptidoglycan recognition protein from the silkworm: expression, characterization and involvement in the prophenoloxidase activation pathway. Dev Comp Immunol 45:1–9. https://doi.org/10.1016/j.dci.2014.01.017
Chen K, Zhou L, Chen F, Peng Y, Lu Z (2016) Peptidoglycan recognition protein-S5 functions as a negative regulator of the antimicrobial peptide pathway in the silkworm, Bombyx mori. Dev Comp Immunol 61:126–135. https://doi.org/10.1016/j.dci.2016.03.023
Chen C, Eldein S, Zhou X, Sun Y, Gao J, Sun Y, Liu C, Wang L (2018a) Immune function of a Rab-related protein by modulating the JAK-STAT signaling pathway in the silkworm, Bombyx mori. Arch Insect Biochem Physiol. https://doi.org/10.1002/arch.21434
Chen J, Zhang DW, Jin X, Xu XL, Zeng BP (2018b) Characterization of the akirin gene and its role in the NF-κB signaling pathway of Sogatella furcifera. Front Physiol 9:1411. https://doi.org/10.3389/fphys.2018.01411
Cheng X, Zhang X, Pflugrath JW, Studier FW (1994) The structure of bacteriophage T7 lysozyme, a zinc amidase and an inhibitor of T7 RNA polymerase. Proc Natl Acad Sci U S A 91:4034–4038. https://doi.org/10.1073/pnas.91.9.4034
Choe KM, Werner T, Stoven S, Hultmark D, Anderson KV (2002) Requirement for a peptidoglycan recognition protein (PGRP) in Relish activation and antibacterial immune responses in Drosophila. Sci 296:359–362. https://doi.org/10.1126/science.1070216
Dawadi B, Wang X, Xiao R, Muhammad A, Hou Y, Shi Z (2018) PGRP-LB homolog acts as a negative modulator of immunity in maintaining the gut-microbe symbiosis of red palm weevil, Rhynchophorus ferrugineus Olivier. Dev Comp Immunol 86:65–77. https://doi.org/10.1016/j.dci.2018.04.021
Dziarski R, Gupta D (2018) A balancing act: PGRPs preserve and protect. Cell Host Microbe 23:149–151. https://doi.org/10.1016/j.chom.2018.01.010
Garver LS, Wu J, Wu LP (2006) The peptidoglycan recognition protein PGRP-SC1a is essential for Toll signaling and phagocytosis of Staphylococcus aureus in Drosophila. Proc Natl Acad Sci U S A 103:660–665. https://doi.org/10.1073/pnas.0506182103
Gendrin M, Zaidman-Remy A, Broderick NA, Paredes J, Poidevin M, Roussel A, Lemaitre B (2013) Functional analysis of PGRP-LA in Drosophila immunity. PLoS ONE 8:e69742. https://doi.org/10.1371/journal.pone.0069742
Gottar M, Gobert V, Michel T, Belvin M, Duyk G, Hoffmann JA, Ferrandon D, Royet J (2002) The Drosophila immune response against Gram-negative bacteria is mediated by a peptidoglycan recognition protein. Nature 416:640–644. https://doi.org/10.1038/nature734
Guan R, Roychowdhury A, Ember B, Kumar S, Boons GJ, Mariuzza RA (2004) Structural basis for peptidoglycan binding by peptidoglycan recognition proteins. Proc Natl Acad Sci U S A 101:17168–17173. https://doi.org/10.1073/pnas.0407856101
Hillyer JF (2016) Insect immunology and hematopoiesis. Dev Comp Immunol 58:102–118. https://doi.org/10.1016/j.dci.2015.12.006
Huang JH, Jing X, Douglas AE (2015) The multi-tasking gut epithelium of insects. Insect Biochem Mol Biol 67:15–20. https://doi.org/10.1016/j.ibmb.2015.05.004
Iatsenko I, Kondo S, Mengin-Lecreulx D, Lemaitre B (2016) PGRP-SD, an extracellular pattern-recognition receptor, enhances peptidoglycan-mediated activation of the Drosophila Imd pathway. Immunity 45:1013–1023. https://doi.org/10.1016/j.immuni.2016.10.029
