
Abstract
Background
The activation of TGF-β pathway can facilitate tumorigenesis. Understanding the TGF-related genes (TRGs) in oral cancer and determining their prognostic value is of utmost importance.
Methods
The TRGs were selected to develop a prognostic model based on lasso regression. Oral cancer patients were classified into high-risk and low-risk groups based on the risk model. Subsequently, multivariate COX regression was employed to identify the prognostic marker. Additionally, the expression of SMURF2 was validated using quantitative real-time polymerase chain reaction (qRT-PCR) and the Human Protein Atlas (HPA) database. To investigate the relationship between SMURF2 expression and immune cell infiltrations, we conducted single-sample Gene Set Enrichment Analysis (ssGSEA) analyses.
Results
We identified 16 differentially expressed TRGs in oral cancer, all of which showed upregulation. From these, we selected eight TRGs as prognostic signatures. Furthermore, the high-risk group demonstrated lower infiltration levels of immune cells, immune score, and higher tumor purity. Interestingly, we also found that SMURF2 serves as an independent prognostic biomarker. SMURF2 was upregulated in oral cancer, as confirmed by public databases and qRT-PCR analysis. Importantly, our results indicate a close association between SMURF2 expression and the immune microenvironment.
Conclusion
The 8-TRG signature prognosis model that we constructed has the ability to predict the survival rate and immune activity of oral cancer patients. SMURF2 could be effective in recognizing prognosis and evaluating immune efficacy for oral cancer.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Cancer incidence and mortality rates have been steadily rising globally. In the year 2018 alone, there were an estimated 18.1 million new cases of cancer and 9.6 million cancer-related deaths worldwide. Among these cases, oral cancer made up approximately 2% of the total (Bray et al. 2018; Siegel et al. 2018). The most prevalent form of oral cancer is oral squamous cell carcinoma (OSCC), which is known for its high morbidity and malignancy (Shi et al. 2019; Huang et al. 2019). Oral squamous cell carcinoma (OSCC) constitutes over 90% of all oral tumor cases and is linked to factors including areca nut chewing, immune deficiencies, alcohol and tobacco use, and other related factors (Markopoulos 2012). Despite notable progress in diagnosis and treatment, the survival rate for OSCC remains low at 50% over a 5-year period, while the likelihood of regional recurrence ranges from 33 to 40% (Vigneswaran and Williams 2014; Bello et al. 2010). The unfavorable prognosis of OSCC can be attributed to its highly invasive nature and tendency to metastasize (Jerjes et al. 2010). Furthermore, the TNM classification system, which encompasses the evaluation of the tumor, lymph node, and metastasis, has gained significant popularity and is regarded as an indispensable instrument for prognosticating the survival prospects of individuals with OSCC. Nevertheless, this system has faced criticism due to its inability to decipher the diverse outcomes observed among patients with identical TNM stages (Costa Ade et al. 2005; Mortensen et al. 2022). Therefore, it is of utmost importance to uncover the biological molecular mechanism that drives the progressive of OSCC and to identify new markers that can accurately predict clinical outcomes.
The signaling pathway of transforming growth factor β is involved in numerous vital biological processes, such as cell migration, apoptosis, differentiation and growth, and even the initiation and development of cancer (Shi and Massagué 2003). The TGF-β protein plays a dual role in the formation of tumors. In the early stages of tumor development, it acts as a suppressor, inducing apoptosis and cell cycle arrest to hinder tumor growth. However, as the tumor advances, the tumor cells gradually lose their sensitivity to TGF-β. Consequently, the TGF-β protein released by the tumor cells promotes immune system suppression, facilitates the formation of new blood vessels to nourish the tumor, and enhances the ability of the tumor to invade surrounding tissues and spread to distant sites (Connolly et al. 2012). Many cancers exhibit increased expression of TGF-β and enhanced activation of intracellular signaling through TGF-β receptors (Massagué 2008). It is worth noting that patients with the poorest prognosis show a significant increase in the activity of the TGF-β pathway (Guinney et al. 2015; Calon et al. 2015). Consequently, the activation of this pathway in cancer cells can lead to a process called epithelial-to-mesenchymal transition, in which the normal polarity and adhesion between epithelial cells are lost, and these cells acquire traits of mobile mesenchymal cells (Nieto et al. 2016). The development of oral cancer is caused by the disruption of TGF-β signaling, leading to changes in the normal physiological processes (Prime et al. 2004; Paterson et al. 2001). Upregulation of TGF-β has been observed in samples obtained from patients with oral cancer who have developed metastases to the bones (Takahashi et al. 2020). Furthermore, according to a recent study, it was found that cancer-associated fibroblasts have the ability to secrete TGF-β1, which can enhance OSCC invasion in vitro (Yang et al. 2022). As a result, genes linked to the TGF-β pathway could potentially be used as markers to forecast the prognosis of oral cancer. Nevertheless, there have been insufficient studies examining the connection between TGF-β pathway-related genes (TRGs) and the outcomes of individuals with oral cancer.
