
Abstract
Transgenic chickens have, in general, been produced by two different procedures. The first procedure is based on viral transfection systems. The second procedure, the non-viral method, is based on genetically modified embryonic cells transferred directly into the recipient embryo. In this review, we analyzed the effectiveness of important elements of the non-viral, cell-based strategy of transgenic chicken production. The main elements of this strategy are: isolation and cultivation of donor embryonic cells; transgene construction; cell transfection in vitro; and chimera production: injection of cells into recipient embryos, raising and identification of germline chimeras, mating germline chimeras, transgene inheritance, and transgene expression. In this overview, recent progress and important limitations in the development of transgenic chickens are presented.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Various methods have been developed to produce successfully transgenic animals, including: direct DNA microinjection into the pronucleus (Brinster et al. 1981); viral vector transfer (Salter et al. 1987); injection of in vitro transfected embryonic stem cells into the blastocyst (Stewart et al. 1985); and cloning of transfected nuclei transferred into enucleated oocytes (Keefer et al. 2001). However, the production of transgenic birds has been hampered by the yolk-laden structure of the ovum and their unique reproductive system. Direct DNA microinjection, a frequently applied technique in mammals, is almost impossible in birds, because fertilization in the hen occurs in the infundibulum of the reproductive tract and fertilization in birds is polyspermic (Perry 1987). Also, identification of the male pronucleus among the supernumerary spermatozoa is difficult, as is the return of the ovum to the oviduct of a fistulated hen (Pancer et al. 1989). Consequently, transgenic chickens have, in general, been produced by two different procedures (reviewed by Song et al. 2010). The first procedure is based on viral transfection systems. The second procedure, the non-viral method, is based on genetically modified embryonic cells transferred directly into the recipient embryo.
Although viral transfection systems allow for efficient introduction and expression of transgenes in chicken dividing and non-dividing cells (McGrew et al. 2004), as well as in the chicken oviduct (Liu et al. 2015; Wu et al. 2015), there are some important limitations to this method. The lentiviral vectors have three main limitations (reviewed by Bednarczyk 2016): (i) restriction of the size of the vector genome to less than 8 to 10 kb (Wu et al. 2010); however, the promoters necessary to drive specific expression in some cells exceed this size; (ii) vector insertion can cause disruption of endogenous genes by insertional mutagenesis or the transactivation of neighboring endogenous genes (Li and Lu 2010); and (iii) integrated lentiviral vectors are subject to positional effects (Yi et al. 2011). Moreover, the viral transduction technique is characterized by high embryonic lethality rates, and relatively low and unpredictable rates of germline transmission and production of transgenic chickens (McGrew et al. 2004; Park and Han 2012b). However, a much more important limitation is public concern, which has questioned the safety of lentivirus-based technology. The fact that the lentiviral vector is derived from lentiviruses, which cause chronic, sometimes life-threatening, illnesses in humans and animals, is a strong argument.
Therefore, over the last few years, some alternative strategies have been developed and the idea of the generation of transgenic chickens through chimeric intermediates described (Petitte et al. 1990). The generation of transgenic chickens has been attempted through chimeric intermediates produced by the transfer of blastodermal cells. The same idea has been implemented in many other experiments, for example, Vick et al. (1993); however, in this instance, primordial germ cells (PGC) were proposed as the vehicle for the introduction of the transgene into the chicken genome. Elaboration of the extended culture method for those cells (van de Lavoir et al. 2006) and the development of assays of their genetic modification (Macdonald et al. 2012; Park and Han 2012a) have culminated in the production of transgenic or chimeric chickens expressing important therapeutic proteins in the oviduct (Lillico et al. 2007; Park et al. 2015). A hen which can secrete a lot of protein in its oviduct and which can regularly, in a 20–24-h cycle, produce eggs is a very attractive vehicle for the recovery of therapeutic proteins, because the sterile contents of the eggs are enveloped in a hard shell. Subsequently, the first therapeutic protein, recombinant human lysosomal acid lipase, produced in the oviduct of a genetically engineered White Leghorn hen was commercialized by Alexion Pharmaceuticals, USA in 2015.
However, the effectiveness of the transgenic method in birds is still very low in many cases. In this review, we analyzed the effectiveness of important elements of the non-viral, cell-based strategy of transgenic chicken production from the literature cited in the Web of Science database, and the experiments that have been carried out in our laboratory. The main elements of the non-viral, cell-based strategy are:
-
Isolation and cultivation of donor embryonic cells;
-
Transgene construction;
-
Cell transfection in vitro; and
-
Chimera production: injection of cells into recipient embryos, raising and identification of germline chimeras, mating germline chimeras, transgene inheritance, and transgene expression.
Isolation and cultivation of donor embryonic cells
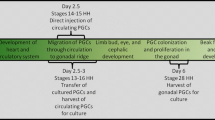
In the non-viral cell-based method, PGC have usually been used to create the transgenic chickens. In contrast to mammals, avian PGC use the circulatory system of a forming embryo to move from the region of the germinal crescent to the future gonads. The unique migratory properties of PGC provide opportunities for the identification/isolation of these cells from: the blastoderm at stage X (Eyal-Giladi and Kochav 1976); blood of 2.5–3-day-old embryos (stages 13–17 of Hamburger and Hamilton 1951; and from the gonads of 5–7-day-old embryos (stages 26–31) (reviewed by Chojnacka-Puchta et al. 2012).
