
Abstract
Background and Objective
MHV370, a dual antagonist of human Toll-like receptors (TLR) 7 and 8, suppresses cytokines and interferon-stimulated genes in vitro and in vivo, and has demonstrated efficacy in murine models of lupus. This first-in-human study aimed to evaluate the safety, tolerability, pharmacokinetics and pharmacodynamics of single and multiple doses of MHV370 in healthy adults, as well as the effects of food consumption on a single dose of MHV370.
Methods
This was a phase 1, randomised, placebo-controlled study conducted in three parts. In part A, participants received (3:1) a single ascending dose (SAD) of 1, 3, 10, 20, 40, 80, 160, 320, 640 and 1000 mg MHV370 or placebo. In part B, participants received (3:1) multiple ascending doses (MAD) of 25, 50, 100, 200 and 400 mg MHV370 twice daily (b.i.d) or placebo for 14 days. In part C, participants received an open-label single dose of 200 mg MHV370 under fasted or fed conditions. Safety, pharmacokinetic and pharmacodynamic parameters were evaluated.
Results
MHV370 was well tolerated, and no safety signal was observed in the study. No dose-limiting adverse events occurred across the dose range evaluated. Plasma concentrations of MHV370 increased with dose (mean [SD] maximum plasma concentrations ranged from 0.97 [0.48] to 1670 [861.0] ng/mL for SAD of 3–1000 mg, 29.5 [7.98] to 759 [325.0] ng/mL for MAD of 25–400 mg b.i.d. on day 1). The intake of food did not have a relevant impact on the pharmacokinetics of MHV370. Pharmacodynamic data indicated time- and dose-dependent inhibition of TLR7-mediated CD69 expression on B cells (100% inhibition at 24 h post-dose starting from SAD 160 mg and MAD 50 mg b.i.d.) and TLR8-mediated TNF release after ex vivo stimulation (>90% inhibition at 24 h post-dose starting from SAD 320 mg and MAD 100 mg b.i.d.).
Conclusion
The safety, pharmacokinetic and pharmacodynamic data support the further development of MHV370 in systemic autoimmune diseases driven by the overactivation of TLR7 and TLR8.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
MHV370, an TLR7/8 antagonist, was well tolerated by healthy participants. |
No relevant effect of food on the plasma pharmacokinetics of MHV370 was observed. |
The pharmacokinetic and pharmacodynamic profile of MHV370 favours a 100 mg b.i.d. dosing regimen. |
1 Introduction
Toll-like receptors (TLRs) are receptors of the innate immune system that recognise conserved pathogen-associated molecular patterns (PAMPs). Upon ligation, TLRs recruit downstream adaptor proteins to initiate transcriptional activity and immune responses against pathogens [1].
TLR7 and TLR8 are expressed in different immune cells and detect a specific type of PAMP, GU-rich single-stranded RNAs (ssRNA) [2], which are present in viruses [3]. As a consequence, activation of TLR7 and TLR8 supports immune protection against viral infections [4]. TLR7 and TLR8 can also recognise endogenous nucleic acids and ribonucleoproteins (RNPs) when released extracellularly, e.g. as autoantigens of systemic autoimmune diseases, such as Ro60 and RNP/Sm [5, 6], and their hyperactivation can result in autoimmunity. Overexpression of TLR7 or human TLR8 in mice consistently drives spontaneous lethal autoimmune disease [7, 8], and polymorphisms resulting in increased expression of TLR7 are associated with an increased risk of developing autoimmune diseases [9]. An important piece of evidence for TLR7 as a disease driver is highlighted by gain-of-function (GOF) mutations observed in patients with autoimmune disease [10].
In preclinical studies, MHV370, a dual antagonist of human TLR7 and TLR8, suppressed inflammatory cytokines (e.g. TNF, IL-6) and interferon (IFN)-stimulated genes (ISGs) in vitro and in vivo and showed efficacy in murine models of lupus. MHV370 also suppressed cytokines and ISGs induced by immune complexes from lupus patient sera and necrotic cell extracts [11]. In contrast to immunosuppressive standard of care medications, MHV370 has the potential to become an orally available, targeted immunomodulatory therapy in patients with autoimmune diseases with limited treatment options.
The primary goal for MHV370 clinical development is to target immune-mediated diseases such as systemic lupus erythematosus (SLE), in which overactivation of TLR7/8 is expected to play a role in immune activation and pathology. These indications have heterogeneous clinical manifestations with high unmet medical needs.
With this phase 1 first-in-human (FIH) study, we had the objective to assess the safety, pharmacokinetics, and pharmacodynamics of single and multiple doses of MHV370 and to explore the effect of food on a single administration of MHV370 in healthy participants.
2 Materials and Methods
2.1 Study Design
This was a phase 1, first-in-human (FIH), randomised, placebo-controlled, investigator- and participant-blinded study. Healthy participants were randomised to receive single or multiple ascending doses of oral MHV370 or placebo. The study consisted of three parts.
