
Abstract
Fusarium proliferatum (Matsushima) Nirenberg is a common pathogen infecting numerous crop plants and occurring in various climatic zones. It produces large amounts of fumonisins, a group of polyketide-derived mycotoxins. Fumonisin biosynthesis is determined by the presence and activity of the FUM cluster, several co-regulated genes with a common expression pattern. In the present work, we analyzed 38 F. proliferatum isolates from different host plant species, demonstrating host-specific polymorphisms in partial sequences of the key FUM1 gene (encoding polyketide synthase). We also studied growth rates across different temperatures and sample origin and tried to establish the relationships between DNA sequence polymorphism and toxigenic potential. Phylogenetic analysis was conducted based on FUM1 and tef-1α sequences for all isolates. The results indicated the greatest variations of both toxigenic potential and growth patterns found across the wide selection of isolates derived from maize. Fumonisin production for maize isolates ranged from 3.74 to 4,500 μg/g of fumonisin B1. The most efficient producer isolates obtained from other host plants were only able to synthesize 1,820–2,419 μg/g of this metabolite. A weak negative rank correlation between fumonisin content and isolate growth rates was observed. All garlic-derived isolates formed a distinct group on a FUM1-based dendrogram. A second clade consisted of tropical and sub-tropical strains (isolated from pineapple and date palm). Interestingly, isolates with the fastest growth patterns were also grouped together and included both isolates originating from rice. The sequence of the FUM1 gene was found to be useful in revealing the intraspecific polymorphism, which is, to some extent, specifically correlated with the host plant.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Fusarium proliferatum (Matsushima) Nirenberg occurs worldwide as a moderately aggressive pathogen of multiple plant species. The pathogen can also survive as an endophyte-like organism, without visible disease symptoms in the host. Apart from F. verticillioides (Saccardo) Nirenberg, the species is considered as the most common maize pathogen, as well as the most effective producer of the polyketide-derived fumonisin mycotoxins (Rheeder et al. 2002). The fumonisins produced are usually B group analogs, with fumonisin B1 (FB1) being the most prevalent. The compound is toxic to both animals and humans due to inhibiting sphingolipid metabolism and cell cycle regulation. In past studies, it has been already associated with esophageal cancer, liver cancer, and neural tube defects (Desjardins 2006). The FUM gene cluster is responsible for the entire fumonisin biosynthetic pathway with the key enzyme, polyketide synthase, encoded by the FUM1 gene. It is responsible for synthesizing the fumonisin backbone subsequently modified by other enzymes. The cluster of the model species, F. verticillioides, has been thoroughly studied and found to consist of 17 co-regulated genes exhibiting a common expression pattern during fumonisin biosynthesis (Brown et al. 2005; Proctor et al. 2003). Notably, the FUM cluster of F. proliferatum was identified and comparative analysis with F. verticillioides was performed, but the sequence of the cluster was not published (Waalwijk et al. 2004).
Among the many molecular markers used for phylogeny reconstruction (e.g., internal transcribed spacers [ITS1/2] region, β-tubulin, calmodulin, and H3 histone genes), the translation elongation factor (tef-1α) sequences appear to be the most useful in taxonomic studies of fungi, especially in the Gibberella fujikuroi species complex, as well as in other members of the Fusarium genus (Geiser et al. 2004; Kristensen et al. 2005). Recently, genes and other sequences directly involved in secondary metabolism gained more attention in phylogenetic studies, as those have the advantage of possible usage in combined approaches to the diagnostics of mycotoxin production abilities (Proctor et al. 2009). Therefore, genes from the FUM cluster merit investigation as a good additional marker for phylogenetic studies of fumonisin-producing Fusarium species (Baird et al. 2008; González-Jaén et al. 2004; Stępień et al. 2011).
The aim of this study was to assess the variability of F. proliferatum isolates coming from different host species on three levels: (i) phylogenetic relationships implied by translation elongation factor 1α (tef-1α) and FUM1 genes sequences, (ii) differences in growth rates at four different temperatures, and (iii) fumonisin B synthesis and accumulation.
