
Abstract
Background and Objectives
Ocedurenone (KBP-5074) is a novel nonsteroidal mineralocorticoid receptor antagonist that has demonstrated safety and efficacy in clinical trials in patients with uncontrolled hypertension and stage 3b/4 chronic kidney disease. The aim of this study was to assess the pharmacokinetics, safety, and tolerability of ocedurenone in individuals with moderate hepatic impairment.
Methods
This study was an open-label, nonrandomized, multi-center study investigating the pharmacokinetics, safety, and tolerability of a single dose of 0.5 mg ocedurenone administered orally in male and female subjects with moderate hepatic impairment (Child–Pugh B, score 7–9) compared with subjects with normal hepatic function. Serial blood samples were obtained from predose through 264 h postdose for analysis of ocedurenone concentrations using a validated liquid chromatography–tandem mass spectrometry method. Free ocedurenone concentrations in plasma were determined ex vivo using equilibrium dialysis.
Results
Following a single oral dose of 0.5 mg ocedurenone administered to subjects with moderate hepatic impairment and subjects with normal hepatic function, ocedurenone was steadily absorbed with median time to peak drug concentration (Tmax) values of 4 and 3 h, respectively. After reaching maximum plasma concentration (Cmax), the disposition of ocedurenone appeared to be biphasic. The geometric mean t1/2 values for the moderate hepatic impairment group and normal hepatic function group were 75.6 and 65.7 h, respectively. Ocedurenone systemic exposure, as assessed by area under the plasma concentration-time curve (AUC) was 23.5–26.6% lower in subjects with moderate hepatic impairment versus subjects with normal hepatic function, whereas Cmax was 41.2% lower. Ocedurenone was determined to be > 99.7% bound to total protein in plasma. Hepatic impairment appeared not to change plasma protein binding or the unbound free fraction. Ocedurenone was safe and well-tolerated in all participants.
Conclusions
Considering the long half-life of ocedurenone and previously completed clinical studies using 0.25 mg and 0.5 mg doses demonstrating efficacy and safety, the observed decreases in AUC and Cmax do not warrant a dose adjustment in patients with moderate hepatic impairment. A single 0.5 mg dose of ocedurenone was safe and well-tolerated when administered to subjects with moderate hepatic impairment and subjects with normal hepatic function.
Clinical Trial Identifier (www.clinicaltrials.gov)
NCT04534699.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
This open-label, nonrandomized study investigating the pharmacokinetics, safety, and tolerability of a single 0.5 mg dose of ocedurenone in participants with or without moderate hepatic impairment determined that no dose adjustment is required in patients with moderate hepatic impairment. |
Ocedurenone was safe and well-tolerated in all participants. |
1 Introduction
The selective inhibition of mineralocorticoid receptors (MRs) is a validated therapeutic approach providing blood pressure control via a mechanism distinct from other antihypertensive agents [1, 2]. In patients with chronic kidney disease (CKD) and type 2 diabetes, treatment with nonsteroidal mineralocorticoid receptor antagonists (MRAs) resulted in lower risks of CKD progression and cardiovascular events [3]. Inhibition of MR signaling leads to increased sodium excretion, resulting in decreased body fluid, lower blood pressure, and less proteinuria. Proteinuria is a sign of abnormal kidney function in patients with CKD [4]. One challenge with inhibition of this pathway is that the excretion of sodium triggered by MR antagonism typically results in the retention of potassium. In many patients with compromised renal function, retention of potassium results in significant elevations in potassium levels, therefore limiting the utility of this class of drugs in patients with advanced CKD and uncontrolled hypertension.
Ocedurenone, a novel nonsteroidal MRA, is currently being investigated for the treatment of uncontrolled hypertension in patients with CKD. Ocedurenone has a higher binding affinity to MRs than steroidal MRAs (e.g., spironolactone and eplerenone) and the nonsteroidal MRA finerenone [5,6,7] and has little or no binding affinity to glucocorticoid or androgen receptors [6]. Hence, this orally available agent may be effective in reducing proteinuria and hypertension by blocking aldosterone from binding to MRs, thereby preventing downstream negative cardiorenal effects, such as target organ inflammation and fibrosis. The phase 2b clinical trial BLOCK-CKD [8] in subjects with CKD stage 3b/4 (estimated glomerular filtration rate 15–44 mL/min/1.73 m2) demonstrated significant blood pressure lowering and showed that neither hyperkalemia nor reductions in kidney function were limiting factors for the use of ocedurenone. To expand on these results, the current study was designed to investigate the pharmacokinetics, safety, and tolerability of ocedurenone in subjects with moderate hepatic impairment compared with subjects with normal hepatic functioning.