PGC can be identified based on certain morphological features, such as: presence of a large spherical nucleus; very well developed Golgi apparatus and endoplasmic reticulum (Fujimoto et al. 1976); and presence of refractive lipids in cytoplasm and numerous grain reserve substances (Zhao and Kuwana 2003). In addition, to identify PGC, the following methods are used: periodic acid–Schiff (PAS) staining (Fujimoto et al. 1976) and immunological markers against cell surface glycoproteins present in PGC, such as SSEA-1 (stage-specific embryonic antigen 1) or CVH (chicken vasa homolog) proteins located in their cytoplasm (reviewed by Nakamura et al. 2013).
Due to the small number of PGC isolated from stage X embryos, this source of cells is not very popular. Blood at stages 13–17 becomes a potential source of PGC for the production of chicken chimeras. In particular, blood coming from stage 14 (Fig. 1) appears to be the most suitable for this purpose, as the concentration of circulating PGC is then the highest (Tajima et al. 1999).

Chicken embryo at stage 14 with 22 somites (50–53 h of incubation), magnification ×16
PGC have been isolated by different purification techniques, including Ficoll, Nycodenz, Percoll density gradient centrifugation, ammonium–chloride–potassium (ACK) lysis buffer, immunomagnetic separation (IMS), and fluorescence-activated cell sorting (FACS) (Chojnacka-Puchta et al. 2015). However, the method of PGC isolation from the embryonic blood was reported to be complicated and resulted in a very small number of PGC being obtained (approximately 0.048% of the total cells in blood) (Yasuda et al. 1992), which limits the use of this method of isolation for more technically advanced transgenic manipulation.
The most practical method of obtaining PGC seems to be their isolation from the gonads of 5–7-day-old embryos. For this purpose, the gonads are fragmented and/or digested using 0.25% trypsin–0.01% EDTA in PBS[−] solution. Compared to the other two methods, the number of PGC per embryo obtained by this method is the highest, thus increasing the success of chimera generation. Nakajima et al. (2011) suggested a new and simple method of collecting highly purified (about 50%) gonadal PGC by the incubation of gonads (at a temperature of 37.8 °C and 5% CO2) from 7-day-old embryos in PBS solution deprived of calcium and magnesium ions (PBS[−]). At present, this is the most effective method of PGC isolation from the gonads of chicken embryos, because it allows, in a short time, the collection of a large number of highly purified and viable PGC (Nakamura et al. 2013).
However, the number of PGC that can be obtained from one embryo is limited (Tajima et al. 1999), regardless of the method of their isolation from the blastoderm, from the blood, or from the gonads. To make effective use of the small number of PGC, it is essential to increase the number by culturing cells in vitro (Fig. 2).

Cultured primordial germ cells (PGC) on feeder cells. PGC proliferated and formed small colonies on the feeder cells after 14 days’ cultivation (left) and were SSEA-1 (stage-specific embryonic antigen 1) positive (right)
Several attempts have been made to maintain and propagate avian PGC in vitro. The first successful long-term culture of PGC was reported by van de Lavoir et al. (2006). PGC were obtained from embryonic blood and were cultured on mouse STO or buffalo rat liver (BRL) feeder cells by BRL-conditioned KnockOut DMEM with basic fibroblast growth factor (bFGF) and stem cell factor (SCF) supplementation. The PGC were cultured for more than 35 days and successfully migrated to the germinal ridges after transfer into the bloodstream of recipient embryos. These cells also differentiated into functional gametes in recipient gonads. The effectiveness of this culture system for PGC was confirmed by Macdonald et al. (2010) and Miyahara et al. (2014). Choi et al. (2010) developed a feeder-free culture system for PGC and found that bFGF is one of the important factors for the proliferation of PGC and also for maintaining the undifferentiated state of these cells. Furthermore, Park and Han (2012a) modified the culture system for PGC using mouse fibroblast feeder cells and produced germline chimeric chickens with more than a 95% germline transmission rate of donor PGC.
These culture systems use xeno-animal cells as feeder cells, and the use of this system may increase the risk of a cross-transfer of animal pathogens from other animal cells. It is, therefore, recommended to use chicken cells as feeder cells for culturing chicken PGC. However, long-term culture of chicken PGC isolated from embryonic blood using feeder cells derived from chicken embryos has not been successful. The propagation of female PGC in this system is less efficient compared with that of male PGC (van de Lavoir et al. 2006; Macdonald et al. 2010). Similarly, in the method developed by Tonus et al. (2016) that allows culture for more than 1 year, cell proliferation, and cryobanking of primary cultures of PGC, all the resulting lines appeared devoid of female cells, despite having been initially pooled from male and female embryos. In contrast, Naito et al. (2015) reported the successful long-term culture of female PGC in vitro and subsequent generation of viable offspring via germline chimeric chickens. In this method, female PGC are present in mixed-sex PGC populations cultured for more than 90 days. Moreover, optimal culture conditions of female PGC are different from those for male PGC.
As a summary of this part of the review, we can conclude that too little is yet known about the effects of long-term PGC cultivation in vitro on cells’ functional ability. Therefore, further investigations are needed to support the higher efficiency of chicken PGC culture and the suppression of cell differentiation by long-term culture in vitro.