The emergence of next-generation sequencing technologies and the availability of the Cancer Genome Atlas (TCGA) datasets have made transcriptomic and genomic data on common cancers widely accessible to the public. As a result, this has created an ideal opportunity to analyze and uncover the prognostic and predictive values of potential biomarkers in the field of precision medicine focused on cancer analysis (Fu et al. 2023; Wang et al. 2023; Zhao et al. 2023). The novelty of our work lies in the comprehensive analysis of TRGs in oral cancer, utilizing a robust methodological approach that combines lasso regression for prognostic model development and multivariate Cox regression to identify key prognostic markers. Additionally, by validating the expression of SMURF2 through quantitative real-time polymerase chain reaction (qRT-PCR) and the Human Protein Atlas database, and examining its relationship with immune cell infiltrations via single-sample Gene Set Enrichment Analysis (ssGSEA), our study not only underscores the prognostic value of SMURF2 but also illuminates its role in shaping the immune landscape of oral cancer. This study aims to provide a deeper understanding of the TGF-β pathway’s involvement in oral cancer, offering novel insights into the prognostic significance of TRGs and highlighting SMURF2 as a potential biomarker for prognosis and immune efficacy evaluation in oral cancer. Through our findings, we aspire to contribute to the ongoing efforts to improve the diagnostic precision and therapeutic outcomes for oral cancer patients, paving the way for more personalized and effective cancer care strategies. The flow chart of this study is presented in Fig. 1.

Flowchart outlining the methodology employed in the present study
Methods
Data collection
The transcriptome expression data of OSCC samples, along with their corresponding clinical data, were downloaded from the TCGA database (https://portal.gdc.cancer.gov) and GEO database (https://www.ncbi.nlm.nih.gov/geo/). We collected a total of 331 OSCC samples and 32 normal samples from the TCGA-OSCC cohort, along with 97 OSCC samples from the GSE41613 dataset. The RNA-seq data in TPM format from TCGA were collected and transformed into log2 [TPM + 1]. TGFP-related genes (TRGs) were collected from the Molecular Signatures Database.
GSEA
Gene set enrichment analysis (GSEA) is a widely used technique for uncovering the biological mechanisms within clinical specimens by examining the patterns of gene expression they display (Han et al. 2019). We conducted a GSEA using the “ClusterProfiler” package in order to determine the most important pathways within the molecular subgroups. We utilized the “h.all.v7.4.symbols.gmt” subset from the Molecular Signatures Database to assess the relevant pathways and molecular mechanisms. Statistical significance was defined as a p value less than 0.05.
Assessment of the score of TGFP by the GSVA
Gene Set Variation Analysis (GSVA), an unsupervised approach, is utilized to assess the diversity of pathway activity among a group of individuals (Hänzelmann et al. 2013). The GSVA algorithm was applied to calculate the score representing the activity of TGFP. The outcomes are illustrated in the form of box plots.
Identification of differentially expressed TRGs
To identify differentially expressed TRGs between normal and tumor samples, we utilized the “limma” package. We set the criteria for significance at p.adjust < 0.05 and |log fold change (FC)| ≥ 1 (Fu et al. 2023). The “pheatmap” package was employed to create the heatmap, while the “ggplot2” package was used to generate the volcano plot.
Enrichment analysis of differentially expressed TRGs
The R package ClusterProfiler was utilized to conduct enrichment analysis for Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG). The outcomes were rendered visually through the ggplot2 package. A term was deemed statistically significant if its p value was below 0.05.
Construction of a prognostic signature based on differentially expressed TRGs
We applied LASSO regression analysis to identify survival-related genes. To avoid overfitting, we employed 1000 rounds of cross-validation to select the penalty parameters. By considering the expression levels and coefficient values of prognosis-related genes, we developed a predictive marker for OSCC patients using the following formula: risk score = (expression level of ACVR1*0.1511) − (expression level of BMP2*0.091) + (expression level of CDK9*0.2191) − (expression level of LTBP2*0.0116) + (expression level of SLC20A1*0.1871) + (expression level of SMURF2*0.0042) + (expression level of TGFB1*0.0976) − (expression level of TGFB3*0.1374). The R “Survminer” package was utilized to determine the median risk score, subsequently dividing OSCC patients into high-risk and low-risk groups. The prognostic efficacy of the risk score was assessed by generating a Kaplan-Meier curve using the “survival” package.