2 Methods
2.1 Study Design and Treatment
This was an open-label, nonrandomized, multicenter, single-dose study (www.clinicaltrials.gov identifier NCT04534699) investigating the pharmacokinetics, safety, and tolerability of orally administered ocedurenone in male and female subjects with moderate hepatic impairment compared with subjects with normal hepatic function. The study was conducted in three research centers: the Orlando Clinical Research Center Inc. in Orlando, Florida; American Research Corporation in San Antonio, Texas; and Clinical Trials of Texas in San Antonio, Texas. A single-dose, parallel design is the standard design used to investigate the pharmacokinetics of a drug in subjects with hepatic impairment. Potential subjects were screened to assess their eligibility (including Child–Pugh classification) to enroll in the study within approximately 28 days before dose administration. Subjects were admitted on day 1 and confined to the study site until discharge on day 12. Subjects received a single oral dose of 0.5 mg ocedurenone with approximately 240 mL of room temperature water on day 1 following an overnight fast of ≥ 10 h. Subjects returned to the study site for a follow-up visit on day 18 of the study (±1 day). Serial blood collections were obtained from 15 min predose through 264 h postdose for analysis of plasma concentrations of ocedurenone. Blood samples for analysis of ocedurenone plasma protein binding were also collected. Safety was monitored by recording adverse events (AEs), clinical laboratory evaluations (clinical chemistry, hematology, and urinalysis), vital sign measurements, 12-lead electrocardiograms (ECGs), and physical examination findings during the study. Age, weight, and sex-matched control subjects with normal hepatic function were enrolled in this study as a reference group for results interpretation and were recruited after participants with hepatic impairment had been identified. Based on nonclinical and clinical data and the known pharmacokinetics profile of ocedurenone, the treatment period duration was considered adequate to achieve study objectives.
2.2 Participants
Inclusion criteria for this study were as follows: age 18–80 years, body mass index (BMI) 18–40 kg/m2, agreement to use contraceptive methods with proven reliability (including abstinence) for the duration of the study, and the ability to comprehend and willingness to sign an informed consent form and to abide by study restrictions.
The degree of hepatic impairment was assessed using the Child–Pugh classification, whereby participants with grade B (moderate, score 7–9) hepatic impairment were eligible for inclusion in the hepatic impairment group [9]. Child–Pugh classification was completed at screening and again at check-in; if the scores at these two time points differed, enrollment was based on the Child–Pugh score at screening. Subjects with hepatic impairment were eligible even if they had abnormal findings not considered to be clinically significant by the investigator, and a single repeat test could be performed to confirm vital signs, electrocardiogram, and clinical laboratory test abnormalities. Subjects with hepatic impairment were required to have a diagnosis of chronic (> 6 months), stable hepatic impairment with no clinically significant changes within 30 days prior to study drug administration on day 1.
Good health was an additional inclusion criterion for subjects with normal hepatic function; good health was defined by no clinically significant findings in medical history, physical examination, 12-lead electrocardiogram, vital sign measurements, or clinical laboratory evaluations at screening and check-in as assessed by the investigator, and a single repeat measurement or test could be performed to confirm good health in control subjects
Exclusion criteria for all participants included the following: (1) significant history or clinical manifestation of any metabolic, allergic, dermatological, renal, hematological, pulmonary, cardiovascular, gastrointestinal, neurological, respiratory, endocrine, or psychiatric disorder as determined by the investigator; (2) history of significant hypersensitivity, intolerance, or allergy to any drug compound, food, component of the investigational medicinal product (IMP), or other substance unless approved by the investigator; (3) history of stomach or intestinal surgery or resection that could potentially alter absorption and/or excretion of any orally administered drugs, excluding uncomplicated appendectomy and hernia repair but not cholecystectomy; (4) congenital nonhemolytic hyperbilirubinemia; and (5) positive urine, breath, or blood test for alcohol or positive urine drug screen at screening and/or check-in (unless explained by permitted medication or diet in study subjects). Additional exclusion criteria can be found in the Supplementary Information. Women of childbearing age were also required to provide a negative result on an approved pregnancy test at screening and check-in; pregnancy and lactating status were both exclusion criteria.