Transgene expression constructs
According to the recommendations of the Center for Genetic Medicine (http://www.cgm.northwestern.edu/cores/ttml/transgenic-projects/transgenic-construct-design.html), standard transgenic constructs should contain all the 5′ and 3′ regulatory elements necessary for transgene expression, the gene of interest and/or marker gene, and restriction sites that allow isolation of a full-length linear transgenic fragment. The addition of sequences that potentially increase the level of transgene expression should be considered. A strategy for the detection of the transgene or its product (i.e., epitope tags on transgenic protein) should also be inherent to the construct design.
The promoter/enhancer elements driving gene expression are of some importance, depending on whether ubiquitous or tissue-specific expression is desired (Chapman et al. 2005). There are several known ubiquitous promoters which are strongly active in a wide range of cells, tissues, and cell cycles, e.g., chicken β-actin, cytomegalovirus (CMV) enhancer (CCAG or CAG promoter), histone H4 promoter, or phosphoglycerol kinase (PGK) promoter. The cytomegalovirus immediate–early gene promoter/enhancer (CMV) is a highly efficient promoter in many vertebrates, but in chickens, it seems to be less efficient than β-actin (reviewed by Chapman et al. 2005).
A number of genes are expressed only in a specific tissue or in response to unique environmental signals. The possibility of tissue-specific transgene expression opens new avenues for biological research and for genetic manipulation in birds. McGrew et al. (2010) investigated the possibility of utilizing rodent regulatory elements to drive transgene expression in the skeletal muscle of chickens, and they demonstrated that myosin light chain promoters/enhancers (MLC) drive skeletal muscle gene expression in transgenic chickens.
The most demanding, but also most explored, area is expression of the ovalbumin gene. The chicken ovalbumin promoter, which produces yields of ovalbumin that constitute about 54% of egg white total protein, is one of the strongest tissue-specific promoters. This expression mechanism induced by estrogen is only found in tubular gland cells of the hen’s oviduct. The chicken ovalbumin promoter has been used to induce oviduct-specific expression of a therapeutic protein in transgenic chicken bioreactors (Chojnacka-Puchta et al. 2015; Harvey and Ivarie 2003; Kwon et al. 2010; Lillico et al. 2007; Liu et al. 2015; Park et al. 2015). The ovalbumin (Ov) gene contains a number of regulatory elements that control its transcriptional activity and restrict expression to the avian oviduct. Lillico et al. (2007) constructed a synthetic oviduct expression vector with 2.8 kb of the 5′-flanking region on the ovalbumin promoter and estrogen-responsive enhancer (ERE) element. Another study (Zhu et al. 2005) also reported that human monoclonal antibody was controlled by the ovalbumin gene. The vectors contain 7.5- or 15-kb sequences on the 5′-flanking region of the ovalbumin promoter and 15.5 kb on its 3′-flanking region.
In our studies (Bednarczyk et al. 2003; Chojnacka-Puchta et al. 2015) of the expression of the human interferon alpha-2a gene (IFNα2a) or hepatitis B virus surface antigen (HBsAg), the specific vectors were constructed under the control of the chicken ovalbumin promoter (OVA), and by strengthening sequences which included the chicken oviduct-specific and enhancer-like (COSE) region and ERE element (Fig. 3).

However, the specific elements associated with the ovalbumin gene that direct oviduct-specific expression have not yet been fully defined, so it is difficult to identify the optimal expression vector(s) providing efficient expression of therapeutic proteins in the chicken oviduct.
Cells transfection in vitro
PGC are characterized by their low ability to induce effective and persistent transfection. Additionally, there are several obstacles to this process, because germ cells are relatively transcriptionally quiescent and prone to switching off transgene expression (Seydoux and Braun 2006). In general, the evaluation of PGC transfection efficiency centers on the selection of a suitable method for the genetic modification of cells, as well as its evaluation system. Usually, transfection efficiency is assessed according to the marker gene encoding the fluorescent protein: EGFP–green, EYFP–yellow, mCherry–red, tdTomato–bright red. Sometimes, the lacZ gene encoding a beta-galactosidase protein is used; however, an enzymatic or histochemical assay is necessary for its product identification.
Among the non-viral systems for the delivery of a gene into PGC, electroporation (van de Lavoir et al. 2006; Oishi 2010) and lipofection (Naito et al. 1998; Furuta et al. 2010; Kaleri et al. 2011) techniques have been used to transfer DNA. The main parameters that affect electroporation effectiveness are: pulse amplitude, pulse duration, number of delivered pulses, osmotic pressure (Kotnik et al. 2003). In lipofection, DNA is shielded and packaged into several different natural or synthetic compounds (carriers) to facilitate cellular uptake and intracellular release (reviewed by Grigsby and Leong 2010). These vectors try to mimic viral vectors in terms of assembly and cellular delivery, but have many advantages over viral vectors, such as their easy large-scale production, large transgene capacity, safety, and simplicity.