Tumor immune microenvironment analysis
We utilized the ESTIMATE to compare the Stromal Score, Immune Score, and ESTIMATE Score between low-risk and high-risk subgroups (Yoshihara et al. 2013). Additionally, the abundance of immune cells in these subgroups was evaluated through ssGSEA (Huang et al. 2021). The results were visualized using a heatmap and box plot. Furthermore, we examined the correlation between SMURF2 expression and immune cell infiltration using the ggplot2 package and presented the findings in a lollipop chart.
Identifying independent prognostic marker
We utilized multivariate Cox regression analysis to further select signature genes. Survival curves were plotted, and the relationship between independent prognostic markers and overall survival was assessed using the ggplot2 and survminer packages. The expression of SMURF2 in oral cancer was analyzed using the TNMplot.com analysis platform (www.tnmplot.com). In addition, the comparison of SMURF2 protein expression between tumor tissue and normal adjacent tissue was performed using the human protein atlas (HPA) website (https://www.proteinatlas.org).
Development and assessment of a prognostic nomogram
A predictive tool, known as a nomogram, was created using the results of a multivariate analysis to forecast the overall survival rate of individuals diagnosed with oral cancer. This nomogram was constructed utilizing the ggplot2 and survival R packages, and calibration curves were generated to ensure accuracy. To assess the reliability of the nomogram, ROC curve was employed. Moreover, a decision curve analysis (DCA) was conducted to evaluate the potential clinical utility of the nomogram.
Cell culture
The oral cancer cell lines (SCC15, SCC9, SCC4, HSC4, and CAL27) and human oral gingival epithelial cell line (HOEC cells) were obtained from the ATCC (Manassas, VA, USA). All cell lines were cultured in high-glucose Dulbecco’s modified Eagle’s medium (Gibco, CA, USA) supplemented with 10% fetal bovine serum and penicillin/streptomycin at 37 °C and 5% CO2.
qRT-PCR
We used Trizol Reagent (Invitrogen, CA, USA) to extract total RNA from cell lines. To assess the purity and concentration of RNA, spectrophotometry at a wavelength of 260/280 nm was employed. Following this, a reverse transcription process was carried out using the reverse transcription kit (Takara). The gene expression levels were detected using SYBR-Green (Takara) and qRT-PCR analysis, with β-ACTIN serving as the internal reference. The primers used are listed in Table S1.
Results
Identification of differentially expressed TRGs
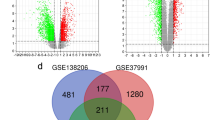
According to the findings in Fig. 2A, the GSEA analysis indicated a significant enrichment of the TGF-β pathway in the tumor group. By employing the GSVA algorithm, we calculated the TGF-β pathway gene set score for each sample. It is important to highlight that the oral cancer group exhibited a significantly higher score for the TGF-β pathway compared to the normal group, as shown in Fig. 2B (p < 0.01). These results imply a potential essential role of the TGF-β pathway gene set in the advancement of oral cancer, underscoring the need for further investigation and analysis of this particular gene set. After conducting a comparative analysis between the groups with oral cancer and normal individuals, it was discovered that a total of 16 TRGs exhibited differential expression. These genes were all observed to be upregulated in the tumor group. Specifically, the upregulated genes included ID3, TGFB3, LTBP2, PMEPA1, BMP2, SERPINE1, TGFB1, TGIF1, SMURF1, SKIL, ACVR1, CDK9, RAB31, TGFBR1, SLC20A1, and SMURF2 (Fig. 2C, D).

Screening of differentially expressed TRGs. A GSEA result showed a significant enrichment of the TGF-β pathway in the tumor group. B A box plot was employed to visually illustrate the TGF-β pathway score comparison between the normal group and the oral cancer group. **p < 0.01. C The volcano plot depicting the expression level of TRGs. D A heatmap was employed to visually display the expression levels of TRGs in both the normal and oral cancer groups
Enrichment analysis of differentially expressed TRGs
As depicted in Fig. 3A, the results from the GO analysis indicated a notable enrichment of these TRGs in the transforming growth factor beta receptor signaling pathway, transmembrane receptor protein serine/threonine kinase signaling pathway, SMAD binding, cellular response to transforming growth factor beta stimulus, etc. KEGG analysis revealed that these TRGs were significantly enriched in the TGF-beta signaling pathway, hippo signaling pathway, cytokine-cytokine receptor interaction, cellular senescence, etc. (Fig. 3B).