In total, 12 participants (6 subjects with moderate hepatic impairment and 6 age-, weight-, and sex-matched subjects without hepatic impairment) were allocated to treatment and completed the study as planned [9]. Each of the six control subjects were matched by age (±10 years), body mass index (BMI, ±20%; data obtained at screening), and sex to an enrolled subject with hepatic impairment.
2.3 Ethics
Before the start of the study, the protocol, informed consent form, investigator’s brochure, and other relevant documents were reviewed and approved by the appropriate Institutional Review Board (Midlands Institutional Review Board in Lenexa, Kansas). The study was conducted in accordance with the protocol and with the consensus ethical principles derived from international guidelines, including the Declaration of Helsinki [10], the Council for International Organizations of Medical Sciences [11], and the International Conference on Harmonization Good Clinical Practice Guideline [12], as well as applicable laws and regulations. All subjects provided informed consent before participation in this study.
2.4 Study Drug
Ocedurenone was administered as a single-dose tablet containing 0.5 mg ocedurenone supplied by SynTheAll Pharmaceutical Co., Ltd., Shanghai, China, in accordance with Good Manufacturing Practice.
2.5 Safety and Tolerability
Safety was monitored by physical examinations, regular measurements of systolic and diastolic blood pressure, heart rate, 12-lead ECGs, and analysis of clinical laboratory chemistry and hematology parameters, including potassium levels. A treatment-emergent adverse event (TEAE) was defined as an AE that started during or after dosing or started prior to dosing and increased in severity after dosing. A treatment-related TEAE was defined as a TEAE with a relationship of possibly related or related to the study treatment, as determined by the investigator. Subjects were encouraged to spontaneously report AEs occurring at any other time during the study. All nonserious AEs, whether reported by the subject voluntarily or upon questioning or noted on physical examination, were recorded from initiation of study drug until study completion. Serious adverse events (SAEs) were recorded from the time the subject signed the informed consent form until study completion. The nature, time of onset, duration, and severity of adverse events were documented together with an investigator’s opinion of the relationship of the event to the study drug. Any clinically significant abnormalities found during the course of the study were followed up until they returned to normal or could be clinically explained. AE definitions, assignment of severity and causality, and procedures for reporting SAEs were defined in the protocol. Additional information about adverse event reporting definitions and assessment of severity can be found in the Supplementary Information.
2.6 Pharmacokinetic Sampling
Blood samples were taken approximately 22 times throughout the course of the study. Blood samples of approximately 10 mL per time point (236 mL total throughout the study) were collected at 15 min predose and 0.5, 1, 2, 4, 6, 8, 12, 24, 48, 72, 96, 120, 144, 168, 192, 216, 240, and 264 h postdose. The allowed sampling window for pharmacokinetics blood samples was the following: within 15 min before dosing for the predose sample timepoint; ±5 min for sampling timepoints ≤ 12 h; ± 30 min for sampling timepoint at 24 h; and ± 60 min for all sampling timepoints ≥ 48 h. For assessment of unbound plasma concentrations of ocedurenone, blood samples were collected at 4 and 12 h postdose.
2.7 Bioanalysis
Plasma concentrations of ocedurenone in human plasma containing K2EDTA as an anticoagulant were determined using protein precipitation followed by a validated high-performance liquid chromatography–tandem mass spectroscopy (HPLC-MS) method [13]. HPLC-MS was completed with an HPLC System Shimazdu 20 Series at Bioanalytical Test Site Covance Laboratories, Inc., in Madison, Wisconsin. The validated range was 0.11 (LLOQ)–100 (ULOQ) ng/mL, and quality control levels were 0.300 ng/mL, 3.50 ng/mL, 40.0 ng/mL, and 80 ng/mL. The sample volume was 50.0 µL and the reference standard was KBP-5314. Five of five runs met the acceptance criteria and a total of 228 samples were analyzed, with 4 samples (1.8%) reassayed. Samples were stored at −60 to −80 ˚C, and the samples analyzed were within known stability.