The transfection efficiency of PGC by synthetic DNA carriers is usually low and transgenes are gradually lost during embryonic development (Naito et al. 1998). After 17 days of incubation following the PGC injection, the lacZ gene is detected in only 14.3% (3/21) of embryos examined. Although the transgene (lacZ gene) has been detected in the gonads of two hatched chicks (11.1%), it has not been detected in the gonads of chimeric chickens at sexual maturity (Naito et al. 1998). However, successful transfer of exogenous genes into chicken PGC has been achieved by lipofection when the gene was introduced into chickens at stage X of development (Furuta et al. 2010).
There are, however, relatively few studies comparing both methods. Hong et al. (1998) compared two methods for PGC transfection. Electroporation was reported to have an 80% efficiency of DNA transfer, whereas transfection with DNA–liposome complexes was only 17% efficient. Our earlier in vitro and in vivo study (Chojnacka-Puchta et al. 2015) aimed to compare the influences of different chicken PGC (isolated from circulating blood or gonads) purification (ACK, Percoll, or trypsin) and transfection methods (electroporation or lipofection) on the expression of transgenes in vitro and the migration of modified donor cells to the recipient gonads. These data confirmed that the combination of PGC purification methods and transfection methods could be an effective strategy for producing transgenic chickens. The highest average frequency of transgene-transfected PGC (75.8%) was achieved with Percoll density gradient centrifugation and electroporation.
Similarly, for human embryonic stem cells, synthetic DNA carriers (lipofectamine) have been considered a successful approach to transient and stable cell line generation; however, the efficiency of this method appeared to be much lower than that of electroporation (Tabar et al. 2015).
Some authors (Macdonald et al. 2012; Park and Han 2012a) have recently proposed the use of transposon elements such as piggyBac, Tol2, and Sleeping Beauty to create a more versatile method to target chicken germline stem cells. Transposons are genetic elements that can relocate between different genomic sites, and the enzyme transposase can excise unique DNA sites and recombine transposons into targeted sites in the genome (Park and Han 2012b). The use of transposon vectors will greatly increase the efficiency of stable genetic modification of PGC (Macdonald et al. 2012). However, it will be necessary to analyze this method further and to explain, for example, the stable expression of the green fluorescent protein (GFP) transgene in multiple tissue types, including heart, brain, liver, intestine, kidney, and gonad, without tissue-specific transgene silencing (Park and Han 2012a). Surprising results have also been derived from an analysis of the progeny of a germline chimera rooster, where only a small number of germ cell-derived offspring were noted: 1 of a total of 518 (Macdonald et al. 2012).
Hitherto, a number of expression vectors have been proposed and numerous techniques have been established for the creation of transgenic birds. To achieve this objective, a reliable in vitro assay system which would serve to verify the efficiency of recombinant gene expression in the oviduct is necessary. The traditional method whereby the transgenic vectors were roughly introduced into the host genome and the tissue-specific protein expression in the egg white from transgenic birds was quantified is both costly and inefficient, because of the lack of vector verification in the target organ, such as the oviduct (Jung et al. 2011). One promising model developed by our group (Kasperczyk et al. 2012) could be the culture of chicken oviduct epithelial cells (COEC) that can be easily transfected with constructs based on oviduct-specific regulatory sequences, for example those derived from the ovalbumin or lysozyme genes. This unique, in-house-developed in vitro culture system for COEC has the potential to produce therapeutic proteins in vitro which would allow the verification of their functions and activity and, most importantly, of the efficiency of recombinant gene expression in the tubular gland cells of the oviduct (Stadnicka et al. 2016).
Chimera production
The formation of chicken chimera involves PGC isolation from donor embryos, cell transplantation into recipient embryos, and the subsequent regular procedure of embryo development. Petitte et al. (1990) were the first to produce a chicken germline chimera by separating blastodermal cells (containing presumptive PGC) from stage X embryos, and these were then injected into the subgerminal cavity of recipient embryos at the same stage of embryonic development. The resulting rooster had functional sperm; however, a low percentage (1.9%) of germline chimeric chicken was observed. In our experiment (Bednarczyk et al. 2002), blastoderm cells from chicken embryos of a donor breed (Green-legged Partridge-like) were transferred to embryos of a recipient breed (White Leghorn), and among 20 putative chimeric chickens, 6 (30%) produced recipient-derived and donor-derived offspring, indicating that they were germline chimeras.
PGC isolated from the blood of 2.5–3-day-old (stages 13–17) embryos, or the gonads of 5.5–6-day-old (stages 26–28) embryos, can be injected through a window opened in the eggshell into the dorsal aorta of recipient embryos. After injection, the eggshell can be sealed and the egg returned to the incubator and incubated until hatching. Sometimes, the recipient embryos are cultured in host eggshells, as described by Naito et al. (1991), to allow precise manipulation. However, the method of direct cell injection and embryo incubation in sealed eggshells (Chojnacka-Puchta et al. 2015) is considerably easier and more efficient than the transfer of embryos to surrogate eggshells, as was used by Naito et al. (1991).