International Aphid Genomics C (2010) Genome sequence of the pea aphid Acyrthosiphon pisum. PLoS Biol 8:e1000313. https://doi.org/10.1371/journal.pbio.1000313
Koh C, Allen SL, Herbert RI, McGraw EA, Chenoweth SF (2018) The transcriptional response of Aedes aegypti with variable extrinsic incubation periods for dengue virus. Genome Biol Evol 10:3141–3151. https://doi.org/10.1093/gbe/evy230
Koyama H, Kato D, Minakuchi C, Tanaka T, Yokoi K, Miura K (2015) Peptidoglycan recognition protein genes and their roles in the innate immune pathways of the red flour beetle, Tribolium castaneum. J Invertebr Pathol 132:86–100. https://doi.org/10.1016/j.jip.2015.09.003
Kurata S (2010) Extracellular and intracellular pathogen recognition by Drosophila PGRP-LE and PGRP-LC. Int Immunol 22:143–148. https://doi.org/10.1093/intimm/dxp128
Laughton AM, Garcia JR, Altincicek B, Strand MR, Gerardo NM (2011) Characterisation of immune responses in the pea aphid, Acyrthosiphon pisum. J Insect Physiol 57:830–839. https://doi.org/10.1016/j.jinsphys.2011.03.015
Lemaitre B, Kromer-Metzger E, Michaut L, Nicolas E, Meister M, Georgel P, Reichhart JM, Hoffmann JA (1995) A recessive mutation, immune deficiency (imd), defines two distinct control pathways in the Drosophila host defense. Proc Natl Acad Sci U S A 92:9465–9469
Leulier F, Parquet C, Pili-Floury S, Ryu JH, Caroff M, Lee WJ, Mengin-Lecreulx D, Lemaitre B (2003) The Drosophila immune system detects bacteria through specific peptidoglycan recognition. Nat Immunol 4:478–484. https://doi.org/10.1038/ni922
Li MF, Zhang M, Wang CL, Sun L (2012) A peptidoglycan recognition protein from Sciaenops ocellatus is a zinc amidase and a bactericide with a substrate range limited to Gram-positive bacteria. Fish Shellfish Immunol 32:322–330. https://doi.org/10.1016/j.fsi.2011.11.024
Li T, Yan D, Wang X, Zhang L, Chen P (2019) Hemocyte changes during immune melanization in Bombyx Mori infected with Escherichia coli. Insects. https://doi.org/10.3390/insects10090301
Liu C, Xu Z, Gupta D, Dziarski R (2001) Peptidoglycan recognition proteins: a novel family of four human innate immunity pattern recognition molecules. J Biol Chem 276:34686–34694. https://doi.org/10.1074/jbc.M105566200
Liu Y, Ye N, Chen M, Zhao H, An J (2020) Structural and functional analysis of PGRP-LC indicates exclusive Dap-type PGN binding in bumblebees. Int J Mol Sci. https://doi.org/10.3390/ijms21072441
Livak KJ, Schmittgen TD (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods 25:402–408. https://doi.org/10.1006/meth.2001.1262
Mellroth P, Steiner H (2006) PGRP-SB1: an N-acetylmuramoyl l-alanine amidase with antibacterial activity. Biochem Biophys Res Commun 350:994–999. https://doi.org/10.1016/j.bbrc.2006.09.139
Mellroth P, Karlsson J, Steiner H (2003) A scavenger function for a Drosophila peptidoglycan recognition protein. J Biol Chem 278:7059–7064. https://doi.org/10.1074/jbc.M208900200
Michel T, Reichhart JM, Hoffmann JA, Royet J (2001) Drosophila Toll is activated by Gram-positive bacteria through a circulating peptidoglycan recognition protein. Nature 414:756–759. https://doi.org/10.1038/414756a
Myllymaki H, Valanne S, Ramet M (2014) The Drosophila imd signaling pathway. J Immunol 192:3455–3462. https://doi.org/10.4049/jimmunol.1303309