Enrichment analysis of differentially expressed TRGs. Functional annotation analyses were performed using GO enrichment analysis (A) and KEGG enrichment analysis (B)
Establishment of a prognostic signature related to TRGs
The LASSO regression analysis was performed using the previously mentioned 16 TRGs. Screening of the data revealed eight TRGs (TGFB3, LTBP2, BMP2, TGFB1, ACVR1, CDK9, SLC20A1, and SMURF2) with non-zero coefficients, indicating their association with the prognosis of oral cancer patients (Fig. 4A, B). The Kaplan-Meier curve demonstrated that patients with OSCC who were classified as high risk experienced significantly reduced overall survival compared to those classified as low risk (Fig. 4C). Furthermore, it was observed that a significant proportion of TRGs were upregulated in the subgroup with a high risk (Fig. 4D).

Establishment of a prognostic signature related to TRGs. A, B LASSO variable trajectory plots were analyzed to identify non-zero variables. C Comparison of survival outcomes between OSCC patients classified as high risk and low risk in the TCGA-OSCC dataset. D A heatmap was employed to visually display the expression levels of TRGs in both the low-risk and high-risk subgroups. **p < 0.01; ***p < 0.001
Immuno-infiltration analysis of the TRG-related signature
By utilizing ssGSEA, we investigated the association between the risk score and immune infiltration and found that patients with a lower risk score showed increased infiltration of immune cells in comparison to those with a higher risk score (Fig. 5A, B). By conducting ESTIMATE algorithm, we demonstrated that the low-risk group had higher ESTIMATE score, stromal score, and immune score, whereas the low-risk group had lower tumor purity compared to the high-risk group (Fig. 5C–F).

Immune infiltration analysis. We utilized the heatmap (A) and box plot (B) to visually illustrate the enrichment fraction of infiltrating cells in both high-risk and low-risk subgroups. Variations in the ESTIMATEScore (C), ImmuneScore (D), StromalScore (E), and tumor purity (F) among patients with high-risk and low-risk statuses. **p < 0.01; ***p < 0.001
GSEA
GSEA results revealed that DNA repair, p53 pathway, glycolysis, G2M checkpoint, PI3K-AKT MTOR signaling, TGF beta signaling, fatty acid metabolism, and apoptosis were significantly enriched in the high-risk subgroup (Fig. 6).

GSEA was conducted to investigate the potential pathways associated with the low-risk and high-risk subgroups
SMURF2 emerged as a significant prognostic indicator for patients diagnosed with oral cancer.
In OSCC patients, a multivariate regression analysis was conducted on the eight TRGs, revealing that SMURF2 emerged as an independent prognostic factor (Table 1). The results from Kaplan-Meier analysis indicated a significant correlation between increased SMURF2 expression and decreased overall survival in patients with oral squamous cell carcinoma (Fig. 7A and Figure S1). The findings from the TCGA-OSCC dataset (Fig. 7B) and analysis conducted on TNMplot.com (Fig. 7C, D) revealed that the mRNA expression level of SMURF2 was significantly higher in the tumor group in comparison to the normal group (p < 0.001 or p < 0.01). In addition, by utilizing the HPA database (Fig. 7E), we have also discovered that the protein expression level of SMURF2 exhibited a marked increase in the tumor group as compared to the normal group. As shown in Figure S2, the comparative analysis of SMURF2 gene expression across various cancer types indicated differential expression patterns between normal and tumor tissues. In the case of adrenocortical carcinoma (ACC), the expression of SMURF2 was significantly lower in tumor samples as compared to normal tissue. Similar patterns of reduced SMURF2 expression in tumor samples were observed in kidney chromophobe (KICH), lung adenocarcinoma (LUAD), lung squamous cell carcinoma (LUSC), ovarian serous cystadenocarcinoma (OV), skin cutaneous melanoma (SKCM), testicular germ cell tumors (TGCT), uterine corpus endometrial carcinoma (UCEC), and uterine carcinosarcoma (UCS), all showing statistical significance (p < 0.05). Conversely, for cholangiocarcinoma (CHOL), colon adenocarcinoma (COAD), lymphoid neoplasm diffuse large B-cell lymphoma (DLBC), esophageal carcinoma (ESCA), glioblastoma multiforme (GBM), head and neck squamous cell carcinoma (HNSC), kidney renal papillary cell carcinoma (KIRP), acute myeloid leukemia (LAML), brain lower grade glioma (LGG), liver hepatocellular carcinoma (LIHC), pancreatic adenocarcinoma (PAAD), rectum adenocarcinoma (READ), stomach adenocarcinoma (STAD), and thymoma (THYM), tumor tissues demonstrated significantly higher expression levels of SMURF2 when compared to normal tissues (p < 0.05).