The production of germline chimeras involves the incorporation of exogenous PGC into the endogenous gonadal tissue of recipient embryos. Therefore, there is a specific competition between the two populations of PGC, endogenous and exogenous, which leads to the production of two types of germ cells: those derived from a donor and those from a recipient. Thus, the proportion of exogenous gametes is determined by the ratio of the number of germ cells of the host to the cells artificially introduced into the gonads of recipient embryos (Nakamura et al. 2013). This number may be affected by two factors. The first is the ability of germ cells to induce mitosis, which varies depending on the chicken breed. For example, chicken breeds such as the White Leghorn, Barred Plymouth Rock, and Fayoumi differ in their ability to accept foreign PGC, with the best results being noted for the first of these breeds and the worst for the Barred Plymouth Rock. The second factor regulating the ratio of the number of host germ cells to artificially introduced cells is the number of exogenous PGC which reach the gonads and colonize them, as well as the number of endogenous PGC already present there. Their relative proportions can be increased by the partial or complete removal of endogenous PGC by sterilization. In order to increase the efficiency of germline transmission and genetic modification, a number of methods of inactivation and removal of endogenous PGC from recipient embryos have been developed. The most important of these include: surgical removal (Naito et al. 1994), inactivation using UV radiation (Reynaud 1976), X-rays (Nakamura et al. 2012) and γ radiation (Carsience et al. 1993), and chemical treatment with busulfan (Nakamura et al. 2009; Lee et al. 2013) and tamoxifen (Mohsen and Ahmed 2002).
Some of these methods are impractical in the routine production of chimeras because they also affect the embryonic development of the recipient (Naito 2003). The mortality of embryos exposed to irradiation combined with PGC injection was higher than that of control embryos and embryos exposed to irradiation (Maeda et al. 1998). Moreover, the same authors demonstrated a reduction in the size of gonads because of the general delay in development induced by γ irradiation. The survival rate of the chick embryos after the removal of a cell cluster from the blastoderm was 59.8% (579 of 969) at day 3 of incubation (Naito et al. 1999).
The sex of donor PGC and recipient PGC is important because the frequency of donor-derived offspring from germline chimeric chickens was significantly higher for the same-sex combinations of donor PGC and recipient embryos when compared with different-sex combinations (reviewed by Naito 2015). When the sex of the donor embryo was the same as that of the recipient embryo, 62.5% to 68.2% of chimeric chickens produced donor-derived offspring. When the sex of the donor embryo differed from that of the recipient embryo, 11.1% to 22.2% of chimeric chickens produced donor-derived offspring (Naito et al. 1999).
Information about the hatchability of manipulated embryos, the raising and identification of germline chimeras, mating germline chimeras, transgene inheritance, and transgene expression is limited. Some of these data are summarized in Table 1.
These data indicate that the efficiency of chimera production is still very low. Some data indicate that germline chimeras exhibit significant alterations in sex hormone levels in the ovary and blood plasma, which may affect their reproductive abilities (Sechman et al. 2006). Also, breeding of the chimeras did not often result in germline transmission of the transgene, indicating that the contribution of the transgenic cells to the germline is either non-existent or very low. In this context, the results published by Trefil et al. (2017) are interesting. They obtained viable genetically modified offspring from male PGC matured in the adult testes of sterilized recipient roosters. This new technique eliminates the germline chimerism of G0 roosters and, therefore, it seems to be faster, more efficient, and requires fewer animals.
Conclusions
The non-viral, cell-based strategy has been used in the generation of transgenic chickens; however, the efficiency of chimera production, transgene inheritance, and transgene expression is still very low. Thus, although this strategy demonstrates that transgenic chickens could successfully produce the humane therapeutic proteins in their oviducts, widespread adoption of this methodology requires additional information. The relationship between embryonic stem cells and germline competency and transgene expression is still an open field of investigation. Presently, very little is known about the effects of primordial germ cells (PGC) manipulation, long-term cultivation in vitro, and different cell/tissue environments in the recipient embryo on the cell phenotypes (morphology, behavior, gene expression, DNA methylation) and on their functional ability. Additionally, more data are required at all cellular, molecular, and biological levels to draw conclusions on the relationship between the reproductive characteristics and physiological state of the chimera, the site genomic integration of the transgene, and the number of its copies, and the level of its expression in the oviduct. Furthermore, the generation of transgenic chickens through chimeric intermediates is a highly skilled and costly process, making it difficult to apply in the general laboratory. From this point of view, it is apparent that the development of simple methods of PGC isolation and genetic modification, and their transfer into recipient embryos, is the essential step for the production of transgenic chickens.