2.8 Pharmacokinetic Evaluation
Human plasma samples obtained from all subjects at 4 and 12 h postdose were incubated in apparatus model HTD96b (HTDialysis LLC, Gales Ferry, Connecticut) for 5 h. The samples were processed prior to HPLC-MS analysis as follows. The donor side samples (plasma) were diluted with Dulbecco’s phosphate-buffered saline (DPBS) and the receiver side samples (DPBS/dialysate) were diluted with unfortified control plasma to provide a common analytical mixed matrix of 90% DPBS and 10% plasma (90:10 DPBS:plasma, v:v). Methanol containing internal standard (KBP-5314) was added to each sample. Samples were vortex mixed and centrifuged at 2250 × g for 5 min. Each supernatant was transferred to a new 96-well plate. Samples were injected in HPLC-MS for analysis of test article according to the method presented above. Plasma protein binding for ocedurenone is 99%, free fraction for AUC and Cmax would be 1% of the total; no impact of hepatic impairment on free fraction was therefore observed. In summary, unbound ocedurenone at 4 and 12 h postdose ranged from 0.115% to 0.296%, and no significant change in free fraction was observed from hepatically impaired subjects or matched control subjects.
The pharmacokinetics analysis was conducted by Covance Early Clinical Biometrics using Phoenix WinNonlin Version 8.1. The pharmacokinetics parameters were determined from the plasma concentrations of ocedurenone using noncompartmental procedures and included AUC0–tlast (AUC from time zero to time of last quantifiable concentration), AUC0–∞ (AUC from time zero extrapolated to infinity), %AUCextrap (percentage of AUC owing to extrapolation from the last quantifiable concentration to infinity), Cmax (maximum observed plasma concentration), Tmax (time of the maximum observed plasma concentration), t1/2 (apparent terminal elimination half-life), CL/F (apparent total oral clearance), and Vz/F (apparent oral volume of distribution). The fraction of unbound drug (fu) was used to calculate unbound ocedurenone pharmacokinetic parameters for each individual subject whenever possible, if deemed appropriate, including AUC0–tlast,u (unbound AUC0–tlast, calculated as AUC0–tlast·fu), AUC0–∞, u (unbound AUC0–∞, calculated as AUC0–∞·fu), Cmax,u (unbound Cmax, calculated as Cmax·fu), CL/F,u (unbound CL/F, calculated as dose/AUC0–∞,u), and Vz/F, u (unbound Vz/F, calculated as CL/F, u/λZ).
2.9 Statistical Analysis
Individual plasma concentrations of ocedurenone and calculated pharmacokinetic parameters were summarized by hepatic function group using descriptive statistics [number of subjects, number of subjects with valid observations, arithmetic mean, arithmetic standard deviation (SD), geometric mean, geometric coefficient of variation (CV), standard error, median, minimum, and maximum] as appropriate. Inferential statistical analysis was performed on log-transformed AUC0–∞, AUC0–tlast, and Cmax only. The analysis planned for this study was to evaluate the pharmacokinetics of KBP-5074 after a single dose in subjects with moderate hepatic impairment (test) compared with subjects with normal hepatic function (reference). The pharmacokinetics parameters AUC0–∞, AUC0–tlast, and Cmax were analyzed on the logarithmic scale using a paired t-test to assess the difference between the test group relative to the reference group. The p-value assessing the difference between the test group relative to the reference group is presented. The 90% confidence interval (CI) for the geometric mean ratio between the test group and the reference group is presented. If the 90% CIs do not overlap, this indicates that there is a trend of statistically significant difference in exposure between subjects with moderate hepatic impairment and healthy subjects with normal hepatic function.
2.10 Sample Size Determination
A total of 12 subjects (6 subjects with moderate hepatic impairment and 6 matched-control healthy subjects with normal hepatic function) were planned to be enrolled in the study. Subjects who withdrew or were withdrawn from the study for reasons other than safety were to be replaced at the discretion of the investigator and the sponsor to ensure that six subjects with moderate impairment and six matched-control healthy subjects completed the study. The sample size was determined without power consideration on the basis of the limited inter-individual variability of pharmacokinetic parameters. A table providing a pharmacokinetic summary of ocedurenone can be found in Supplementary Information.
3 Results
3.1 Demographics
Demographic data for subjects receiving study treatment are summarized in Table 1 and a summary of the matched subjects’ demographics is presented in Table 2. Three male and three female subjects were included in the moderate hepatic impairment group. Subjects were aged 54–69 years, inclusive, with BMI ranging between 26.6 and 38.1 kg/m2, inclusive; all subjects with moderate hepatic impairment were white, and two subjects were Hispanic or Latino. Each subject in the normal function group was demographically matched by age (± 10 years), BMI (± 20%), and sex to an enrolled subject with hepatic impairment. Five subjects with normal hepatic function were white, one subject was Black or African-American, and three were Hispanic or Latino.