SMURF2 was a significant predictor of prognosis in patients with oral cancer. A Comparison of survival outcomes between OSCC patients classified as high-SMURF2 and low-SMURF2 in the TCGA-OSCC dataset. The expression of SMURF2 was examined in both the TCGA-OSCC dataset (B) and through analysis on the TNMplot.com platform (C, D) to compare the levels between normal and tumor groups. E The protein expression level of SMURF2 in both normal and tumor tissue
Development of nomogram
We have developed a nomogram to forecast the overall survival rates at 1, 3, and 5 years for patients diagnosed with oral cancer. The nomogram, depicted in Fig. 8A, includes the expression of the SMURF2 gene. The calibration curve, shown in Fig. 8B, demonstrates a strong correlation between the predicted outcomes and the actual results observed in oral cancer patients. Furthermore, Fig. 8C highlights the high diagnostic accuracy of the SMURF2 gene, with an area under the curve (AUC) of 0.838. The findings of DCA demonstrated that SMURF2 exhibited a favorable overall benefit and possessed the ability to forecast the overall survival of individuals diagnosed with oral cancer, both in the short and long term (Fig. 8D–F).

Development and assessment of the nomogram. A The nomogram provides the ability to assess the likelihood of survival at 1-year, 3-year, and 5-year time points for individuals with oral cancer. B The accuracy of the nomogram in predicting oral cancer patient outcomes is evident in its well-calibrated curve. C The diagnostic value of SMURF2 as a marker for oral cancer is shown through the ROC curve analysis. D–F Survival rates of oral cancer patients were analyzed using DCA curves for 1, 3, and 5 years, focusing on the SMURF2 gene
Single-gene GSEA
Single-gene GSEA results revealed that PI3K-AKT MTOR signaling, glycolysis, G2M checkpoint, TGF beta signaling, hypoxia, WNT beta catenin signaling, DNA repair, IL2 STAT5 signaling, p53 pathway, inflammatory response, IL6-JAK-STAT3 signaling, and TNFA signaling via NFKB were significantly enriched in high-SMURF2 subgroup (Fig. 9).

Single-gene GSEA was conducted to investigate the potential pathways associated with the low-SMURF2 and high-SMURF2 subgroups
Investigation of the immune cell infiltration patterns within the subgroups of SMURF2
As shown in Fig. 10A, B, the ssGSEA results demonstrated significantly different levels of immune cell infiltration between the high and low SMURF2 subgroups. Correlation analysis demonstrated a positive correlation between the SMURF2 gene and the infiltration level of Th2 cells, T helper cells, NK cells, Tcm, eosinophils, Tem, macrophages, and Tgd. On the other hand, a negative correlation was observed between the SMURF2 gene and the infiltration level of Th17 cells, cytotoxic cells, pDC, NK CD56dim cells, T cells, and B cells (Fig. 10C).

Investigation of the immune cell infiltration patterns within the subgroups of SMURF2. The visual representation of the 24 immune cell types in both the low-SMURF2 and high-SMURF2 groups was presented using a heatmap (A) and box plot (B). C To illustrate the correlation between SMURF2 expression and immune cell infiltration levels, a Lollipop plot was employed. *p < 0.05; **p < 0.01; ***p < 0.001
Validation of SMURF2 expression by qRT-PCR analysis
As shown in Fig. 11, the qRT-PCR findings demonstrated that the expression level of SMURF2 was increased in SCC15, SCC9, SCC4, HSC4, and CAL27 cells compared to HOEC cells (p < 0.05 or p < 0.001). These results align with our bioinformatics analyses.

Cell experiments were conducted to validate the expression of SMURF2. The expression level of SMURF2 was compared between various oral cancer cell lines (SCC15, SCC9, SCC4, HSC4, and CAL27) and a human oral gingival epithelial cell line (HOEC cells). *p < 0.05; ***p < 0.001
Discussion
Oral cancer has emerged as one of the most lethal diseases globally. Over the past four decades, there has been little improvement in the outcomes for patients as they continue to suffer from advanced disease, leading to locoregional recurrence and unfavorable results (Gormley et al. 2022). The treatment methods for oral cancer are both expensive and lengthy, making them financially burdensome for many individuals. Therefore, it is crucial to accurately and promptly predict the outcomes of oral cancer, as it can greatly influence the available therapeutic choices and enhance prognosis. The field of diagnosis and prognosis at the individual level holds great promise with the utilization of high-throughput genomics technologies, paving the way for a promising future (Fu et al. 2022; Zhao et al. 2021). The progression of oral cancer involves a complex series of events, in which the TGF-β pathway plays a crucial role (Paterson et al. 2001; Guo et al. 2023). Therefore, the primary objective of this study was to discover a specific signature related to the TGF-β pathway and evaluate the infiltration of immune cells using bioinformatics methods.