References
Bednarczyk M (2016) Some aspects of poultry biotechnology: a review. Slovak J Anim Sci 49(4):157–159
Bednarczyk M, Łakota P, Słomski R, Pławski A, Lipinski D, Siemieniako B et al (2002) Reconstitution of a chicken breed by inter se mating of germline chimeric birds. Poult Sci 81:1347–1453
Bednarczyk M, Płucienniczak G, Płucienniczak A, Łakota P, Sochanik A, Dłużniewska P et al (2003) Expression of exogenous genes in blastodermal cells of chicken in vitro. Folia Biol (Krakow) 51(3–4):189–194
Brinster RL, Chen HY, Trumbauer M, Senear AW, Warren R, Palmiter RD (1981) Somatic expression of herpes thymidine kinase in mice following injection of a fusion gene into eggs. Cell 27:223–231
Carsience RS, Clark ME, Verrinder Gibbins AM, Etches RJ (1993) Germline chimeric chickens from dispersed donor blastodermal cells and compromised recipient embryos. Development 117:669–675
Chapman SC, Lawson A, MacArthur WC, Wiese RJ, Loechel RH, Burgos-Trinidad M et al (2005) Ubiquitous GFP expression in transgenic chickens using a lentiviral vector. Development 132:935–940
Choi JW, Kim S, Kim TM, Kim YM, Seo HW, Park TS et al (2010) Basic fibroblast growth factor activates MEK/ERK cell signaling pathway and stimulates the proliferation of chicken primordial germ cells. PLoS One 5(9):e12968. https://doi.org/10.1371/journal.pone.0012968
Chojnacka-Puchta L, Kasperczyk K, Płucienniczak G, Sawicka D, Bednarczyk M (2012) Primordial germ cells (PGCs) as a tool for creating transgenic chickens. Pol J Vet Sci 15(1):181–188
Chojnacka-Puchta L, Sawicka D, Lakota P, Płucienniczak G, Bednarczyk M, Płucienniczak A (2015) Obtaining chicken primordial germ cells used for gene transfer: in vitro and in vivo results. J Appl Genet 56:493–504
Eyal-Giladi H, Kochav S (1976) From cleavage to primitive streak formation: a complementary normal table and a new look at the first stages of the development of the chick. I. General morphology. Dev Biol 49:321–337
Fujimoto T, Ukeshima A, Kiyofuji R (1976) The origin, migration and morphology of the primordial germ cells in the chick embryo. Anat Rec 185:139–153
Furuta H, Sanada T, Takayanagi R, Yoshida T (2010) Introduction of exogenous gene into chicken PGCs via blastoderm. J Anim Vet Adv 9:748–750
Grigsby CL, Leong KW (2010) Balancing protection and release of DNA: tools to address a bottleneck of non-viral gene delivery. J R Soc Interface 7(Suppl 1):S67–S82
Hamburger V, Hamilton HL (1951) A series of normal stages in the development of the chick embryo. J Morphol 88:49–92
Harvey AJ, Ivarie R (2003) Validating the hen as a bioreactor for the production of exogenous proteins in egg white. Poult Sci 82:927–930
Hong YH, Moon YK, Jeong DK, Han JY (1998) Improved transfection efficiency of chicken gonadal primordial germ cells for the production of transgenic poultry. Transgenic Res 7:247–252
Jung JG, Park TS, Kim JN, Han BK, Lee SD, Song G et al (2011) Characterization and application of oviductal epithelial cells in vitro in Gallus domesticus. Biol Reprod 85(4):798–807
Kaleri HA, Xiang L, Aniwashi J, Xu S (2011) Oviduct-specific expression of tissue plasminogen activator in laying hens. Genet Mol Biol 34:231–236
Kasperczyk K, Bajek A, Joachimiak R, Walasik K, Marszałek A, Drewa T et al (2012) In vitro optimization of the Gallus domesticus oviduct epithelial cells culture. Theriogenology 77:1834–1845
Keefer CL, Baldassarre H, Keyston R, Wang B, Bhatia B, Bilodeau AS et al (2001) Generation of dwarf goat (Capra hircus) clones following nuclear transfer with transfected and nontransfected fetal fibroblasts and in vitro-matured oocytes. Biol Reprod 64:849–856
Kotnik T, Pucihar G, Rebersek M, Miklavcic D, Mir LM (2003) Role of pulse shape in cell membrane electropermeabilization. Biochim Biophys Acta 1614:193–200
Kwon SC, Choi JW, Jang HJ, Shin SS, Lee SK, Park TS et al (2010) Production of biofunctional recombinant human interleukin 1 receptor antagonist (rhIL1RN) from transgenic quail egg white. Biol Reprod 82:1057–1064
Lee HC, Kim SK, Park TS, Rengaraj D, Park KJ, Lee HJ et al (2013) Compensatory proliferation of endogenous chicken primordial germ cells after elimination by busulfan treatment. Stem Cell Res Ther 4:136. http://stemcellres.com/content/4/6/136
Li JJ, Lu LZ (2010) Recent progress on technologies and applications of transgenic poultry. Afr J Biotechnol 9(24):3481–3488
Lillico SG, Sherman A, McGrew MJ, Robertson CD, Smith J, Haslam C et al (2007) Oviduct-specific expression of two therapeutic proteins in transgenic hens. Proc Natl Acad Sci U S A 104:1771–1776
Liu T, Wu H, Cao D, Li Q, Zhang Y, Li N et al (2015) Oviduct-specific expression of human neutrophil defensin 4 in lentivirally generated transgenic chickens. PLoS One 10(5):e0127922. https://doi.org/10.1371/journal.pone.0127922
Macdonald J, Glover JD, Taylor L, Sang HM, McGrew MJ (2010) Characterisation and germline transmission of cultured avian primordial germ cells. PLoS One 5(11):e15518. https://doi.org/10.1371/journal.pone.0015518