Part A was the single-ascending dose (SAD) arm of the study (Fig. 1A). Participants were randomised (3:1) to receive a single oral dose of MHV370 or matching placebo in ten consecutive cohorts of eight participants each, named cohorts A1 to A10 (1, 3, 10, 20, 40, 80, 160, 320, 640 and 1000 mg). The starting dose of 1 mg was chosen based on the calculation of the maximum recommended starting dose (MRSD). Each cohort consisted of a screening visit, a baseline visit, a treatment visit and follow-up visits during specified time periods (Fig. 1B). As is standard practice for drugs in early development with no food-effect data, participants fasted overnight for at least 9 h prior to the administration of MHV370 and continued to fast for at least 4 h post-dose. No fluid consumption was permitted for 2 h before and after dosing. Every cohort consisted of six MHV370-treated and two placebo-treated participants. A sentinel dosing scheme was adopted for each SAD cohort to mitigate the potential risk to participants: two sentinel participants, one receiving MHV370 and the other a placebo, were treated on the first dosing day. The remaining six participants were dosed a minimum of 48 h later. Participants were required to stay at the study site from day − 1 until day 3, corresponding to the MHV370 washout time. The next SAD cohort was initiated following a supportive review of all clinical safety and pharmacokinetic data until study day 6 by the sponsor clinical team and the investigator.

Dose escalation scheme (A) and study design (B). b.i.d. twice daily
Part B was the multiple-ascending dose (MAD) part of the study (Fig. 1A). It was started after the completion of cohort A8 of the SAD. Participants were randomised (3:1) to receive multiple oral doses of MHV370 or matching placebo in five cohorts of eight participants each, named cohorts B1 to B5 (25, 50, 100, 200, and 400 mg twice daily [b.i.d.]). Every cohort consisted of a screening visit, a baseline evaluation, a 14-day in-house treatment period, and ambulatory follow-up visits (Fig. 1B). Participants received two daily doses of the study medication for 13 days and a single dose on day 14. The morning dose was administered after an overnight fast of at least 9 h, and participants continued to fast for at least 4 h after dosing. For the evening doses, participants fasted for at least 2 h before and after compound administration. The first two sentinel participants of each cohort were dosed on day 1 (one received MHV370, one received placebo), and the remaining six participants were dosed a minimum of 4 days later. The subsequent MAD cohort was initiated following a safety review of all the clinical and pharmacokinetic data until at least study day 19 by the sponsor clinical team and the investigator.
Part C was the food effect part of the study, with a single-dose administration of MHV370 in an open-label, randomised, crossover design (Fig. 1A). The cohort consisted of a screening period, an in-house period, and a safety follow-up period (Fig. 1B). A single dose of 200 mg MHV370 was given under fasted or fed (i.e., 5 min after a high-fat breakfast) conditions, and participants crossed over between both phases after a minimum of 72 h for MHV370 washout. In each treatment period, participants were domiciled at the study site from approximately 24 h prior to dosing until 24 h after dosing. The 200 mg dose in the food effect study was selected, as this dose level (given as b.i.d.) had demonstrated full efficacy in terms of CD69 expression and TNF release after ex vivo stimulation in the multiple ascending dose (MAD) arm of the study.
The study was conducted at a single clinical site (Charité Research Organization, Berlin, Germany) in accordance with the ethical principles of the Declaration of Helsinki, Good Clinical Practice standards, and applicable regulatory requirements. All participants provided written informed consent before the study initiation, and the study protocol was reviewed by the independent ethics committee (Landesamt für Gesundheit und Soziales (LAGeSo); Geschäftsstelle der Ethik-Kommission des Landes, Berlin). The study was registered on EudraCT (EudraCT number: 2017-004559-21).
2.2 Study Objectives
The primary study objective was to assess the safety and tolerability of single and multiple oral doses of MHV370 in healthy volunteers. Secondary objectives included the analysis of the plasma pharmacokinetics of MHV370, particularly under fed and fasted conditions. Exploratory objectives included pharmacodynamic effects.
2.3 Study Participants
Inclusion and exclusion criteria were in line with the standard criteria for a healthy volunteer study. Men and women aged 18 to 55 years with a body weight of ≥ 55 kg and a body mass index of 18–30 kg/m2 who were assessed to be healthy based on their medical history, physical examination, vital signs, electrocardiogram, and laboratory tests at screening were eligible. Women of childbearing potential, pregnant or nursing women were not eligible. Participants were excluded if they had clinically significant disease or any clinically relevant laboratory abnormalities at screening or baseline.
2.4 Safety and Tolerability Assessments
Safety and tolerability were assessed throughout the study. Safety assessments for the decision on dose escalation were based on physical examination, clinical laboratory testing, vital signs, 12-lead ECG, online telemetry until 4 h post-dose and 24-h Holter ECG at day − 1 and day 1 (part A) and day − 1, day 1, day 2 and day 14 (part B). The incidence rates of adverse events (AEs) and serious adverse events (SAE) were also assessed. To evaluate a potential impact of MHV370 on suicidal ideation or behaviour, all participants were interrogated using the Columbia-Suicide Severity Rating Scale (CSSRS) on day − 1, day 1 (part A) and up to day 19 in part B. The severity of AEs was graded by the investigators based on the Common Toxicity Criteria AE grade (CTCAE version 4.03). To investigate a potential effect of MHV370 on the measured glomerular filtration rate (mGFR), 5 mL of iohexol solution (300 mg/mL iodine) was intravenously administered to participants in cohort B5 and flushed with 10 mL of normal saline solution [12, 13]. Blood samples were taken at 3 h, 4 h and 5 h after injection as per the standard process for mGFR measurement [14, 15].