The findings from our study indicated that the TGF-β pathway was significantly more active in the oral cancer group compared to the normal group, as demonstrated by the GSVA results. Through an analysis of the TCGA database, we identified 16 specific TRGs that were differentially expressed in patients with OSCC. Furthermore, our study unveiled the close relationship between TGF-β pathway-associated risk subgroups and the prognosis as well as the tumor immune microenvironment of OSCC. These findings are consistent with previous studies. In oral cancer patients, the function of TGF-β signaling is intricate due to its dual effects on tumor development. Initially, TGF-β signaling plays a critical role in suppressing tumor formation. However, in later stages of the tumor, TGF-β signaling can stimulate cancer cell proliferation or facilitate metastatic growth, thus accelerating cancer development (Sun et al. 2008; Qiao et al. 2010). The immune system plays a vital role in the development of cancer. TGF-β and IL-10 are well-known immunosuppressive cytokines that have the ability to dampen immune responses, thereby aiding neoplastic cells in evading detection by the immune system (Arantes et al. 2016; Thomas and Massagué 2005). In OSCC, the immune evasion of NK cells is influenced by the upregulation of TGF-β and IL-10, as well as the downregulation of immune-activating cytokines like NK receptors and IL-2 (Gaur et al. 2014; Dutta et al. 2015). Hence, the impact of the TGF-β pathway on the development of oral cancer could potentially be influenced by its ability to control immune reactions.
Out of the eight TRGs studied, SMURF2 has emerged as a promising marker with prognostic potential in oral cancer. Examining data from the TCGA database and conducting cell experiments, we discovered that SMURF2 is significantly upregulated in oral cancer tissues. Importantly, this elevated expression of SMURF2 was found to correlate with a poorer prognosis in patients with oral cancer. Smad ubiquitin regulatory factor 2 (SMURF2), a factor involved in the regulation of ubiquitin protein modification, is primarily found within the nucleus. Its essential function lies in the inhibitory control of TGF-β signaling pathways (David et al. 1835). Increasing evidence suggested that SMURF2 plays a vital role in regulating tissue homeostasis, establishing cell polarity, maintaining the stability of the genome, and promoting tumorigenesis (Koganti et al. 2018). SMURF2 plays a crucial functional role in inhibiting cancer cell proliferation and tumorigenesis, achieved by promoting ubiquitination and degradation of several vital cellular proteins (Yu et al. 2020; Li et al. 2019). Elevated SMURF2 expression levels have been observed in connection with various forms of cancer and have been linked to unfavorable prognosis (Jin et al. 2009; Klupp et al. 2019; Fukuchi et al. 2002). Our study revealed that SMURF2, as demonstrated through the analysis of various databases and cell experiments, exhibited increased expression in oral cancer. Furthermore, our findings indicated a direct correlation between elevated SMURF2 levels and decreased overall survival time in oral cancer patients. These results align with the previously mentioned studies, reinforcing the significant impact of SMURF2 on prognosis.
Nonetheless, there are limitations to our study. Despite validating the expression level of SMURF2 through cell experiments, the inclusion of a more extensive array of clinical samples would improve the credibility of the findings. Furthermore, the prognostic efficacy of SMURF2 warrants validation in broader cohorts of oral cancer patients.
Conclusion
To summarize, we have successfully created a prognostic model consisting of eight TRGs for patients with oral cancer. Additionally, this model can also be utilized as a tool to predict prognosis and changes in the immune microenvironment. Furthermore, our research has identified SMURF2 as a promising prognostic biomarker, showing a significant association with immune cell infiltration in oral cancer. These findings offer a solid foundation for future investigations into the involvement of SMURF2 in the development and progression of oral cancer.
Data availability
The website we offer provides access to all the data, and the corresponding author can provide them upon request.