Macdonald J, Taylor L, Sherman A, Kawakami K, Takahashi Y, Sang HM et al (2012) Efficient genetic modification and germ-line transmission of primordial germ cells using piggyBac and Tol2 transposons. Proc Natl Acad Sci U S A 109:1466–1472
Maeda T, Clark ME, Etches RJ (1998) Mortality, size of the gonads, and ultrastructure of primordial germ cell in chick embryos treated with γ-irradiation or injected with donor cells. Poult Sci 77:905–907
McGrew MJ, Sherman A, Ellard FM, Lillico SG, Gilhooley HJ, Kingsman AJ et al (2004) Efficient production of germline transgenic chickens using lentiviral vectors. EMBO Rep 5:728–733
McGrew MJ, Sherman A, Lillico SG, Taylor L, Sang H (2010) Functional conservation between rodents and chicken of regulatory sequences driving skeletal muscle gene expression in transgenic chickens. BMC Dev Biol 10:26. https://doi.org/10.1186/1471-213X-10-26
Miyahara D, Mori T, Makino R, Nakamura Y, Oishi I, Ono T et al (2014) Culture conditions for maintain propagation, long-term survival and germline transmission of chicken primordial germ cell-like cells. J Poult Sci 51:87–95
Miyahara D, Oishi I, Makino R, Kurumisawa N, Nakaya R, Ono T et al (2016) Chicken stem cell factor enhances primordial germ cell proliferation cooperatively with fibroblast growth factor 2. J Reprod Dev 62:143–149
Mohsen GAM, Ahmed RAL (2002) Tamoxifen inhibits the migration of the primordial germ cells of the chick embryos with less mortality rate. Pakistan J Biol Sci 5:1060–1062
Naito M (2003) Development of avian embryo manipulation techniques and their application to germ cell manipulation. Anim Sci J 74:157–168
Naito M (2015) Embryo manipulation in chickens. J Poult Sci 52:7–14
Naito M, Watanabe M, Kinutani M, Nirasawa K, Oishi T (1991) Production of quail-chick chimaeras by blastoderm cell transfer. Br Poult Sci 32:79–86
Naito M, Tajima A, Yasuda Y, Kuwana T (1994) Production of germline chimeric chickens, with high transmission rate of donor-derived gametes, produced by transfer of primordial germ cells. Mol Reprod Dev 39:153–161
Naito M, Sakurai M, Kuwana T (1998) Expression of exogenous DNA in the gonads of chimaeric chicken embryos produced by transfer of primordial germ cells transfected in vitro and subsequent fate of the introduced DNA. J Reprod Fertil 113:137–143
Naito M, Matsubara Y, Harumi T, Tagami T, Kagami H, Sakurai M et al (1999) Differentiation of donor primordial germ cells into functional gametes in the gonads of mixed-sex germline chimaeric chickens produced by transfer of primordial germ cells isolated from embryonic blood. J Reprod Fertil 117:291–298
Naito M, Harumi T, Kuwana T (2015) Long-term culture of chicken primordial germ cells isolated from embryonic blood and production of germline chimaeric chickens. Anim Reprod Sci 153:50–61
Nakajima Y, Minematsu T, Naito M, Tajima A (2011) A new method for isolating viable gonadal germ cells from 7-day-old chick embryos. J Poult Sci 48:106–111
Nakamura Y, Usui F, Atsumi Y, Otomo A, Teshima A, Ono T et al (2009) Effects of busulfan sustained-release emulsion on depletion and repopulation of primordial germ cells in early chicken embryos. J Poult Sci 46:127–135
Nakamura Y, Usui F, Miyahara D, Mori T, Ono T, Kagami H et al (2012) X-irradiation removes endogenous primordial germ cells (PGCs) and increases germline transmission of donor PGCs in chimeric chickens. J Reprod Dev 58:432–437
Nakamura Y, Kagami H, Tagami T (2013) Development, differentiation and manipulation of chicken germ cells. Dev Growth Differ 55:20–40
Oishi I (2010) Improvement of transfection efficiency in cultured chicken primordial germ cells by Percoll density gradient centrifugation. Biosci Biotechnol Biochem 74:2426–2430
Pancer Z, Snapir N, Robinzon B, Friedman A, Eyalgiladi H (1989) Recovery of ova and their re-insertion into the hen’s oviduct through a fistula. Br Poult Sci 30:953–957
Park TS, Han JY (2012a) piggyBac transposition into primordial germ cells is an efficient tool for transgenesis in chickens. Proc Natl Acad Sci U S A 109:9337–9341
Park TS, Han JY (2012b) Genetic modification of chicken germ cells. Ann N Y Acad Sci 1271(1):104–109
Park TS, Lee HG, Moon JK, Lee HJ, Yoon JW, Yun BN et al (2015) Deposition of bioactive human epidermal growth factor in the egg white of transgenic hens using an oviduct-specific minisynthetic promoter. FASEB J 29:2386–2396
Perry MM (1987) Nuclear events from fertilisation to the early cleavage stages in the domestic fowl (Gallus domesticus). J Anat 150:99–109
Petitte JN, Clark ME, Liu G, Verrinder Gibbins AM, Etches RJ (1990) Production of somatic and germline chimeras in the chicken by transfer of early blastodermal cells. Development 108:185–189
Reynaud G (1976) Localization study of the primordial germ cells in embryos of the Japanese quail by means of ultraviolet irradiation. C R Hebd Seanc Acad Sci D 282:1195–1198
Salter DW, Smith EJ, Hughes SH, Wright SE, Crittenden LB (1987) Transgenic chickens: insertion of retroviral genes into the chicken germ line. Virology 157(1):236–240