All enrolled subjects satisfied the inclusion and exclusion criteria prior to study entry. There were no findings in the medical history of clinical concern for any subjects that were not consistent with their hepatic function classification. In addition, there were no baseline signs or symptoms that were not consistent with a hepatically impaired or unimpaired population prior to dosing for any subjects.
3.2 Pharmacokinetics
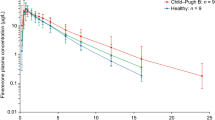
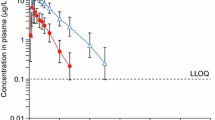
Owing to the low unbound drug fraction (< 0.3%) and little difference between subjects with normal hepatic function and moderate hepatic impairment, it was not deemed appropriate to calculate unbound pharmacokinetic parameters. Arithmetic mean (+SD) plasma concentration-time profiles for ocedurenone following a single oral dose of 0.5 mg are presented in Fig. 1. Plasma pharmacokinetic parameters of ocedurenone following a single oral doses of 0.5 mg are summarized in Table 3, and the statistical analysis of pharmacokinetic data is presented in Table 4.

Arithmetic mean (+SD) plasma concentrations of ocedurenone following a single oral dose (linear and semilogarithmic scale)
Following a single oral dose of 0.5 mg ocedurenone in subjects with moderate hepatic impairment (n = 6) or normal hepatic function (n = 6), ocedurenone was steadily absorbed with median Tmax values of 4 h (range 2–12) and 3 h (range 2–24), respectively. After reaching Cmax, the disposition of ocedurenone appeared to be biphasic. The geometric mean t1/2 values for the moderate hepatic impairment and normal hepatic function groups were 75.6 h (range 63.1–90.1) and 65.7 h (range 56.7–73.1), respectively (Table 3).
Systemic exposure, as measured by geometric mean ocedurenone AUC0–∞, AUC0–tlast, and Cmax values, was lower in the moderate hepatic impairment group compared with the normal hepatic function group. The ratios of geometric least squares means (90% CI) of AUC0–∞, AUC0–tlast, and Cmax were 76.5 (66.1, 88.4), 73.4 (64.0, 84.2), and 58.8 (50.3, 68.8) for the moderate hepatic impairment to normal hepatic function groups, respectively (Table 4). Variability (as measured by geometric CV%) was low, with AUC0–∞, AUC0–tlast, and Cmax CV% values of 16.8%, 16.6%, and 22.9% and 14.0%, 12.9%, and 12.3% in the moderate hepatic impairment group and normal hepatic function group, respectively. None of the 90% CIs spanned unity (100%), potentially indicating a trend of statistically significant differences in exposure between subjects with moderate hepatic impairment versus healthy subjects with normal hepatic function, although the clinical implications of such a finding require further evaluation.
3.3 Safety and Tolerability
Treatment with a single, oral, 0.5 mg dose of ocedurenone was safe and well-tolerated in subjects with either moderate hepatic impairment or no hepatic impairment. No fatal TEAEs were reported, and none of the subjects discontinued treatment prematurely owing to an AE. One subject in the moderate hepatic impairment group experienced one treatment-related TEAE of nausea, which was mild in severity. The event had a duration of 8 h, and the investigator considered it to be related to the study drug. The event resolved without intervention, and no concomitant medication was administered. No TEAEs were reported by any subjects in the normal hepatic function group. Safety laboratory assessments revealed no relevant changes of medical concern. Blood pressure, heart rate, and ECG parameters were not affected by ocedurenone in any subjects with or without hepatic impairment. Overall, ocedurenone was well tolerated and considered safe.
4 Discussion
This was an open-label, nonrandomized, multicenter, single-dose study with the primary objective of assessing the plasma pharmacokinetics profiles of a single dose of ocedurenone in subjects with moderate hepatic impairment compared with matched control subjects with normal hepatic function. The secondary objective of the study was to evaluate the safety and tolerability of a single dose of ocedurenone in subjects with moderate hepatic impairment compared with matched control subjects with normal hepatic function.
Following a single oral dose of 0.5 mg ocedurenone administered to subjects with moderate hepatic impairment and subjects with normal hepatic function, ocedurenone was steadily absorbed. After reaching Cmax, the disposition of ocedurenone appeared to be biphasic. Ocedurenone systemic exposure, as assessed by AUC, was lower in subjects with moderate hepatic impairment than in subjects with normal hepatic function and the difference in exposure based on Cmax, and AUC was statistically significant.