Materials and methods
Fusarium strains, media, and growth rate measurement
Thirty-eight F. proliferatum isolates, collected from various host plant species, were chosen for the analyses of growth speed in relation to the temperature and fumonisin B1–B3 content. All isolates were stored in the KF Fusarium collection at the Institute of Plant Genetics, Polish Academy of Sciences, Poznań, Poland. The isolates are summarized (including data on fumonisin content and growth) in Table 1. The colony measurements were performed at four different temperatures, 20°C, 25°C, 30°C, and 35°C, on 90-mm plates with potato dextrose agar (PDA) medium (Oxoid, Basingstoke, Hampshire, UK), with growth measured as the colony diameter in 24-h intervals. Two replicates were done for each isolate/temperature combination.
PCR primers, cycling profiles, and DNA sequencing
For the amplification of tef-1α and FUM1 gene fragments, Ef728M (CATCGAGAAGTTCGAGAAGG)/Tef1R (GCCATCCTTGGAGATACCAGC) and Fum1F1 (CACATCTGTGGGCGATCC)/Fum1R2 (ATATGGCCCCAGCTGCATA) primers were used, respectively, designed and tested in the previous work (Stępień et al. 2011). The polymerase chain reaction (PCR) was done in 25-μl aliquots using PTC-200 thermal cycler (MJ Research, Watertown, MA, USA). Each sample contained 1 unit of Platinum HotStart Taq DNA polymerase (Invitrogen, Carlsbad, CA, USA), 2.5 μl of 10× PCR buffer, 12.5 pmol of forward/reverse primers, 2.5 mM of each dNTP, and about 20 ng of fungal DNA. The PCR conditions were as follows: 15 min at 95°C, 35 cycles of (30–60 s at 94°C, 30–60 s at 58-63°C, 1–2 min at 72°C), and 10 min at 72°C. Amplicons were electrophoresed in 1.5% agarose gels (Invitrogen) with ethidium bromide.
For sequence analysis, PCR-amplified DNA fragments were purified with exonuclease I (Epicentre, Madison, WI, USA) and shrimp alkaline phosphatase (Promega, Madison, WI, USA) using the following program: 30 min at 37°C, followed by 15 min at 80°C. In the case of multiple amplification fragments, the bands were cut out of the 0.8% agarose gels and purified using PureLink® Quick Gel Extraction Kit (Invitrogen). We labeled both strands using the BigDyeTerminator 3.1 kit (Applied Biosystems, Foster City, CA, USA), according to Błaszczyk et al. (2005) and the manufacturer’s instructions. To remove the remains of the reagents, labeled fragments were precipitated with ethanol. Sequence reading was performed using Applied Biosystems equipment.
Sequence analysis and phylogeny reconstruction
The sequences were initially aligned with ClustalW and subsequently realigned separately for intron and exon regions using MUSCLE (Edgar 2004). Phylogenetic relationships were reconstructed using MEGA4 (Tamura et al. 2007). Both tef-1α and FUM1 gene sequences were analyzed using the maximum parsimony approach (closest neighbor interchange heuristics set to level 3) for phylogeny reconstruction. No positions containing gaps were considered in the phylogeny analysis. All reconstructions were tested by bootstrapping with 1,000 replicates.
Fumonisin content analysis
Fumonisin B1, B2, and B3 standards and all reagents were purchased from Sigma (St. Louis, MO, USA). Water for the high-performance liquid chromatography (HPLC) mobile phase was purified using a Milli-Q system (Millipore, Bedford, MA, USA).