References
Arantes DA, Costa NL, Mendonça EF, Silva TA, Batista AC (2016) Overexpression of immunosuppressive cytokines is associated with poorer clinical stage of oral squamous cell carcinoma. Archiv oral biol 61:28–35
Bello IO, Soini Y, Salo T (2010) Prognostic evaluation of oral tongue cancer: means, markers and perspectives (II). Oral Oncol 46:636–643
Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A (2018) Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA: a cancer j clin 68:394–424
Calon A, Lonardo E, Berenguer-Llergo A, Espinet E, Hernando-Momblona X, Iglesias M, Sevillano M, Palomo-Ponce S, Tauriello DVF, Byrom D, Cortina C, Morral C, Barceló C, Tosi S, Riera A, Attolini CS-O, Rossell D, Sancho E, Batlle E (2015) Stromal gene expression defines poor-prognosis subtypes in colorectal cancer. Nature Genet 47:320–329
Connolly EC, Freimuth J, Akhurst RJ (2012) Complexities of TGF-β targeted cancer therapy. Int j biolog sci 8:964–978
Costa Ade L, Araújo Júnior RF, Ramos CC (2005) Correlation between TNM classification and malignancy histological feature of oral squamous cell carcinoma. Brazil j otorhinolaryngol 71:181–187
David D, Nair SA, Pillai MR (1835) Smurf E3 ubiquitin ligases at the cross roads of oncogenesis and tumor suppression. Biochimica et biophysica acta 2013:119–128
Dutta A, Banerjee A, Saikia N, Phookan J, Baruah MN, Baruah S (2015) Negative regulation of natural killer cell in tumor tissue and peripheral blood of oral squamous cell carcinoma. Cytokine 76:123–130
Fu H, Zhao D, Sun L, Huang Y, Ma X (2022) Identification of autophagy-related biomarker and analysis of immune infiltrates in oral carcinoma. J clin laborat anal 36:e24417
Fu J, Lin J, Zeng X, Li G, Wei Y, Xian L (2023) GABRP is a promising prognostic biomarker and associated with immune cell infiltration in lung squamous cell carcinoma. Pharmacogen personal med 16:357–371
Fukuchi M, Fukai Y, Masuda N, Miyazaki T, Nakajima M, Sohda M, Manda R, Tsukada K, Kato H, Kuwano H (2002) High-level expression of the Smad ubiquitin ligase Smurf2 correlates with poor prognosis in patients with esophageal squamous cell carcinoma. Cancer res 62:7162–7165
Gaur P, Singh AK, Shukla NK, Das SN (2014) Inter-relation of Th1, Th2, Th17 and Treg cytokines in oral cancer patients and their clinical significance. Human immunol 75:330–337
Gormley M, Gray E, Richards C, Gormley A, Richmond RC, Vincent EE, Dudding T, Ness AR, Thomas SJ (2022) An update on oral cavity cancer: epidemiological trends, prevention strategies and novel approaches in diagnosis and prognosis. Commun dental health 39:197–205
Guinney J, Dienstmann R, Wang X, de Reyniès A, Schlicker A, Soneson C, Marisa L, Roepman P, Nyamundanda G, Angelino P, Bot BM, Morris JS, Simon IM, Gerster S, Fessler E, De Sousa EMF, Missiaglia E, Ramay H, Barras D et al (2015) The consensus molecular subtypes of colorectal cancer. Nature med 21:1350–1356
Guo Y, Xu T, Chai Y, Chen F (2023) TGF-β signaling in progression of oral cancer. Int j molecul sci 24:10263
Han H, Lee S, Lee I (2019) NGSEA: Network-based gene set enrichment analysis for interpreting gene expression phenotypes with functional gene sets. Molecules cells 42:579–588
Hänzelmann S, Castelo R, Guinney J (2013) GSVA: gene set variation analysis for microarray and RNA-seq data. BMC bioinform 14:7
Huang L, Wu C, Xu D, Cui Y, Tang J (2021) Screening of important factors in the early sepsis stage based on the evaluation of ssGSEA algorithm and ceRNA regulatory network. Evolution bioinformat online 17:11769343211058463
Huang TH, Li KY, Choi WS (2019) Lymph node ratio as prognostic variable in oral squamous cell carcinomas: systematic review and meta-analysis. Oral Oncol 89:133–143
Jerjes W, Upile T, Petrie A, Riskalla A, Hamdoon Z, Vourvachis M, Karavidas K, Jay A, Sandison A, Thomas GJ, Kalavrezos N, Hopper C (2010) Clinicopathological parameters, recurrence, locoregional and distant metastasis in 115 T1-T2 oral squamous cell carcinoma patients. Head neck oncol 2:9
Jin C, Yang YA, Anver MR, Morris N, Wang X, Zhang YE (2009) Smad ubiquitination regulatory factor 2 promotes metastasis of breast cancer cells by enhancing migration and invasiveness. Cancer res 69:735–740
Klupp F, Giese C, Halama N, Franz C, Lasitschka F, Warth A, Schmidt T, Kloor M, Ulrich A, Schneider M (2019) E3 ubiquitin ligase Smurf2: a prognostic factor in microsatellite stable colorectal cancer. Cancer manag res 11:1795–1803
Koganti P, Levy-Cohen G, Blank M (2018) Smurfs in protein homeostasis, signaling, and cancer. Front oncol 8:295