Neyen C, Runchel C, Schupfer F, Meier P, Lemaitre B (2016) The regulatory isoform rPGRP-LC induces immune resolution via endosomal degradation of receptors. Nat Immunol 17:1150–1158. https://doi.org/10.1038/ni.3536
Paredes JC, Welchman DP, Poidevin M, Lemaitre B (2011) Negative regulation by amidase PGRPs shapes the Drosophila antibacterial response and protects the fly from innocuous infection. Immunity 35:770–779. https://doi.org/10.1016/j.immuni.2011.09.018
Ramet M (2012) The fruit fly Drosophila melanogaster unfolds the secrets of innate immunity. Acta Paediatr 101:900–905. https://doi.org/10.1111/j.1651-2227.2012.02740.x
Ramet M, Manfruelli P, Pearson A, Mathey-Prevot B, Ezekowitz RA (2002) Functional genomic analysis of phagocytosis and identification of a Drosophila receptor for E. coli. Nature 416:644–648. https://doi.org/10.1038/nature735
Ramirez JL, Muturi EJ, Barletta ABF, Rooney AP (2019) The Aedes aegypti IMD pathway is a critical component of the mosquito antifungal immune response. Dev Comp Immunol 95:1–9. https://doi.org/10.1016/j.dci.2018.12.010
Reiser JB, Teyton L, Wilson IA (2004) Crystal structure of the Drosophila peptidoglycan recognition protein (PGRP)-SA at 1.56 A resolution. J Mol Biol 340:909–917. https://doi.org/10.1016/j.jmb.2004.04.077
Royet J, Gupta D, Dziarski R (2011) Peptidoglycan recognition proteins: modulators of the microbiome and inflammation. Nat Rev Immunol 11:837–851. https://doi.org/10.1038/nri3089
Takehana A, Katsuyama T, Yano T, Oshima Y, Takada H, Aigaki T, Kurata S (2002) Overexpression of a pattern-recognition receptor, peptidoglycan-recognition protein-LE, activates imd/relish-mediated antibacterial defense and the prophenoloxidase cascade in Drosophila larvae. Proc Natl Acad Sci U S A 99:13705–13710. https://doi.org/10.1073/pnas.212301199
Tanaka H, Sagisaka A (2016) Involvement of peptidoglycan recognition protein L6 in activation of immune deficiency pathway in the immune responsive silkworm cells. Arch Insect Biochem Physiol 92:143–156. https://doi.org/10.1002/arch.21326
Tanaka H, Ishibashi J, Fujita K, Nakajima Y, Sagisaka A, Tomimoto K, Suzuki N, Yoshiyama M, Kaneko Y, Iwasaki T, Sunagawa T, Yamaji K, Asaoka A, Mita K, Yamakawa M (2008) A genome-wide analysis of genes and gene families involved in innate immunity of Bombyx mori. Insect Biochem Mol Biol 38:1087–1110. https://doi.org/10.1016/j.ibmb.2008.09.001
Valanne S, Wang JH, Ramet M (2011) The Drosophila Toll signaling pathway. J Immunol 186:649–656. https://doi.org/10.4049/jimmunol.1002302
Wang S, Beerntsen BT (2015) Functional implications of the peptidoglycan recognition proteins in the immunity of the yellow fever mosquito, Aedes aegypti. Insect Mol Biol 24:293–310. https://doi.org/10.1111/imb.12159
Wang ZM, Li X, Cocklin RR, Wang M, Wang M, Fukase K, Inamura S, Kusumoto S, Gupta D, Dziarski R (2003) Human peptidoglycan recognition protein-l is an N-acetylmuramoyl-l-alanine amidase. J Biol Chem 278:49044–49052. https://doi.org/10.1074/jbc.M307758200
Wang L, Tang N, Gao X, Chang Z, Zhang L, Zhou G, Guo D, Zeng Z, Li W, Akinyemi IA, Yang H, Wu Q (2017) Genome sequence of a rice pest, the white-backed planthopper (Sogatella furcifera). Gigascience 6:1–9. https://doi.org/10.1093/gigascience/giw004
Wang YH, Chang MM, Wang XL, Zheng AH, Zou Z (2018) The immune strategies of mosquito Aedes aegypti against microbial infection. Dev Comp Immunol 83:12–21. https://doi.org/10.1016/j.dci.2017.12.001