For toxin quantification, rice cultures were prepared for individual Fusarium isolates (Stępień et al. 2011). Long-grain white rice samples were used (50 g per flask, with the addition of 12.5 μl of sterile water), left overnight, and sterilized by autoclaving the next day. The rice samples were subsequently inoculated with 4 cm2 of 7-day-old mycelium on PDA medium. The culture humidity was kept around 30% for 14 days. Then, the cultures were dried at room temperature and samples (5 g) of each strain were homogenized for 3 min in 10 ml of methanol–water (3:1, v/v) and filtered through Whatman no. 4 filter paper according to the method described by Sydenham et al. (1990). A detailed description of procedure of the extraction and purification of FB1 was reported earlier (Waśkiewicz et al. 2010). The fumonisin B1, B2, and B3 standard (5 μl) or extracts (20 μl) were derivatized with 20 or 80 μl of the OPA reagent. After 3 min, the reaction mixture (10 μl) was injected onto an HPLC column. Methanol–sodium dihydrogen phosphate (0.1 M in water) solution (77:23, v/v) adjusted to pH 3.35 with o-phosphoric acid, after filtration through a 0.45-μm Waters HV membrane, was used as the mobile phase with a flow rate of 0,6 ml min−1. A Waters 2695 apparatus (Waters Division of Millipore, Milford, MA, USA), with a C-18 Nova Pak column (3.9 × 150 mm) and a Waters 2475 fluorescence detector (λEx = 335 nm and λEm = 440 nm), were used in the metabolite quantitative determination. The detection limits were 10 ng g−1 for FB1, FB2, and FB3. Positive results (on the basis of retention time) were confirmed by HPLC analysis and compared with the relevant calibration curve (the correlation coefficients for FB1, FB2, and FB3 were 0.9967, 0.9983, and 0.9966, respectively). Recoveries for FB1, FB2, and FB3 were 93, 96, and 87% respectively, which were measured in triplicate by extracting the mycotoxins from blank samples spiked with 10–100 ng g−1 of the compound. The relative standard deviations were less than 8%.
Results
Growth rates
The observed growth curves for isolates from different species are presented in Fig. 1. The optimal growth temperature for all isolates was found to be 25°C, with marked (reversible) inhibition of growth observed at both 30 and 35°C. The relationship between total fumonisin content and colony diameter at 25°C is summarized in Fig. 2. Four isolates proved to be notable outliers, capable of moderate growth, even at 35°C. The same isolates, two isolates from rice (KF 422_1.1 and KF 1329_2), one from maize (KF 925), and one from asparagus (KF 3662), displayed the fastest growth at all of the temperatures tested. Although the temperature of 35°C limited the growth rates severely, after decreasing to 20–25°C, high speed of growth of the analyzed isolates was restored (results not shown).

Growth rate curves based on the mean colony diameter of all strains of Fusarium proliferatum originating from one host plant. Potato dextrose agar (PDA) medium was used, and measurements were performed in 24-h intervals. Four temperatures were tested: 20, 25, 30, and 35°C. Abbreviations of host species: Ac Ananas comosus, Ao Asparagus officinalis, As Allium sativum, Ca Cambria sp., Os Oryza sativa, Pd Phoenix dactylifera, So Saccharum officinarum, Ta Triticum aestivum, Zm Zea mays, NA unknown host

The relationship between total fumonisin content (B1 + B2 + B3) and colony diameter after 7 days at 25°C for 37 isolates of Fusarium proliferatum (the logarithmic scale was used, see Table 1 for details of the fumonisin biosynthesis for individual isolates). The host origin has been marked by symbols. Abbreviations of host species: Ac Ananas comosus, Ao Asparagus officinalis, As Allium sativum, Ca Cambria sp., Os Oryza sativa, Pd Phoenix dactylifera, So Saccharum officinarum, Ta Triticum aestivum, Zm Zea mays, NA unknown host
Fumonisin biosynthesis levels
The fumonisins B1–B3 content was assayed in controlled conditions using rice cultures (Kostecki et al. 1999). Based on the results of the analysis, three groups can be distinguished among 38 analyzed isolates: low-producers (nine isolates yielding below 20 μg/g of FB1), medium-producers (five isolates yielding between 20 and 200 μg/g of FB1), and high-producers (24 isolates yielding over 200 μg/g FB1). Sixteen isolates produced more than 1 mg/g of fumonisin B1 and no relationship was found between host species and the amount of toxin produced, as the most effective FB-producing isolates originated not only from maize, but also from asparagus, pineapple, and garlic. The fumonisin biosynthesis by individual isolates is summarized in Table 1 and (in relation to the growth rate and host plant) in Fig. 2.