2.5 Pharmacokinetic Assessments for MHV370
Concentrations of MHV370 were determined in plasma (in parts A and B), or in plasma and urine (in part C) by a validated liquid chromatography-tandem mass spectrometry (LC-MS/MS) method. The lower limit of quantification (LLOQ) was 1 ng/mL. All pharmacokinetic parameters were determined using the actual recorded sampling times and non-compartmental method(s) under both fasting (parts A and B) and fasting vs fed conditions (part C) with Phoenix WinNonlin (version 6.4).
The analyses were performed by Eurofins ADME Bioanalyses (Vergèze, France; method 18-007) in accordance with the European Medicines Agency (EMA) reflection paper for laboratories that perform the analysis or evaluation of clinical trial samples, the United States Food and Drug Administration and EMA guidelines on bioanalytical method validation, and the requirements of Good Laboratory Practices [16,17,18,19]. The acceptance criteria for intra- and inter-day accuracy and precision were a mean bias at each concentration above the LLOQ of ± 15%, a mean precision at each concentration above the LLOQ of ≤ 15%, a mean bias at the LLOQ of ± 20%, and a mean precision at the LLOQ of ≤ 20%.
The pharmacokinetic parameters assessed in all parts of the study included the following: maximum plasma concentration (Cmax), time to Cmax (Tmax), area under the plasma concentration–time curve (AUC) from time zero (dosing time) to the time of the last quantifiable concentration (AUClast), AUC from time zero to infinity (AUCinf), terminal half-life (T1/2), apparent volume of distribution during the terminal elimination phase following extravascular administration (Vz/F), and apparent systemic clearance from plasma following extravascular administration (CL/F).
In part A, serial blood samples for pharmacokinetic assessments were collected 2 h pre-dose and 0.25, 0.5, 0.75, 1, 1.5, 2, 2.5, 3, 3.5, 4, 6, 8, 12, 24, 36 and 48 h post-dose.
In part B, blood samples were collected 2 h pre-dose and 0.25, 0.5, 0.75, 1, 1.5, 2, 2.5, 3, 4, 6, 8 and 12 h (as a pre-evening dose) after the morning dose on day 1, pre-morning and pre-evening doses on day 2, pre-morning doses on days 3–13, and 2 h pre-dose and 0.25, 0.5, 0.75, 1, 1.5, 2, 2.5, 3, 4, 6, 8, 12, 24, 48, 72 and 120 h post-dose on day 14.
In part B, the following additional parameters were determined: Cmax following drug administration at steady state (Cmax,ss), Tmax at steady state (Tmax,ss), AUC during a dosing interval at steady state (AUCtau,ss), minimum plasma concentration observed during a dosing interval at steady state (Cmin,ss), AUC from time zero to the end of the dosing interval tau (AUCtau), average steady-state plasma concentration during multiple dosing (Cav,ss), percent fluctuation (Fluc), apparent volume of distribution at steady state during the terminal elimination phase following extravascular administration (Vz,ss/F), apparent systemic clearance from plasma at steady state following extravascular administration (CLss/F), T1/2 after the last dose (day 14), and the accumulation ratio (Racc).
In part C, blood samples were collected 2 h pre-dose and 0.25, 0.5, 0.75, 1, 1.5, 2, 2.5, 3, 4, 6, 8, 12, 24, 36 and 48 h post-dose in treatment periods 1 and 2, and at 72 h post-dose in treatment period 2. In part C, urine samples were also collected 2 h pre-dose and 3, 8, 24 and 48 h post-dose in treatment periods 1 and 2, and at 72 h post-dose in treatment period 2.
In addition to the plasma exposure and pharmacokinetic parameters, the amount of MHV370 excreted into the urine from time zero to 24 h (Ae0–24h) was determined by multiplying measured urine concentrations by measured urine volumes excreted, and renal clearance (CLr) was calculated by dividing Ae0–24h by the plasma AUC0–24h.
In all parts of the study, a 3 mL blood sample was collected in K2-EDTA tubes at each timepoint. All samples were then stored on dry ice at approximately − 70 °C until the time of analysis.
2.6 Pharmacodynamic Assessments
In part A, blood for pharmacodynamic marker assessment was collected at screening, 2 h pre-dose, and 1, 3, 8, 24, 48 and 120 h post-dose. In part B, blood was collected at screening, 2 h pre-dose, and 1, 3 and 8 h post-dose on day 1, pre-dose on day 2, post-evening dose on days 3, 4, 7 and 11, and 2 h before the morning dose and 1, 3, and 8 h post-dose on day 14.