In this study single doses of ocedurenone appeared to be safe and well tolerated when administered to subjects with moderate hepatic impairment and subjects with normal hepatic function. One subject in the moderate hepatic impairment group experienced one treatment-related TEAE of nausea, which was mild in severity and resolved without intervention. No TEAEs were reported by any subjects in the normal hepatic function group. There were no SAEs during the study, and no TEAE led to premature discontinuation of a subject from the study. There were no clinically significant findings in the clinical laboratory evaluations, vital sign data, 12-lead ECG, or physical examination data during the study.
The phase 2b BLOCK-CKD study with ocedurenone in subjects with uncontrolled hypertension and CKD stage 3b/4 demonstrated that ocedurenone was highly effective for lowering blood pressure, safe and well-tolerated across doses of 0.25 mg and 0.5 mg, and associated with a low incidence of AEs, such as hyperkalemia and decreased glomerular filtration rate [6]. Considering the long half-life and previously completed clinical studies using 0.25 mg and 0.5 mg doses that demonstrated efficacy and safety, the observed decrease in AUC and Cmax does not warrant a dose adjustment in patients with moderate hepatic impairment. A single 0.5 mg dose of ocedurenone was safe and well tolerated when administered to subjects both with and without moderate hepatic impairment.
This study had two primary limitations. Only 12 subjects (6 subjects with moderate hepatic impairment and 6 matched-control healthy subjects with normal hepatic function) were enrolled in the study, and no data were collected for participants with severe hepatic impairment (Child–Pugh C). Based on previous experience, ocedurenone pharmacokinetic parameters have limited interindividual variability. It was therefore anticipated that six subjects would provide accurate assessments for ocedurenone pharmacokinetic properties and the effects of moderate hepatic impairment.
5 Conclusions
In summary, ocedurenone systemic exposure, as assessed by AUC, was lower in subjects with moderate hepatic impairment compared with subjects with normal hepatic function, and Cmax was also lower. The difference in exposure between subjects with moderate hepatic impairment and subjects with normal hepatic function based on Cmax and AUCs was statistically significant. The median Tmax of ocedurenone was later or delayed in subjects with moderate hepatic impairment compared with subjects with normal hepatic function. The geometric mean t1/2 of ocedurenone was longer in subjects with moderate hepatic impairment than in subjects with normal hepatic function. Ocedurenone was determined to be highly plasma protein bound in both the hepatically impaired and unimpaired groups. No difference in unbound drug concentration was observed between hepatically impaired and unimpaired subjects. Moderate hepatic impairment did not appear to significantly impact ocedurenone exposure, but hepatic impairment may lead to decreased absorption.
References
Williams B, MacDonald TM, Morant S, Webb DJ, Sever P, McInnes G, Ford I, Cruickshank JK, Caulfield MJ, Salsbury J, Mackenzie I, Padmanabhan S, Brown MJ. Spironolactone versus placebo, bisoprolol, and doxazosin to determine the optimal treatment for drug-resistant hypertension (PATHWAY-2): a randomised, double-blind, crossover trial. Lancet. 2015;386(10008):2059–68. https://doi.org/10.1016/s0140-6736(15)00257-3.
Zhou X, Crook MF, Sharif-Rodriguez W, Zhu Y, Ruben Z, Pan Y, Urosevic-Price O, Wang L, Flattery AM, Forrest G, Szeto D, Zhao H, Roy S, Forrest MJ. Chronic antagonism of the mineralocorticoid receptor ameliorates hypertension and end organ damage in a rodent model of salt-sensitive hypertension. Clin Exp Hypertens. 2011;33(8):538–47. https://doi.org/10.3109/10641963.2011.566956.
Bakris GL, Agarwal R, Anker SD, Pitt B, Ruilope LM, Rossing P, Kolkhof P, Nowack C, Schloemer P, Joseph A, Filippatos G. Effect of finerenone on chronic kidney disease outcomes in type 2 diabetes. N Engl J Med. 2020;383(23):2219–29. https://doi.org/10.1056/NEJMoa2025845.
Agarwal R, Kolkhof P, Bakris G, Bauersachs J, Haller H, Wada T, Zannad F. Steroidal and non-steroidal mineralocorticoid receptor antagonists in cardiorenal medicine. Eur Heart J. 2021;42(2):152–61. https://doi.org/10.1093/eurheartj/ehaa736.