Werner T, Liu G, Kang D, Ekengren S, Steiner H, Hultmark D (2000) A family of peptidoglycan recognition proteins in the fruit fly Drosophila melanogaster. Proc Natl Acad Sci USA 97:13772–13777. https://doi.org/10.1073/pnas.97.25.13772
Yang J, Wang W, Wei X, Qiu L, Wang L, Zhang H, Song L (2010) Peptidoglycan recognition protein of Chlamys farreri (CfPGRP-S1) mediates immune defenses against bacterial infection. Dev Comp Immunol 34:1300–1307. https://doi.org/10.1016/j.dci.2010.08.006
Yang DQ, Su ZL, Qiao C, Zhang Z, Wang JL, Li F, Liu XS (2013) Identification and characterization of two peptidoglycan recognition proteins with zinc-dependent antibacterial activity from the cotton bollworm, Helicoverpa armigera. Dev Comp Immunol 39:343–351. https://doi.org/10.1016/j.dci.2012.12.006
Yang J, Wang X, Tang S, Shen Z, Wu J (2015) Peptidoglycan recognition protein S2 from silkworm integument: characterization, microbe-induced expression, and involvement in the immune-deficiency pathway. J Insect Sci 15:20. https://doi.org/10.1093/jisesa/iev007
Yang PJ, Zhan MY, Ye C, Yu XQ, Rao XJ (2017) Molecular cloning and characterization of a short peptidoglycan recognition protein from silkworm Bombyx mori. Insect Mol Biol 26:665–676. https://doi.org/10.1111/imb.12330
Yang LL, Zhan MY, Zhuo YL, Pan YM, Xu Y, Zhou XH, Yang PJ, Liu HL, Liang ZH, Huang XD, Yu XQ, Rao XJ (2018) Antimicrobial activities of a proline-rich proprotein from Spodoptera litura. Dev Comp Immunol 87:137–146. https://doi.org/10.1016/j.dci.2018.06.011
Yang PJ, Zhan MY, Yang LL, Liu QQ, Xu Y, Pan YM, Rao XJ (2019) Characterization of PGRP-S1 from the oriental armyworm, Mythimna separata. Dev Comp Immunol 90:121–129. https://doi.org/10.1016/j.dci.2018.09.009
Zaidman-Remy A, Herve M, Poidevin M, Pili-Floury S, Kim MS, Blanot D, Oh BH, Ueda R, Mengin-Lecreulx D, Lemaitre B (2006) The Drosophila amidase PGRP-LB modulates the immune response to bacterial infection. Immunity 24:463–473. https://doi.org/10.1016/j.immuni.2006.02.012
Zaidman-Remy A, Poidevin M, Herve M, Welchman DP, Paredes JC, Fahlander C, Steiner H, Mengin-Lecreulx D, Lemaitre B (2011) Drosophila immunity: analysis of PGRP-SB1 expression, enzymatic activity and function. PLoS ONE 6:e17231. https://doi.org/10.1371/journal.pone.0017231
Zhan MY, Yang PJ, Rao XJ (2018) Molecular cloning and analysis of PGRP-L1 and IMD from silkworm Bombyx mori. Comp Biochem Physiol B Biochem Mol Biol 215:19–30. https://doi.org/10.1016/j.cbpb.2017.10.002
Zheng H, Powell JE, Steele MI, Dietrich C, Moran NA (2017) Honeybee gut microbiota promotes host weight gain via bacterial metabolism and hormonal signaling. Proc Natl Acad Sci U S A 114:4775–4780. https://doi.org/10.1073/pnas.1701819114
Acknowledgements
This research was supported by the National Natural Science Foundation of China (No. 31860505) and the Joint Project from Zunyi science and Technology Bureau and Zunyi Normal University Natural Science Foundation (No.ZSKH HZ (264)).
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Yu, Y., Luo, C., Zhang, D. et al. Characterization of PGRP-LB and immune deficiency in the white-backed planthopper Sogatella furcifera (Hemiptera: Delphacidae). Appl Entomol Zool 57, 1–14 (2022). https://doi.org/10.1007/s13355-021-00750-w
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13355-021-00750-w