Since TLR7 and TLR8 are not endogenously activated in healthy volunteers, pharmacodynamic biomarkers for MHV370 were measured following ex vivo stimulation of the TLR7/8 pathway. Whole blood samples from all participants in parts A and B were collected at the indicated time points and stimulated ex vivo with the TLR7/8 agonist R848. Expression of CD69 on B cells was used as a pharmacodynamic marker for TLR7, since B cells express TLR7 but not TLR8. Secretion of TNF was used as a pharmacodynamic marker for TLR8, since the most prominent TNF-secreting cells in blood are monocytes, which express TLR8 but not TLR7 [8, 11].
For the pharmacodynamic assay on TLR7, samples were analysed in duplicate. Heparinised whole blood was collected into S-Monovette tubes (Sarstedt). Whole blood (190 mL) was activated in 96 deep-well plates with 2 mg/mL R848 (Enzo) for 24 h in an incubator at 37 °C and 5% CO2. After stimulation, erythrocytes were fixed/lysed in 900 mL lysis buffer (BD FACS lysing solution) per well, mixed, and an additional 900 mL lysis buffer was added per well for 20 min at RT. Plates were frozen overnight at − 75 °C. After thawing, plates were briefly centrifuged (460g, 5 min) and the supernatants removed. Plates were washed in cell staining buffer (500 mL, Biolegend), and 50 mL blocking buffer (Human Trustain FcX 10× [Biolegend] diluted in cell staining buffer) was added to each well for 10 min. Cells were stained with the addition of 50 mL of CD69 staining cocktail, i.e. 1.5 mg/mL each of anti-human CD3, CD19, CD45 and CD69 antibodies (Clones sk7, HIB19, 2D1 and FN50 from Biolegend) in cell staining buffer. Plates were incubated for 30 min at RT in the dark and washed with cell staining buffer. Samples were fixed in 1% PFA in PBS, acquired on a FACS Canto II, and analysed using FACS DIVA software (version 8.0.2). Molecules of Equivalent Soluble Fluorochrome (MESF) data for CD69-positive CD19 cells were recorded.
For the pharmacodynamic assay on TLR8, whole blood was collected in Na-heparin S-Monovette tubes (Sarstedt). Immediately after collection, tubes were inverted at least eight times. Samples were diluted 9:1 into X-vivo medium (Lonza) and stimulated in duplicate with R848 at 1.5 mg/mL in 96-well plates and incubated for 22 ± 2 h at 37 °C and 5% CO2. Plates were placed on ice for 10 min and centrifuged (1000g, 10 min at 4 °C). Supernatants (50–100 mL) were transferred to Micronic U tubes and frozen at − 80 °C for at least 24 h prior to analysis. TNF was quantified using the MSD V-PLEX human TNF-ECLIA Kit (Meso Scale Discovery) according to the manufacturer’s protocol [20]. The LLOQ of TNF was 10.8 pg/mL.
2.7 Statistical Analysis
All the background and demographic variables and safety assessments were summarised using descriptive statistics. The safety analysis set included all the participants who received any study medication. For pharmacokinetics, the dose proportionality of single doses of MHV370 was assessed using data from part A. A power model was fitted for each of the pharmacokinetic parameters, including Cmax and AUClast (i.e. ln[PK] = m + β × ln[Dose] + e).
The accumulation of MHV370 was assessed using data from part B. The AUC0–12h from day 1 was compared to AUCtau,ss on day 14 using a linear fixed effect model. In part C, the food effect evaluation was performed using a linear fixed effect model containing fixed effects for sequence, treatment (fed and fasted), period, and participants within the sequence for log-transformed Cmax and AUClast.
The relationship between the doses of MHV370 and the pharmacokinetic parameters was determined from the estimate of the slope and the corresponding 90% confidence interval. The pharmacokinetic analysis set included all participants with at least one available and valid pharmacokinetic concentration measurement who received any study medication with no protocol deviations that impacted pharmacokinetic data. The percentage inhibition of TNF secretion and B-cell activation (compared to the baseline) are presented. The pharmacodynamic analysis set included all participants with available pharmacodynamic data and no protocol deviations with a relevant impact on pharmacodynamic data. The placebo participants in parts A and B were pooled together into one placebo group within each part.
3 Results
3.1 Participant Disposition
The study was conducted between June 25, 2018 and March 29, 2021. In total, 80 participants in part A (60 in the MHV370 and 20 in the placebo groups), 44 in part B (33 in the MHV370 and 11 in the placebo groups), and 12 in part C (6 each in the fed/fasted and fasted/fed groups) were enrolled and assigned to a study treatment. All participants in parts A and C completed the study; in part B, 39 of 44 (88.6%) completed the study and 5 discontinued due to the physician’s decision (n = 2), AE (n = 1), the sponsor’s decision (n = 1) and the participant’s decision (n = 1). All participants who enrolled in parts A, B and C were males, the mean age varied between 31 years and 41 years, and the mean BMI varied between 22 and 27 kg/m2 across the cohorts (Tables 1, 2). No relevant medical history was reported.
3.2 Safety and Tolerability
In parts A, B and C, 17 of 80 (21.3%), 20 of 44 (45.5%) and 4 of 12 (33.3%) participants experienced at least one AE. The most common AEs were nasopharyngitis, dry skin and headache of grade 1 severity (Table 3).