Phylogenetic analysis
The partial sequences of the translation elongation factor tef-1α and FUM1 genes from 38 isolates studied were amplified by PCR using Ef728M/Tef1R and Fum1F1/Fum1R2 primers, respectively. PCR products were used directly for sequencing the fragments. In one case (KF 1327), we were not able to obtain an expected amplicon and in another sample (KF 3404 isolated from pineapple), we were not successful in obtaining the FUM1 sequence of good quality, despite having the fragment amplified (results not shown). Obtained sequences were used for phylogeny reconstruction (Figs. 3 and 4). Regarding the comparison of cladograms based on tef-1α and FUM1 sequences, bootstrapping of the latter yields clearer separation between isolates coming from different hosts. The tef-1α-based tree relied on 15 parsimony informative residues (out of 250) with consistency and retention indices for the most parsimonious tree equal to, respectively, 0.791 and 0.921. The FUM1 reconstruction relied on 15 parsimony informative residues (out of 428), yielding consistency and retention indices of, respectively, 0.857 and 0.969. The FUM1 region used for analysis was mapped to the reference F. verticillioides gene and was found to correspond to the coding sequence (partial linker region between dehydratase and methyltransferase domains).

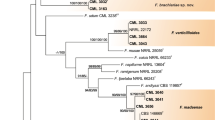
Consensus phylogenetic tree for 38 Fusarium proliferatum isolates, based on the translation elongation factor 1α (tef-1α) sequences. As an outgroup, an F. verticillioides strain was included. The tree was reconstructed using the maximum parsimony approach and tested by bootstrapping (1,000 replicates). Only branches with 50% and above support from bootstrapping were shown. The letters L, M, and H stand for low, medium, and high content of fumonisins, respectively (see the Results section). Abbreviations used for pathogen/host code: Fp F. proliferatum, Fv F. verticillioides, Ac Ananas comosus, Ao Asparagus officinalis, As Allium sativum, Ca Cambria sp., Os Oryza sativa, Pd Phoenix dactylifera, So Saccharum officinarum, Ta Triticum aestivum, Zm Zea mays

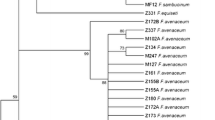
Consensus phylogenetic tree for 36 Fusarium proliferatum isolates, based on the partial sequence of the FUM1 gene. As an outgroup, an F. verticillioides strain was included. The tree was reconstructed using the maximum parsimony approach and tested by bootstrapping (1,000 replicates). Only branches with 50% and above support from bootstrapping were shown. The letters L, M, and H stand for low, medium, and high content of fumonisins, respectively, and the vertical bar marks the clade of fastest-growing isolates (see the Results section). Abbreviations used for pathogen/host code: Fp F. proliferatum, Fv F. verticillioides, Ac Ananas comosus, Ao Asparagus officinalis, As Allium sativum, Ca Cambria sp., Os Oryza sativa, Pd Phoenix dactylifera, So Saccharum officinarum, Ta Triticum aestivum, Zm Zea mays
Discussion
F. proliferatum is a common pathogen infecting multiple crop plants from various climatic zones. However, little is known about its variability at the phenotypic, metabolic, and genetic levels, especially in the context of the host. Also, intraspecific differences in the mycotoxigenic abilities of individual isolates from different hosts have not been extensively studied. Besides several polymorphic regions, mycotoxin biosynthetic genes have gained significant attention as useful tools for studies of the diversity and phylogenetic relationships among Fusarium species. Therefore, apart from the tef-1α gene, we decided to use the sequence of the FUM1 gene in our study. The tef-1α gene has been used successfully in numerous studies of fungal molecular taxonomy, also concerning Fusarium species (Geiser et al. 2004; Kristensen et al. 2005; Stępień et al. 2011). The limited intraspecies divergence of the tef-1α sequences did not allow for distinguishing clear clades of individual isolates in relation to different countries of origin/host plant (Fig. 3); however, there were groups of several more closely related groups of isolates. It applies specifically to garlic-derived isolates, which formed a separate branch on the dendrogram (Fig. 3). The remaining isolates were dispersed across all groupings with no clear correlation to the host origin. This stays in good agreement with previous studies with a number of F. proliferatum strains from different hosts. Especially when comparing maize- and asparagus-derived strains, remarkable sequence similarities in the tef-1α region were recorded (Jurado et al. 2010).