Sechman A, Łakota P, Wojtysiak D, Hrabia A, Mika M, Lisowski M et al (2006) Sex steroids level in blood plasma and ovarian follicles of the chimeric chicken. J Vet Med A Physiol Pathol Clin Med 53:501–508
Seydoux G, Braun RE (2006) Pathway to totipotency: lessons from germ cells. Cell 127:891–904
Song G, Park TS, Kim TM, Han JY (2010) Avian biotechnology: insights from germ cell-mediated transgenic systems. J Poult Sci 47:197–207
Stadnicka K, Bodnar M, Marszałek A, Bajek A, Drewa T, Płucienniczak G et al (2016) Efficient source of cells in proximal oviduct for testing non-viral expression constructs in the chicken bioreactor model and for other in vitro studies. Folia Biol (Krakow) 64:37–46
Stewart CL, Vanek M, Wagner EF (1985) Expression of foreign genes from retroviral vectors in mouse teratocarcinoma chimaeras. EMBO J 4(13B):3701–3709
Tabar MS, Hesaraki M, Esfandiari F, Samani FS, Vakilian H, Baharvand H (2015) Evaluating electroporation and lipofectamine approaches for transient and stable transgene expressions in human fibroblasts and embryonic stem cells. Cell J 17(3):438–450
Tajima A, Hayashi H, Kamizumi A, Ogura J, Kuwana T, Chikamune T (1999) Study on the concentration of circulating primordial germ cells (cPGCs) in early chick embryos. J Exp Zool 284:759–764
Tonus C, Cloquette K, Ectors F, Piret J, Gillet L, Antoine N et al (2016) Long term-cultured and cryopreserved primordial germ cells from various chicken breeds retain high proliferative potential and gonadal colonisation competency. Reprod Fertil Dev 28(5):628–639
Trefil P, Aumann D, Koslová A, Mucksová J, Benešová B, Kalina J et al (2017) Male fertility restored by transplanting primordial germ cells into testes: a new way towards efficient transgenesis in chicken. Sci Rep 7(1):14246
Tyack SG, Jenkins KA, O’Neil TE, Wise TG, Morris KR, Bruce MP et al (2013) A new method for producing transgenic birds via direct in vivo transfection of primordial germ cells. Transgenic Res 22:1257–1264
van de Lavoir MC, Diamond JH, Leighton PA, Mather-Love C, Heyer BS, Bradshaw R et al (2006) Germline transmission of genetically modified primordial germ cells. Nature 441:766–769
Vick L, Li Y, Simkiss K (1993) Transgenic birds from transformed primordial germ cells. Proc Biol Sci 251:179–182
Wu Z, Yang H, Colosi P (2010) Effect of genome size on AAV vector packaging. Mol Ther 18:80–86
Wu H, Cao D, Liu T, Zhao J, Hu X, Li N (2015) Purification and characterization of recombinant human lysozyme from eggs of transgenic chickens. PLoS One 10(12):e0146032. https://doi.org/10.1371/journal.pone.0146032
Yasuda Y, Tajima A, Fujimoto T, Kuwana T (1992) A method to obtain avian germ-line chimaeras using isolated primordial germ cells. J Reprod Fertil 96:521–528
Yi Y, Noh MJ, Lee KH (2011) Current advances in retroviral gene therapy. Curr Gene Ther 11:218–228
Zhao DF, Kuwana T (2003) Purification of avian circulating primordial germ cells by Nycodenz density gradient centrifugation. Br Poult Sci 44:30–35
Zhu L, van de Lavoir MC, Albanese J, Beenhouwer DO, Cardarelli PM, Cuison S et al (2005) Production of human monoclonal antibody in eggs of chimeric chickens. Nat Biotechnol 23:1159–1169
Acknowledgements
We acknowledge Dr. Luiza Chojnacka-Puchta from the Institute of Biotechnology and Antibiotics (IBA, Warsaw) for delivering visualization material incorporated in Fig. 3.
Author information
Authors and Affiliations
Contributions
MB, PL, and AS made substantial contributions to the conception and design of this manuscript. TK, IK, and AS acquired and interpreted the data reported. MB supervised the project and writing. TK and AS performed experiments to provide photographs for Figs. 1 and 2. MB, IK, PL, AS, KS, and TK provided the original draft. MB and KS reviewed and edited the final manuscript.
Corresponding author
Ethics declarations
Ethical standards
The in vitro experiment was performed to produce the original Fig. 2 in this manuscript. The use of modified chicken embryonic cells is regulated by the Polish Ministry of Environment, decision no. 149/2016. The research conducted on animals in this report is regulated by Directive 2010/63/EU and the approvals of the Polish Local Ethical Commission, Bydgoszcz, Poland, decision nos. 23/2014 and 35/2012.
Grant support
The work reported herein was supported by the National Center for Research and Development, grant no. PBS3/A8/30/2015 and partially by the National Science Centre, grant no. UMO-2011/03/N/NZ9/03814.
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Communicated by: Maciej Szydlowski
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Bednarczyk, M., Kozłowska, I., Łakota, P. et al. Generation of transgenic chickens by the non-viral, cell-based method: effectiveness of some elements of this strategy. J Appl Genetics 59, 81–89 (2018). https://doi.org/10.1007/s13353-018-0429-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13353-018-0429-6