No deaths were reported in the study. No SAEs occurred in parts A and C of the study. In part B, two SAEs were reported. Both were considered unrelated to the study medication by the investigator. One participant who was enrolled in the lowest-dose cohort of part B (25 mg b.i.d.) reported during a follow-up phone call that he was hospitalised due to treatment of ureteric stone that occurred after the end of the study visit. A second SAE occurred in a participant enrolled in the second highest MAD cohort (200 mg b.i.d.) after the administration of the first dose. The participant experienced a panic attack during the night-time, which was treated accordingly. It was decided to discontinue dosing and to discharge the participant from the study unit in a mentally stable condition. During follow-up, the participant was hospitalised due to an anxiety disorder. The treating psychiatrist reported a pre-existing anxiety disorder that was not disclosed by the participant at the screening visit. Of note, the standardised assessment of all participants by CSSRS did not indicate any impact of MHV370 on suicidal ideation or behaviour.
No clinically relevant trends in vital signs, clinical chemistry or haematology parameters were observed. One sentinel participant in the 400 mg b.i.d. cohort in part B presented an elevated serum creatinine level (> 0.3 mg/dL) on day 3. Since a creatinine increase of > 0.3 mg/dL was a predefined discontinuation criterion, dose administration was stopped. The creatinine levels returned to normal on day 5. A creatinine increase could be the result of the inhibition of multidrug and toxic compound extrusion (MATE) transporters, as shown in pre-clinical testing (Supplementary Fig. S1). To rule out a potential impact of MHV370 on kidney function, creatinine-independent measures of kidney function (i.e. serum cystatin C and the glomerular filtration rate [mGFR] using iohexol infusion [13]) were introduced in the highest dose of the MAD cohort B5 (400 mg b.i.d.). The data confirmed that the observed mild serum creatinine elevation was not linked to a decrease in kidney function (Supplementary Figs. S2 and S3).
3.3 Pharmacokinetics of MHV370
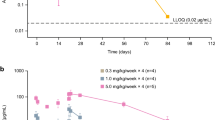
In part A, following single oral administration of MHV370 at a dose ranging from 1 to 1000 mg, the plasma concentrations of MHV370 increased with dose and achieved peak concentrations at a median Tmax ranging from 1.5 to 7 h after administration (Fig. 2A and Table 4). Following Cmax, the plasma concentration declined in a biphasic manner. The mean terminal half-life (T1/2) ranged from 8.75 to 14.6 h. The mean total systemic clearance (CL/F) ranged from 61.7 to 101 L/h. The mean volume of distribution (Vz/F) ranged from 867 to 1690 L. The Cmax and AUClast of MHV370 increased with increasing dose, but not in a dose-proportional manner at lower doses. For the higher doses (20–1000 mg), both parameters were dose proportional (Supplementary Fig. S4A and S4B).

Arithmetic mean (SD) plasma concentration–time profiles for MHV370 following A a single dose (part A)—semi-logarithmic scale and B multiple doses (part B)—semi-logarithmic scale; C food effect evaluation (part C)—linear scale. b.i.d. twice daily
In part B, following repeated oral administration of MHV370 over a period of 14 days from 25 to 400 mg b.i.d., the plasma concentrations of MHV370 increased with dose on both day 1 and day 14 and achieved peak concentrations at a median Tmax ranging from 1 to 2.5 h on day 1 and from 1.26 to 2 h on day 14 (Table 5). Following Cmax, the plasma concentrations declined in a biphasic manner (Fig. 2B). The mean terminal half-life (T1/2) ranged from 10.9 to 13.7 h. On day 14, mean plasma clearance (CL/F) at steady state ranged from 70.8 to 164 L/h and increased with increasing dose. The mean volume of distribution (Vss) at steady state on day 14 ranged from 1300 to 2570 L and increased with increasing dose. The adjusted geometric means of AUCtau day 14 and AUCtau day 1 increased with increasing dose, and the ratio of adjusted means, reflecting the rate of accumulation from day 1 to day 14, decreased with increasing dose and ranged from 2.53 to 0.804 (Table 5, Supplementary Fig. S4C and S4D).
In part C, the food effect was studied in 12 participants after the administration of 200 mg MHV370 together with a high-fat meal or under fasting conditions. The arithmetic mean plasma concentration–time plot per treatment is displayed in Fig. 2C. When MHV370 was administered after a high-fat breakfast, the median Tmax was delayed (fasted: 1.5 h and fed: 2.75 h), while T1/2 was comparable in the fasted state (11.3 h) and in the fed state (10.6 h). The statistical analysis of the effect of food on MHV370 pharmacokinetics is presented in Table 6. Although there was an increase in AUClast and Cmax in the fed vs fasted conditions, along with a delayed Tmax, those effects were not considered therapeutically significant.
3.4 Pharmacodynamics of MHV370
Time- and dose-dependent inhibition of CD69 expression on B cells was observed following ex vivo stimulation of blood of MHV370-dosed participants with the TLR7/8 agonist R848. This parameter was used as a TLR7-dependent pharmacodynamic biomarker, since B cells express TLR7 but not TLR8. At 24 h post-dose, 100% inhibition was observed at doses of MHV370 higher than 160 mg in part A (SAD cohort), and 50 mg b.i.d. in part B (MAD cohort) (Fig. 3A, B).