The comparison of the partial FUM1 sequences of 36 isolates studied originating from different hosts allowed to differentiate the host-related groups more clearly than in the case where the tef-1α sequences were used (Fig. 4). The group of isolates characteristic for tropical/subtropical climatic zones, i.e., recovered from hosts like pineapple (Ananas comosus (L.) Merr.) or date palm (Phoenix dactylifera L.) formed a separate well-established clade of the consensus phylogenetic tree (Fig. 4). Similar results were reported for F. proliferatum strains isolated from date palm and banana, even when based on the analysis of the tef-1α gene, but using a significantly longer fragment of the gene (Jurado et al. 2010). On the other hand, genes from the FUM cluster (particularly FUM1) were already accepted as good targets for phylogenetic studies and also for designing markers useful in the diagnostics of the fumonisin production ability of species from the G. fujikuroi species complex (Baird et al. 2008; von Bargen et al. 2009; González-Jaén et al. 2004), as well as for distinguishing individual species belonging to the G. fujikuroi species complex (Stępień et al. 2011). Of all of the isolates we used in the FUM1 analysis, the best distinguished group consisted of all six strains originating from garlic (Allium sativum L.). All of them were also classified into the group of high-producers of fumonisins (Table 1 and Fig. 4). Interestingly, three of the four fastest-growing isolates (regardless of the temperature used), i.e., KF 422_1.1, KF 925, and KF 1329_2, were found to share FUM1 polymorphisms and formed a separate clade (marked on Fig. 4 with a vertical line), along with an additional maize isolate of lower toxigenic ability and growth speed (KF 2227).
Like that observed while analyzing the tef-1α dendrogram, in the case of the FUM1 analysis, maize- and asparagus-derived isolates formed a common group (Fig. 4). Recently, similar regularity was reported by other research groups (Jurado et al. 2010). The possible explanation for such a wide distribution of maize isolates across the whole species variability is the fact of systemic colonization of the plant by the pathogen without visible infection symptoms. A similar phenomenon was already observed for closely related F. verticillioides (Desjardins et al. 2007). The abundance of maize as a crop plant and its worldwide distribution, along with the ease of seed transfer (possibly infected with pathogens from the G. fujikuroi species complex, like F. proliferatum), justifies enough the fact that maize isolates are diverse to such extent (de Oliveira Rocha et al. 2011).
Unsurprisingly, the highest variability in both fumonisin content and growth rates occurred among 11 isolates derived from maize (Fig. 2). Similar variability was not observed in either pineapple- and asparagus-derived samples (seven isolates each), characterized by both high fumonisin content and stable growth rates (with the exception of the fast-growing KF 3362 isolate from asparagus). Having analyzed all 38 isolates, we observed only weak negative rank correlation between fumonisin accumulation and growth speed (see Fig. 2 for a graphical presentation of those two traits combined, wherein for the growth rate, we mean the colony diameter after 7 days at 25°C). The correlation hypothesis was finally rejected based on the rank test of Spearman’s correlation coefficient ρ = −0.304 with a significance of p = 0.06. While it should be expected that secondary metabolite synthesis in vitro comes at some cost to primary metabolic processes (Chen et al. 2011; Jurado et al. 2008; Marín et al. 1995) and, hence, growth speed, the observed relationship between fumonisin content and growth rates at 25°C suggests that this cost in the case of in vitro fumonisin production by F. proliferatum isolates is relatively low.