Pitt B, Jaisser F, Bakris G. An evaluation of KBP-5074 in advanced chronic kidney disease with uncontrolled hypertension. Expert Opin Investig Drugs. 2021;30(10):1017–23. https://doi.org/10.1080/13543784.2021.1985462.
Chow CP, Liu JR, Tan XJ, Huang ZH. Pharmacological profile of KBP-5074, a novel nonsteroidal mineralocorticoid receptor antagonist for the treatment of cardiorenal diseases. J Drug Res Dev. 2017. https://doi.org/10.16966/2470-1009.137.
Pitt B, Filippatos G, Gheorghiade M, Kober L, Krum H, Ponikowski P, Nowack C, Kolkhof P, Kim SY, Zannad F. Rationale and design of ARTS: a randomized, double-blind study of BAY 94–8862 in patients with chronic heart failure and mild or moderate chronic kidney disease. Eur J Heart Fail. 2012;14(6):668–75. https://doi.org/10.1093/eurjhf/hfs061.
Bakris G, Pergola PE, Delgado B, Genov D, Doliashvili T, Vo N, Yang YF, McCabe J, Benn V, Pitt B. Effect of KBP-5074 on blood pressure in advanced chronic kidney disease: results of the BLOCK-CKD study. Hypertension. 2021;78(1):74–81. https://doi.org/10.1161/hypertensionaha.121.17073.
Pugh RN, Murray-Lyon IM, Dawson JL, Pietroni MC, Williams R. Transection of the oesophagus for bleeding oesophageal varices. Br J Surg. 1973;60(8):646–9. https://doi.org/10.1002/bjs.1800600817.
World Medical Association. World Medical Association Declaration of Helsinki: ethical principles for medical research involving human subjects. JAMA. 2013;310(20):2191–4. https://doi.org/10.1001/jama.2013.281053.
Council for International Organizations of Medical Sciences (CIOMS). International ethical guidelines for health-related research involving humans. Geneva, Switzerland; 2016.
Dixon JR (198) The guideline. Qual Assur 6(2):65–74. Doi: https://doi.org/10.1080/105294199277860.
Wang P, Liu J, Tan X, Yang F, McCabe J, Zhang J. Pharmacokinetics and drug-drug interaction of ocedurenone (KBP-5074) in vitro and in vivo. Eur J Drug Metab Pharmacokinet. 2023;48(4):397–410. https://doi.org/10.1007/s13318-023-00837-5.
Acknowledgements
Writing and editorial assistance were provided by Terri Levine, Ph.D., in accordance with Good Publication Practice (GPP 2022) guidelines. The authors thank the volunteers who participated in this study.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
Funding for this study and manuscript was provided by KBP BioSciences USA, Inc., Princeton, NJ, USA.
Conflict of Interest
Authors were employees of KBP Biosciences USA, Inc., Princeton, NJ, but have no other relevant financial or non-financial interests to disclose.
Availability of Data and Materials
Data and materials information can be made available upon reasonable request.
Code Availability
Not applicable.
Consent for Publication
Not applicable.
Ethical Approval
Before the start of the study, the protocol, informed consent form, investigator’s brochure, and other relevant documents were reviewed and approved by the appropriate Institutional Review Board. The study was conducted in accordance with the protocol and with the consensus ethical principles derived from international guidelines, including the Declaration of Helsinki [10], the Council for International Organizations of Medical Sciences [11], and the International Conference on Harmonization Good Clinical Practice Guideline [12], as well as applicable laws and regulations.
Informed Consent
All subjects provided informed consent before participation in this study.
Author contributions
Jay Zhang, James McCabe, and Fred Yang participated in the research design. Jay Zhang and James McCabe performed data analysis. James McCabe, Vincent Benn, Fred Yang, and Jay Zhang wrote or contributed to the writing of the manuscript. James McCabe provided medical oversight. All authors read and approved the final manuscript.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
McCabe, J., Zhang, J., Yang, F. et al. Pharmacokinetics of the Novel Nonsteroidal Mineralocorticoid Receptor Antagonist Ocedurenone (KBP-5074) in Individuals with Moderate Hepatic Impairment. Eur J Drug Metab Pharmacokinet 49, 229–237 (2024). https://doi.org/10.1007/s13318-024-00879-3
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13318-024-00879-3