Arithmetic mean (SE) cellular biomarker time profiles: percentage inhibition of CD69 expression on B cells in part A of study (A) and part B of study (B), and TNF levels in part A of study (C) and part B of study (D). b.i.d. twice daily, MAD multiple ascending dose, SAD single ascending dose, TNF tumour necrosis factor, SE standard error
Inhibition of R848-mediated TNF release was used as a TLR8-dependent pharmacodynamic biomarker, since the main TNF producers in blood and monocytes expressed TLR8 but not TLR7. The inhibition of this marker was also time and dose dependent, with > 90% inhibition observed at 24 h post-dose with doses of MHV370 higher than 320 mg in part A (SAD) and 100 mg b.i.d. in part B (MAD) (Fig. 3C, D). Together, these results showed that MHV370 could be dosed in humans such that TLR7 and TLR8 are fully inhibited.
4 Discussion
TLRs are important for recognising PAMPs and, as such, play an important role in both the innate and adaptive immune systems to defend against pathogens. However, abnormal activation of them has been shown to play a role in the development of systemic autoimmune diseases (sAIDs). The most direct evidence implying TLR7 as a disease driver is from patients expressing TLR7 gain-of-function (GOF) mutations. One mutation in TLR7 (Y264H) has been identified in a child suffering from an autoimmune disease, and transgenic expression of this mutation in mice causes spontaneous severe lupus-like disease [4].
Antagonists of TLR7/8 and of downstream signalling nodes, e.g. IRAK4, are being evaluated as potential treatments for various autoimmune diseases. TLR7/8 antagonists in early stages of clinical development include MHV370, enpatoran (M5049), afimetoran (BMS-986256) and E6742 [15, 21,22,23]. Preclinical data with the TLR7/8 antagonist MHV370 demonstrate that it suppresses the expression of ISGs and lupus-like disease in vivo upon therapeutic dosing. Moreover, MHV370 blocks cytokine secretion from cells following stimulation with immune complexes of lupus patient sera and necrotic cell extract [11]. These preclinical data supported further investigation of MHV370 in a clinical setting.
This phase 1 study was undertaken to evaluate the safety and tolerability as well as the pharmacokinetics and pharmacodynamics of single and multiple ascending doses of MHV370 in healthy participants. The effect of food on the pharmacokinetics was also assessed after a single dose of MHV370. In the higher-dose cohorts of the MAD study, an increase in serum creatinine, which was still within the normal range, was observed in a subset of participants. Moreover, in participants who received a single dose of MHV370, serum creatinine increased towards the upper limit of the normal range yet normalised during the course of the study. No clinical symptoms or abnormalities in urinary analyses were apparent. Consequently, the protocol was amended to include creatinine-independent measures of kidney function, such as cystatin C and GFR measurements. The increase in creatinine, likely due to the inhibition of MATE transporters, was shown to be not associated with a decrease in glomerular filtration, as MHV370 did not affect cystatin c levels or iohexol clearance.
MHV370 was well tolerated and not associated with any unexpected safety concerns across the dose levels tested or with dose-limiting toxicity. TLR7-deficient individuals, unlike patients suffering from other primary immunodeficiency syndromes, do not suffer from a general susceptibility to infections, suggesting that inhibition of TLR7/8 signalling might confer inflammation control without broadly compromising host defence against pathogens [24]. While there is an association between the TLR7 loss of function mutation and severe COVID-19 [25], pharmacological inhibition differs substantially from genetic loss of function. Notably, a clinical trial for the treatment of patients with COVID-19 pneumonia with TLR7/8 inhibition (NCT04448756) has shown a numerical improvement in the treatment arm, though the primary efficacy objective was not met [26]. The data currently available are not considered to be a confirmation of either a benefit or risk of MHV370 use in COVID-19 disease.
Following oral administration of a single dose of up to 1000 mg MHV370 in part A and multiple doses of up to 400 mg MHV370 b.i.d. in part B, the plasma concentrations of MHV370 increased with dose. For the higher doses in part A (i.e. 20–1000 mg), the exposures were dose proportional, while the exposures were near or below LLOQ for the lower doses (i.e. 1–10 mg), so no full time–concentration courses were obtained. The biphasic decline of the plasma exposures may point to an initial tissue distribution with subsequent elimination. In part B, exposures after 14 days showed underproportional increases with higher doses. Total systemic clearance and volume of distribution at steady state on day 14 increased with increasing doses, suggesting increased metabolism, thus eventually leading to the underproportional exposures. In part C of the study, food intake was shown not to have a therapeutically relevant impact on the pharmacokinetic parameters of MHV370. Consequently, the drug can be administered orally regardless of food intake status.
Pharmacodynamic ex vivo analyses indicated time- and dose-dependent inhibition of TLR7-mediated CD69 expression on B cells and TLR8-mediated TNF release. An inhibition of > 90% of both pathways at trough serum exposure was achieved with doses of 100 mg MHV370 b.i.d. or above.