In the present study, using a method of polymorphism detection inside the FUM1 sequence among a number of strains from different hosts, we did not find the correlation between the diversity of fumonisin biosynthesis and the structure of the FUM1 gene and, in some cases, in one clade grouped isolates of high and low fumonisin production (Fig. 4). This stays in agreement with the results of our previous studies, where some non-producing mutants were identified, despite having at least a part of the FUM cluster present (Stępień et al. 2011). Remarkably, F. proliferatum is a species with relatively strong (if varied) mycotoxin biosynthetic potential, regardless of the host plant and year of isolation (see Table 1 for details). The widespread and variable toxigenic potential possibly results from naturally occurring genetic variance in the fungal population (non-producing mutants were recorded) and not yet clearly known directions of selection pressure, although the role of fumonisin in the process of plant infection by F. verticillioides has already been proven (Glenn et al. 2008; Proctor et al. 2006). Another reason for this variation is, undoubtedly, a vast number of regulatory factors affecting the mycotoxin production in natural conditions. The analysis of the FUM gene transcription level could be used as a complementary analysis for future studies focused on establishing a link between fumonisin content and activity of the cluster in different isolates and conditions. This approach has already been utilized for collections of F. verticillioides and F. proliferatum isolates, relating the increased level of the FUM1 gene transcript with high amounts of mycotoxin produced; however, not all isolates gave a clear, well-supported correlation (Jurado et al. 2008 and 2010; López-Errasquín et al. 2007). This suggests the possible incidence of the additional level of regulation that still remains to be confirmed.
References
Baird R, Abbas HK, Windham G, Williams P, Baird S, Ma P, Kelley R, Hawkins L, Scruggs M (2008) Identification of select fumonisin forming Fusarium species using PCR applications of the polyketide synthase gene and its relationship to fumonisin production in vitro. Int J Mol Sci 9:554–570
Błaszczyk L, Tyrka M, Chełkowski J (2005) PstIAFLP based markers for leaf rust resistance genes in common wheat. J Appl Genet 46:357–364
Brown DW, Cheung F, Proctor RH, Butchko RAE, Zheng L, Lee Y, Utterback T, Smith S, Feldblyum T, Glenn AE, Plattner RD, Kendra DF, Town CD, Whitelaw CA (2005) Comparative analysis of 87,000 expressed sequence tags from the fumonisin-producing fungus Fusarium verticillioides. Fungal Genet Biol 42:848–861
Chen F, Zhang J, Song X, Yang J, Li H, Tang H, Liao YC (2011) Combined metabonomic and quantitative real-time PCR analyses reveal systems metabolic changes of Fusarium graminearum induced by Tri5 gene deletion. J Proteome Res 10(5):2273–2285
de Oliveira Rocha L, Reis GM, da Silva VN, Braghini R, Geraldes Teixeira MM, Corrêa B (2011) Molecular characterization and fumonisin production by Fusarium verticillioides isolated from corn grains of different geographic origins in Brazil. Int J Food Microbiol 145:9–21
Desjardins AE (2006) Fusarium, mycotoxins: chemistry, genetics, and biology. APS Press, St. Paul, MN
Desjardins AE, Busman M, Muhitch M, Proctor RH (2007) Complementary host-pathogen genetic analyses of the role of fumonisins in the Zea mays–Gibberella moniliformis interaction. Physiol Mol Plant Pathol 70:149–160
Edgar RC (2004) MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res 32:1792–1797
Geiser DM, Jiménez-Gasco MdM, Kang S, Makalowska I, Veeraraghavan N, Ward TJ, Zhang N, Kuldau GA, O’Donnell K (2004) FUSARIUM-ID v. 1.0: a DNA sequence database for identifying Fusarium. Eur J Plant Pathol 110:473–479
Glenn AE, Zitomer NC, Zimeri AM, Williams LD, Riley RT, Proctor RH (2008) Transformation-mediated complementation of a FUM gene cluster deletion in Fusarium verticillioides restores both fumonisin production and pathogenicity on maize seedlings. Mol Plant Microbe Interact 21:87–97