Li Y, Yang D, Tian N, Zhang P, Zhu Y, Meng J, Feng M, Lu Y, Liu Q, Tong L, Hu L, Zhang L, Yang JY, Wu L, Tong X (2019) The ubiquitination ligase SMURF2 reduces aerobic glycolysis and colorectal cancer cell proliferation by promoting ChREBP ubiquitination and degradation. J biolog chem 294:14745–14756
Markopoulos AK (2012) Current aspects on oral squamous cell carcinoma. Open dentistry J 6:126–130
Massagué J (2008) TGFbeta in cancer. Cell 134:215–230
Mortensen J, Mirian C, Ovesen T (2022) Limits and potential implementation of the present pN classification for oral squamous cell carcinoma. Curr opin otolaryngol head neck surg 30:94–98
Nieto MA, Huang RY-J, Jackson RA, Thiery JP (2016) EMT: 2016. Cell 166:21–45
Paterson IC, Matthews JB, Huntley S, Robinson CM, Fahey M, Parkinson EK, Prime SS (2001) Decreased expression of TGF-beta cell surface receptors during progression of human oral squamous cell carcinoma. J pathol 193:458–467
Prime SS, Davies M, Pring M, Paterson IC (2004) The role of TGF-beta in epithelial malignancy and its relevance to the pathogenesis of oral cancer (part II). Crit rev oral biol med: an official public Am Assoc Oral Biolog 15:337–347
Qiao B, Johnson NW, Gao J (2010) Epithelial-mesenchymal transition in oral squamous cell carcinoma triggered by transforming growth factor-beta1 is Snail family-dependent and correlates with matrix metalloproteinase-2 and -9 expressions. Int j oncol 37:663–668
Shi J, Bao X, Liu Z, Zhang Z, Chen W, Xu Q (2019) Serum miR-626 and miR-5100 are promising prognosis predictors for oral squamous cell carcinoma. Theranostics 9:920–931
Shi Y, Massagué J (2003) Mechanisms of TGF-beta signaling from cell membrane to the nucleus. Cell 113:685–700
Siegel RL, Miller KD, Jemal A (2018) Cancer statistics, 2018. CA: a cancer j clin 68:7–30
Sun L, Diamond ME, Ottaviano AJ, Joseph MJ, Ananthanarayan V, Munshi HG (2008) Transforming growth factor-beta 1 promotes matrix metalloproteinase-9-mediated oral cancer invasion through snail expression. Molecular cancer res: MCR 6:10–20
Takahashi K, Akatsu Y, Podyma-Inoue KA, Matsumoto T, Takahashi H, Yoshimatsu Y, Koinuma D, Shirouzu M, Miyazono K, Watabe T (2020) Targeting all transforming growth factor-β isoforms with an Fc chimeric receptor impairs tumor growth and angiogenesis of oral squamous cell cancer. J biolog chem 295:12559–12572
Thomas DA, Massagué J (2005) TGF-beta directly targets cytotoxic T cell functions during tumor evasion of immune surveillance. Cancer cell 8:369–380
Vigneswaran N, Williams MD (2014) Epidemiologic trends in head and neck cancer and aids in diagnosis. Oral maxillof surg clin North Am 26:123–141
Wang B, Huang L, Ye S, Zheng Z, Liao S (2023) Identification of novel prognostic biomarkers that are associated with immune microenvironment based on GABA-related molecular subtypes in gastric cancer. Pharmacogen personal med 16:665–679
Yang W, Zhang S, Li T, Zhou Z, Pan J (2022) Single-cell analysis reveals that cancer-associated fibroblasts stimulate oral squamous cell carcinoma invasion via the TGF-β/Smad pathway. Acta biochimica et biophysica Sinica 55:262–273
Yoshihara K, Shahmoradgoli M, Martínez E, Vegesna R, Kim H, Torres-Garcia W, Treviño V, Shen H, Laird PW, Levine DA, Carter SL, Getz G, Stemke-Hale K, Mills GB, Verhaak RG (2013) Inferring tumour purity and stromal and immune cell admixture from expression data. Nature commun 4:2612
Yu L, Dong L, Li H, Liu Z, Luo Z, Duan G, Dai X, Lin Z (2020) Ubiquitination-mediated degradation of SIRT1 by SMURF2 suppresses CRC cell proliferation and tumorigenesis. Oncogene 39:4450–4464
Zhao R, Wei W, Zhen L (2023) WGCNA-based identification of potential targets and pathways in response to treatment in locally advanced breast cancer patients. Open medicine (Warsaw, Poland) 18:20230651
Zhao XT, Zhu Y, Zhou JF, Gao YJ, Liu FZ (2021) Development of a novel 7 immune-related genes prognostic model for oral cancer: a study based on TCGA database. Oral Oncol 112:105088
Author information
Authors and Affiliations
Contributions
Lu Deng wrote the main manuscript text. Chuanxi Sun performed bioinformatics analysis and prepared figures. Zhe Liu edited the article. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interets
All authors declared that there was no conflict of interest.
Additional information
Communicated by: Ewa Ziętkiewicz
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Communicated by: Ewa Ziętkiewicz
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Deng, L., Wu, Z., Sun, C. et al. Transcriptome analysis revealed SMURF2 as a prognostic biomarker for oral cancer. J Appl Genetics (2024). https://doi.org/10.1007/s13353-024-00869-w
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s13353-024-00869-w