An important limitation of the study is that all data were generated in healthy participants; hence, the findings may not be generalised to patients with autoimmune disease having preactivated TLR7 and TLR8 pathways. In addition, the pharmacodynamic data after ex vivo stimulation by R848 may only be an imperfect approximation of the in vivo potency of MHV370 in humans.
Based on the data presented here, a phase 2 proof-of-concept study of MHV370 has been initiated to test its safety and efficacy in patients with Sjögren’s syndrome and mixed connective tissue disease (ClinicalTrials.gov Identifier: NCT04988087).
5 Conclusions
No safety signal was observed in healthy participants receiving MHV370 via up to 1000 mg single and up to 400 mg b.i.d. administration for 14 days. There is no relevant effect of food on the plasma pharmacokinetics of MHV370. The observed pharmacokinetic profile and the demonstration of the full pharmacodynamic inhibition of both the TLR7 and TLR8 pathways with a dose of 100 mg b.i.d. provide a rationale for conducting further studies to assess MHV370 as a treatment option for systemic autoimmune diseases.
References
Kawai T, Akira S. Signaling to NF-kappaB by Toll-like receptors. Trends Mol Med. 2007;13(11):460–9.
Akira S, Takeda K, Kaisho T. Toll-like receptors: critical proteins linking innate and acquired immunity. Nat Immunol. 2001;2(8):675–80.
Zhang SY, Jouanguy E, Sancho-Shimizu V, von Bernuth H, Yang K, Abel L, et al. Human Toll-like receptor-dependent induction of interferons in protective immunity to viruses. Immunol Rev. 2007;220(1):225–36.
Farrugia M, Baron B. The role of toll-like receptors in autoimmune diseases through failure of the self-recognition mechanism. Int J Inflam. 2017;2017:8391230.
Kelly-Scumpia KM, Nacionales DC, Scumpia PO, Weinstein JS, Narain S, Moldawer LL, et al. In vivo adjuvant activity of the RNA component of the Sm/RNP lupus autoantigen. Arthritis Rheum. 2007;56(10):3379–86.
Roers A, Hiller B, Hornung V. Recognition of endogenous nucleic acids by the innate immune system. Immunity. 2016;44(4):739–54.
Deane JA, Pisitkun P, Barrett RS, Feigenbaum L, Town T, Ward JM, et al. Control of toll-like receptor 7 expression is essential to restrict autoimmunity and dendritic cell proliferation. Immunity. 2007;27(5):801–10.
Guiducci C, Gong M, Cepika AM, Xu Z, Tripodo C, Bennett L, et al. RNA recognition by human TLR8 can lead to autoimmune inflammation. J Exp Med. 2013;210(13):2903–19.
Junt T, Barchet W. Translating nucleic acid-sensing pathways into therapies. Nat Rev Immunol. 2015;15(9):529–44.
Brown GJ, Cañete PF, Wang H, Medhavy A, Bones J, Roco JA, et al. TLR7 gain-of-function genetic variation causes human lupus. Nature. 2022;605(7909):349–56.
Hawtin S, André C, Collignon-Zipfel G, Appenzeller S, Bannert B, Baumgartner L, et al. Preclinical characterization of the Toll-like receptor 7/8 antagonist MHV370 for lupus therapy. Cell Rep Med. 2023;4(5): 101036.
Gaspari F, Thakar S, Carrara F, Perna A, Trillini M, Aparicio MC, et al. Safety of iohexol administration to measure glomerular filtration rate in different patient populations: a 25-year experience. Nephron. 2018;140(1):1–8.
Delanaye P, Ebert N, Melsom T, Gaspari F, Mariat C, Cavalier E, et al. Iohexol plasma clearance for measuring glomerular filtration rate in clinical practice and research: a review. Part 1: how to measure glomerular filtration rate with iohexol? Clin Kidney J. 2016;9(5):682–99.
Gaspari F, Perico N, Ruggenenti P, Mosconi L, Amuchastegui CS, Guerini E, et al. Plasma clearance of nonradioactive iohexol as a measure of glomerular filtration rate. J Am Soc Nephrol. 1995;6(2):257–63.
Nakai K, Yasuda S, Chang M-K, Matijevic M, McGrath S, Yang H, et al. Safety, pharmacokinetics, and pharmacodynamics in first-in-human study of a novel compound E6742, a Toll-like receptor 7 and 8 antagonist. In: WCP2018 (18th World Congress of Basic and Clinical Pharmacology); 2018 Jul 1–6; Kyoto, Japan (WCP2018:PO1-11-3).
European Medicines Agency. Reflection paper for laboratories that perform the analysis or evaluation of clinical trial samples (EMA/INS/GCP/532137/2010). 2012. https://www.ema.europa.eu/en/documents/regulatory-procedural-guideline/reflection-paper-laboratories-perform-analysis-evaluation-clinical-trial-samples_en.pdf. Accessed 22 June 2023.
US Food and Drug Administration. Guidance for industry—bioanalytical method validation. 2018. https://www.fda.gov/files/drugs/published/Bioanalytical-Method-Validation-Guidance-for-Industry.pdf. Accessed 22 June 2023.