González-Jaén MT, Mirete S, Patiño B, López-Errasquín E, Vázquez C (2004) Genetic markers for the analysis of variability and for production of specific diagnostic sequences in fumonisin-producing strains of Fusarium verticillioides. Eur J Plant Pathol 110:525–532
Jurado M, Marín P, Magan N, González-Jaén MT (2008) Relationship between solute and matric potential stress, temperature, growth, and FUM1 gene expression in two Fusarium verticillioides strains from Spain. Appl Environ Microbiol 74:2032–2036
Jurado M, Marín P, Callejas C, Moretti A, Vázquez C, González-Jaén MT (2010) Genetic variability and fumonisin production by Fusarium proliferatum. Food Microbiol 27:50–57
Kostecki M, Wiśniewska H, Perrone G, Ritieni A, Goliński P, Chełkowski J, Logrieco A (1999) The effects of cereal substrate and temperature on production of beauvericin, moniliformin and fusaproliferin by Fusarium subglutinans ITEM-1434. Food Addit Contamin 16:361–365
Kristensen R, Torp M, Kosiak B, Holst-Jensen A (2005) Phylogeny and toxigenic potential is correlated in Fusarium species as revealed by partial translation elongation factor 1 alpha gene sequences. Mycol Res 109:173–186
López-Errasquín E, Vázquez C, Jiménez M, González-Jaén MT (2007) Real-time RT-PCR assay to quantify the expression of fum1 and fum19 genes from the fumonisin-producing Fusarium verticillioides. J Microbiol Methods 68:312–317
Marín S, Sanchis V, Magan N (1995) Water activity, temperature, and pH effects on growth of Fusarium moniliforme and Fusarium proliferatum isolates from maize. Can J Microbiol 41:1063–1070
Proctor RH, Brown DW, Plattner RD, Desjardins AE (2003) Co-expression of 15 contiguous genes delineates a fumonisin biosynthetic gene cluster in Gibberella moniliformis. Fungal Genet Biol 38:237–249
Proctor RH, Plattner RD, Desjardins AE, Busman M, Butchko RAE (2006) Fumonisin production in the maize pathogen Fusarium verticillioides: genetic basis of naturally occurring chemical variation. J Agric Food Chem 54:2424–2430
Proctor RH, McCormick SP, Alexander NJ, Desjardins AE (2009) Evidence that a secondary metabolic biosynthetic gene cluster has grown by gene relocation during evolution of the filamentous fungus Fusarium. Mol Microbiol 74:1128–1142
Rheeder JP, Marasas WFO, Vismer HF (2002) Production of fumonisin analogs by Fusarium species. Appl Environ Microbiol 68:2101–2105
Stępień Ł, Koczyk G, Waśkiewicz A (2011) FUM cluster divergence in fumonisins-producing Fusarium species. Fungal Biol 115:112–123
Sydenham EW, Gelderblom WCA, Thiel PG, Marasas WFO (1990) Evidence for the natural occurrence of fumonisin B1, a mycotoxin produced by Fusarium moniliforme, in corn. J Agric Food Chem 38:285–290
Tamura K, Dudley J, Nei M, Kumar S (2007) MEGA4: Molecular Evolutionary Genetics Analysis (MEGA) software version 4.0. Mol Biol Evol 24:1596–1599
von Bargen S, Martinez O, Schadock I, Eisold A-M, Gossmann M, Büttner C (2009) Genetic variability of phytopathogenic Fusarium proliferatum associated with crown rot in Asparagus officinalis. J Phytopathol 157:446–456
Waalwijk C, van der Lee T, de Vries I, Hesselink T, Arts J, Kema GHJ (2004) Synteny in toxigenic Fusarium species: the fumonisin gene cluster and the mating type region as examples. Eur J Plant Pathol 110:533–544
Waśkiewicz A, Goliński P, Karolewski Z, Irzykowska L, Bocianowski J, Kostecki M, Weber Z (2010) Formation of fumonisins and other secondary metabolites by Fusarium oxysporum and F. proliferatum: a comparative study. Food Addit Contamin Part A Chem Anal Control Expo Risk Assess 27:608–615
Acknowledgments
The research was partially supported by the National Science Centre (NCN) project NN 310 732440.
Grzegorz Koczyk has been supported by the National Centre for Research and Development (NCBiR) grant LIDER 19/113/L-1/09/NCBiR/2010 “Modelling, prediction and verification of toxigenic potential in fungi”.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Stępień, Ł., Koczyk, G. & Waśkiewicz, A. Genetic and phenotypic variation of Fusarium proliferatum isolates from different host species. J Appl Genetics 52, 487–496 (2011). https://doi.org/10.1007/s13353-011-0059-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13353-011-0059-8