European Medicines Agency. Guideline on bioanalytical method validation (EMEA/CHMP/EWP/192217/2009 Rev. 1 Corr. 2). 2011. https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-bioanalytical-method-validation_en.pdf. Accessed 22 June 2023.
OECD. OECD series on principles of good laboratory practice and compliance monitoring, number 1 (ENV/MC/CHEM(98)17). OECD principles on good laboratory practice. 1998. https://one.oecd.org/document/ENV/MC/CHEM(98)17/en/pdf. Accessed 22 June 2023.
Meso Scale Discovery (MSD). Multi Spot Assay System manual: Proinflammatory Panel 1 (Human) kits. Document # 18094-v5-2020Jan 2. 2020. https://www.mesoscale.com/~/media/files/product%20inserts/proinflammatory%20panel%201%20human%20insert.pdf. Accessed 22 June 2023.
Dudhgaonkar S RA, Ranade S, Subramani S, Nagar J, Karunanithi P, Bhutani P, Kurawattimath V, Zhang R, Qiu H, Dyckman A, Schieven G. Inhibition of toll-like receptor 7 (TLR7) with the potent and selective inhibitor of human TLR7 and TLR8 BMS-986256 provides robust efficacy in murine lupus models, reversing established disease [abstract #0470]. Arthritis Rheumatol. 2021;73 (suppl 10).
Port A, Shaw JV, Klopp-Schulze L, Bytyqi A, Vetter C, Hussey E, et al. Phase 1 study in healthy participants of the safety, pharmacokinetics, and pharmacodynamics of enpatoran (M5049), a dual antagonist of toll-like receptors 7 and 8. Pharmacol Res Perspect. 2021;9(5): e00842.
Vlach J, Bender AT, Przetak M, Pereira A, Deshpande A, Johnson TL, et al. Discovery of M5049: a novel selective toll-like receptor 7/8 inhibitor for treatment of autoimmunity. J Pharmacol Exp Ther. 2021;376(3):397–409.
Fillatreau S, Manfroi B, Dörner T. Toll-like receptor signalling in B cells during systemic lupus erythematosus. Nat Rev Rheumatol. 2021;17(2):98–108.
Asano T, Boisson B, Onodi F, Matuozzo D, Moncada-Velez M, MagloriusRenkilaraj MRL, et al. X-linked recessive TLR7 deficiency in ~1% of men under 60 years old with life-threatening COVID-19. Sci Immunol. 2021;6(62):eabl4348.
Mckinnon JE, Santiaguel J, Murta C, Yu D, Khursheed M, Moreau F, et al. Phase II trial of enpatoran in patients hospitalized with COVID-19 pneumonia. Ann Rheum Dis. 2022;81:971–2.
Acknowledgements
The authors thank Ramji Narayanan, M.Pharm, and Divya Chandrasekhar, PhD (Novartis Healthcare Pvt Ltd, Hyderabad, India), for providing medical writing support, which was funded by Novartis Pharma AG, Basel, Switzerland, in accordance with Good Publication Practice (GPP3) guidelines (http://www.ismpp.org/gpp3).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Author Contributions
Conception and design: TS, SH, PG, RF, TJ, AA. Collection of data: TS, RF, MGP, JL, AA. Data analysis and interpretation: TS, SH, RF, TJ, PG, RD, RS. Manuscript writing and reviewing: TS, JS, SH, RF, TJ, MGP, JL, RS. Final approval of manuscript: all authors.
Funding
The study was funded by Novartis Pharma AG.
Data Availability Statement
Novartis is committed to sharing with qualified external researchers access to patient-level data and supporting clinical documents from eligible studies. These requests are reviewed and approved by an independent review panel on the basis of scientific merit. All data provided are anonymised to respect the privacy of participants who have participated in the trial in line with applicable laws and regulations.
Conflict of Interest
TS, RF, TJ, SH, JS, AA, RD, PM, RS and PG are currently employees of Novartis Pharma AG. MGP and JL were employees of Charité Research Organisation GmbH. MGP is currently an employee of Scirent Clinical Research and Science. JL is currently an employee of Pfizer.
Registration
The study was registered on EudraCT (EudraCT number: 2017-004559-21).
Ethics Approval
The study protocol was reviewed and approved by the independent ethics committee (Landesamt für Gesundheit und Soziales (LAGeSo); Geschäftsstelle der Ethik-Kommission des Landes, Berlin) (approval date: 17 May 2018).
Consent to Participate
The study was conducted at a single clinical site (Charité Research Organization, Berlin, Germany) in accordance with the ethical principles of the Declaration of Helsinki, Good Clinical Practice standards, and applicable regulatory requirements. All participants provided written informed consent before the study initiation.
Consent for Publication
Not applicable.
Code Availability
Not applicable.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Shisha, T., Posch, M.G., Lehmann, J. et al. First-in-Human Study of the Safety, Pharmacokinetics, and Pharmacodynamics of MHV370, a Dual Inhibitor of Toll-Like Receptors 7 and 8, in Healthy Adults. Eur J Drug Metab Pharmacokinet 48, 553–566 (2023). https://doi.org/10.1007/s13318-023-00847-3
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13318-023-00847-3